

Abstract
The proline-rich homeodomain protein, PRH/HEX, participates in the early development of the brain, thyroid, and liver and in the later regenerative processes of damaged liver, vascular endothelial, and hematopoietic cells. A virulent strain of lymphocytic choriomeningitis virus (LCMV-WE) that destroys hematopoietic, vascular, and liver functions also alters the transcription and subcellular localization of PRH. A related virus (LCMV-ARM) that does not cause disease in primates can infect cells without affecting PRH. Biochemical experiments demonstrated the occurrence of binding between the viral RING protein (Z) and PRH, and genetic experiments mapped the PRH-suppressing phenotype to the large (L) segment of the viral genome, which encodes the Z and polymerase genes. The Z protein is clearly involved with PRH, but other viral determinants are needed to relocate PRH and to promote disease. By down-regulating PRH, the arenavirus is able to eliminate the antiproliferative effects of PRH and to promote liver cell division. The interaction of an arenavirus with a homeodomain protein suggests a mechanism for viral teratogenic effects and for the tissue-specific manifestations of arenavirus disease.
The proline-rich homeodomain (PRH) protein, also known as hematopoietically expressed homeobox (HEX), belongs to a family of transcription factors that regulate development. In mice, PRH is essential for the development of the embryonic forebrain, thyroid, lungs, and liver and is expressed in the adult thyroid, lungs, and liver (6, 28, 43). Gene targeting studies with mice identified PRH as being among the earliest genes required for liver development (22, 43, 47, 65). PRH belongs to the subset of homeobox genes that are expressed in adult tissues (33, 41) and that play a major role in the regenerative processes of vascularization and hematopoiesis (2, 15, 27, 46, 60). Within the hematopoietic compartment, PRH is expressed in pre-B cells and myeloid and erythroid cells, but not in T-cell lineages (5, 7, 14, 40, 42). PRH is expressed preferentially in myelocytes and hepatocytes during liver injury and regeneration (1, 24, 48, 57). It is remarkable that these same liver, vascular endothelial, and hematopoietic cells are the major target cells for hemorrhagic fever viruses (55).
The infection of primates with the WE strain of lymphocytic choriomeningitis virus (LCMV-WE) leads to a disease resembling Lassa hemorrhagic fever in human beings (38). LCMV infections often involve several systems, e.g., the central nervous system, the respiratory system, and the hematopoietic system (11), but the liver is the site of the highest rate of virus replication (26, 38) and the most prominent necropsy finding in LCMV-WE-infected monkeys and Lassa fever patients (38, 44).
Although there is insufficient histological damage in the liver to account for fatality, liver-derived molecules affect hematopoiesis and coagulation, and thus, small hepatic malfunctions have systemic consequences (1, 39, 65).
We used an experimental monkey model to study the molecular basis of hematopoietic failure and liver disease after LCMV-WE infection (38, 39, 50). Our working hypothesis is that virulent arenaviruses cause liver dysfunction by inhibiting physiological repair processes. An acute arenavirus infection is accompanied by liver cell proliferation, similar to the response after hepatectomy (39). Our experiments for this study revealed the inhibitory effect of viral infection on PRH expression, which is needed for differentiation and repair in both liver and hematopoietic systems.
Although the studies described here used liver cells, they follow previous studies of myeloid cells that linked PRH suppression to the control of cellular proliferation. PRH has direct effects on transcription (57), but its ability to regulate posttranscriptional events impacts cell division (62). Like several other homeodomain proteins, PRH interacts directly with nuclear eukaryotic initiation factor 4E (eIF4E) to suppress the eIF4E-mediated transport of cyclin D1 mRNA (62). The down-regulation of PRH activates eIF4E-mediated mRNA transport and initiates cellular proliferation. Elevated eIF4E is found in many human malignancies, including primary leukemias and lymphomas, and indicates a proliferative state (56, 61). The proliferative state precludes maturation and results in the accumulation of undifferentiated cells, e.g., immature B cells accumulate in Hex knockout mice (7), and liver development stops in Hex-negative embryos and chimeric mice (6, 24, 28, 43). Homeodomain proteins such as PRH may be critical targets for viruses that affect major organ systems.
Arenaviruses are bisegmented negative-strand RNA viruses that encode five proteins, including a nucleocapsid protein (NP), two envelope glycoproteins (GP-1 and GP-2), an RNA polymerase (L protein), and a small zinc-binding protein (Z protein) (55). The Z protein binds zinc through its RING domain, which is highly conserved among the arenaviruses (10, 17) and is involved in virus structure, replication, and assembly (20, 25, 36, 53, 54). Yeast two-hybrid and coprecipitation analyses demonstrated that the viral Z protein binds PRH (59). For this study, we used two viral strains and reassortants that contain a genome segment from each strain in order to map the PRH-suppressing effects to the large genome segment (encoding the Z protein and the polymerase).
For the present study, we examined the subcellular localization, expression, and function of PRH in hepatic cell lines and in monkey livers infected with virulent and nonvirulent isolates of LCMV. Our results show that the viral suppression of PRH coincides with the liver disease observed during arenaviral hemorrhagic fever and that this suppression provides a likely disease mechanism.
MATERIALS AND METHODS
Virus stocks and virus titration.
Isolates of the arenaviruses LCMV-ARM (Armstrong 53b strain) and LCMV-WE were plaque purified and stored at 107 to 108 PFU/ml. The reassortant viruses WE/ARM (carrying the L segment of WE and the S segment of Armstrong) and ARM/WE (carrying the L segment of WE and the S segment of Armstrong) were also plaque purified and were stored at 2 × 107 and 5 × 106 PFU/ml, respectively (49). All virus stocks were titrated by plaque assays on Vero E6 cells as described previously (50). To assess virus production in hepatic cells, we plated cells at 2 × 105/well in six-well plates for 5 h at 37°C and then infected them with virus at a multiplicity of infection of 1 PFU per cell. At various times, the culture medium was freeze-thawed and centrifuged to remove cell debris. The supernatants were serially diluted, and virus titers were determined by a plaque assay on Vero E6 cells and were expressed as PFU per milliliter of supernatant.
Rhesus macaques, LCMV inoculations, and collection of blood and tissue.
Animals were inoculated intravenously with 103 PFU of the benign LCMV-ARM or with a lethal dose of LCMV-WE (103 PFU) as previously described (38). Blood samples and liver biopsies were obtained from infected animals, and tissue samples were obtained at necropsy. Biosafety and animal use protocols were approved by University of Maryland internal review boards and were compliant with the American Veterinary Medicine Association Panel on Euthanasia. Paraffin-embedded liver samples were used for the immunohistochemical localization of vascular endothelial growth factor (VEGF), PRH, Ki-67, or viral antigen. Both blood and liver specimens were used for total RNA and protein isolations as described below.
Cell culture.
Human hepatoma Huh7 (American Type Culture Collection, Rockville, Md.) and HepG2 (ATCC HB-8065) cells were maintained in minimal essential medium (GIBCO-BRL, Grand Island, N.Y.) supplemented with penicillin G (100 U/ml), streptomycin (100 U/ml), glutamine (2 mM), and 10% heat-inactivated fetal bovine serum (FBS) at 37°C in a humidified atmosphere of 5% CO2. Vero E6 cells were cultivated in Dulbecco's modified Eagle's medium (GIBCO-BRL) supplemented with 10% FBS, penicillin (100 U/ml), streptomycin (100 U/ml), and l-glutamine (2 mM). Peripheral blood mononuclear cells were purified from EDTA-treated blood of monkeys on Ficoll-Hypaque and then were cultivated in six-well culture plates in RPMI 1640 with 10% fetal calf serum. Cell lines were also plated in 12-well plates (105 cells/well) for transient transfections and β-galactosidase expression assays or in T25 flasks (2 × 106 cells/flask) for the isolation of total cell extracts and RNAs. Normal human hepatocytes were obtained from Clonetics (Gaithersburg, Md.) and were cultivated according to the manufacturer's protocol. For cell growth experiments, 2 × 105 cells per well were plated in six-well plates in minimal essential medium with 10% FBS and then were counted. Cells were grown on coverslips for immunofluorescence analysis.
Immunofluorescence and laser scanning confocal microscopy.
Microscopy was performed as described previously (13, 60). Briefly, hepatic cells grown on coverslips were rinsed two times in 1× phosphate-buffered saline (PBS; 9.1 mM dibasic sodium phosphate, 1.7 mM monobasic sodium phosphate, 150 mM NaCl, pH 7.4). Adherent cells were fixed either by submersion in 100% acetone at room temperature for 1 min or by exposure to 3.7% paraformaldehyde at room temperature, followed by permeabilization with blocking buffer (10% fetal bovine serum and 0.1% Tween 20 in 1× PBS). Both fixation protocols gave identical results. After blocking, the coverslips were incubated with primary antibodies diluted in blocking buffer for 2 h at room temperature. The primary antibodies were affinity-purified rabbit anti-PRH (1:50), affinity-purified rabbit anti-Z (1:50), mouse monoclonal anti-eIF4E (1:20; Transduction Laboratories), and mouse monoclonal anti-promyelocytic leukemia protein or PML (MAb 5E10; 1:20). After incubation with primary antibodies, the cells were washed three times with 1× PBS and then probed with secondary antibodies diluted in blocking buffer for 1 h at room temperature. The secondary antibodies were fluorescein isothiocyanate (FITC)-conjugated donkey anti-rabbit (Dakopatts, Stockholm, Sweden, or Jackson Laboratory, West Grove, Pa.), Texas Red-conjugated donkey anti-rabbit (Dakopatts), FITC-conjugated donkey anti-mouse, and Cy5-conjugated donkey anti-mouse (Jackson Laboratory). For triple PML-Z-PRH staining, we used Cy5-conjugated, affinity-purified rabbit anti-PRH diluted in blocking buffer (1:20) overnight at 4°C. Later, the antibody was conjugated by the use of a FluoroLink Antibody Cy5 labeling kit (Amersham) according to the manufacturer's instructions. After incubation with secondary antibodies, the cells were washed three times with 1× PBS, dried, mounted in Vectashield mounting medium with DAPI (4′,6′-diamidino-2-phenylindole; Vector Laboratories), and sealed.
Fluorescence was observed at a magnification of ×100 with a zoom of 2, unless indicated otherwise, under a Leica TCS-SP (UV) confocal microscope with excitation at 488, 568, 633, or 351/364 nm. All channels were detected separately, with no cross talk between channels. Micrographs represent single sections with a thickness of 300 nm. Each confocal microscope experiment was performed at least twice, and the number of cells in each sample exceeded 500. Images were overlaid in Photoshop and enlarged for counting of the PML, PRH, and Z bodies found within each nucleus and for recording of the percentages of bodies that colocalized. One can distinguish whether one or both antigens are present by overlaying confocal microscope images: nuclear bodies that contain only one antigen stain red or green, and nuclear bodies that contain both antigens appear yellow.
Immunohistochemistry to detect VEGF and PRH antigens in tissues.
Immunohistochemistry was performed as previously described (39). Briefly, formalin-fixed, paraffin-embedded tissue sections were deparaffinized, unmasked with heat, blocked with normal human serum, and incubated with anti-human VEGF (R&D Systems, Minneapolis, Minn.) (1:200 dilution) or affinity-purified anti-PRH (HEX, or P-21; Santa Cruz Biotechnology, San Diego, Calif.) (1:100 dilution). Hyperimmune guinea pig serum (1:50 dilution) was used as a primary antibody to detect LCMV antigens. Ki-67 is a nuclear antigen that marks cells undergoing division (39), so we used polyclonal rabbit anti-Ki-67 (Zymed, San Francisco, Calif.) at a 1:50 dilution to detect proliferating cells. Sections were washed in PBS and incubated for 1 h at room temperature with a biotin-labeled goat anti-guinea pig or goat anti-rabbit serum (Sigma Chemical Co., St. Louis, Mo.). Secondary antibodies were visualized with streptavidin-conjugated peroxidase according to the manufacturer's recommendations for use of a cell and tissue staining kit (R&D Systems).
RT-PCR analysis.
Total RNAs were prepared from infected or uninfected hepatic cells (a maximum of 2 × 106 cells) or from 50 to 100 mg of liver tissue. Cultured cells were subjected to Trizol extraction (GIBCO-BRL), whereas tissues were homogenized by the use of 16-ml glass tissue grinders (VWR, Bridgeport, N.J.) and then were extracted with Trizol. A Qiagen RNase-free DNase supplement kit was used to ensure that the RNAs had no DNA contamination. RNA quantities were determined by measuring the absorbance at 260 nm. Amplification of the first cDNA was performed and analyzed by real-time PCR as described previously (18, 38). In this study, we show for the first time that the rhesus PRH gene can be amplified by the use of human PRH gene primers (GenBank accession no. L16499) (5′-AGGAAAGGCGGCCAGGTGAG-3′ and 5′-TTATTGCTTTGAGGGTTCTCCTG-3′). Specific primer pairs for the LCMV-WE glycoprotein (covering nucleotides 55 to 73 and 342 to 323; GenBank accession no. M22138) and the LCMV-ARM glycoprotein (covering nucleotides 118 to 137 and 364 to 344; GenBank accession no. M22138) were designed to discriminate the possible cross-contamination of viral RNA. Primers for a housekeeping gene, human glyceraldehyde-3-phosphate dehydrogenase (GAPDH; GenBank accession no. M17851) (sense strand, 5′-GTTGCCATCAATGACCCCTTCATTG-3′; antisense strand, 5′-CAGCCTTCTCCATGGTGGTG-3′), were used to control for the integrity and amount of RNA in each sample. Oligonucleotide primers that anneal to 18S rRNA served as an additional internal control. Reverse transcription-PCR (RT-PCR) products were analyzed by 1% agarose gel electrophoresis.
The level of PRH mRNA was evaluated by the use of SYBR green technology, and amplification plots were expressed as CT values to be analyzed with 5700 SDS software (Perkin-Elmer Systems). The CT is the reaction cycle at which PCR products or amplicons reach a threshold level of detection: the lower the CT value, the more abundant the substrate. CT values were normalized by using GAPDH or rRNA 18S amplicons as standards. Dissociation analysis of the PCR products was used to confirm specificity.
Western blot analysis.
Guinea pig polyclonal anti-LCMV and rabbit anti-Z antibodies were produced as described previously (54). For Western blot analysis, 3 × 106 cells were lysed in 0.5 ml of lysis buffer (PBS containing 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, and protease inhibitors). Each sample was sonicated to reduce its viscosity. Western blot analysis was performed according to standard procedures (23). For each lane, 30 μg of protein (8 × 104 cell equivalents) was fractionated by sodium dodecyl sulfate-10% polyacrylamide gel electrophoresis, followed by Coomassie blue staining or semidry electroblotting onto a polyvinylidene difluoride membrane (Millipore). Membranes were blocked with 5% (wt/vol) nonfat milk for 30 min at room temperature. Guinea pig polyclonal anti-LCMV or mouse anti-PML antibodies and affinity-purified anti-PRH antibodies were diluted 1:1,000 in Tris-buffered saline (TBS) containing 0.2% (vol/vol) Tween 20 and were used as primary antibodies. Blots were incubated with primary antibodies overnight at 4°C and were then washed twice in TBS-Tween. The secondary antibody, which was a 1:10,000 dilution of anti-mouse immunoglobulin G (IgG) or anti-guinea pig IgG conjugated to horseradish peroxidase (Sigma), was incubated with blots for 60 min. Blots were washed twice in TBS-Tween and then visualized by enhanced chemiluminescence (Pierce). Band intensities were measured by densitometry on a densitometer and were analyzed with ImageQuant software (Molecular Dynamics, Sunnyvale, Calif.).
Plasmid constructs and transfections.
Plasmids containing the full-length LCMV-ARM, LCMV-WE, and Lassa Z open reading frames were constructed by ligating virus-derived cDNAs into pCRII (Invitrogen, Carlsbad, Calif.) and then excising them with HindIII and XhoI for subcloning into pcDNA3 (Invitrogen). The plasmid pGFP (Invitrogen), from which a green fluorescent protein (GFP) reporter gene can be expressed from a cytomegalovirus (CMV) promoter (pGFP) in pCDNA3, was used as a transfection control. For assays of PRH transcription function, we used the plasmid p4×HRE-βGAL, containing four copies of the PRH/HEX response element (HRE), as previously described (16). The HRE was derived from the promoter of the sodium-dependent bile acid transporter (ntcp) gene. Thus, the plasmid used to assess the PRH transcription function consisted of the ntcp promoter and the reporter gene encoding β-galactosidase (β-Gal).
Cells were transfected at 50% confluence by use of the GenePORTER reagent as specified by the manufacturer (Gene Therapy Systems, San Diego, Calif.). Briefly, cells were transfected with 0.5 μg of plasmid for 24 h at 37°C and then were infected for the times indicated in the figure legends. Data are given as means ± standard errors of at least three independent experiments, each of which was performed in duplicate wells.
Assay of PRH function.
The bile acid transporter (ntcp) gene promoter is a target of PRH and is thus a useful tool for evaluating its function. To determine whether LCMV infection affects PRH functions, we assayed infected hepatic cells that had been transfected with reporter constructs for β-Gal expression. HepG2 cells were grown in 12-well plates (2 × 105 cells/well) for transient transfections and β-Gal expression assays. The cells were transfected, as described above, with 500 ng of p4×HRE-βGAL or a control plasmid, pGL3 (containing a simian virus 40 promoter and β-Gal; Promega, Madison, Wis.). Total protein aliquots were stored at −70°C until they were assayed. β-Galactosidase assays were done according to the manufacturer's instructions (Promega). The amounts of cell extracts used for activity assays were adjusted for the optimal assay range of β-Gal activity.
RESULTS
Virulent LCMV-WE and nonvirulent LCMV-ARM replicate similarly in hepatic cell lines.
Our experimental approach was to document viral effects on PRH expression, localization, and function. To relate these events to hepatic disease, we compared the effects of virulent and nonvirulent strains of LCMV. Virulence was operationally defined as the ability to cause acute disease in guinea pigs and primates. It was first necessary to determine whether primary liver cells and hepatic cell lines were similarly capable of supporting virus replication. Cell growth rates (not shown) slowed down in the same way upon infection by either virus, and virus production (Table 1) was supported by all hepatic cells infected with either LCMV-WE or LCMV-ARM.
TABLE 1.
Replication of LCMV-WE and LCMV-ARM in hepatic cellsa
| Virus | Time of infection (h) | Titer (PFU/ml) in cellsb
|
Titer (PFU/g) in liverc | ||
|---|---|---|---|---|---|
| HepG2 | Huh7 | NHep | |||
| LCMV-ARM | 16 | Not done | Not done | 3.6 × 103 | |
| 24 | 3.8 × 105 | 1.0 × 106 | 3.2 × 104 | ||
| 48 | 7.0 × 106 | 1.0 × 107 | 5.8 × 104 | ||
| 72 | 7.0 × 107 | 6.4 × 106 | Not done | ||
| Day 11 or 12 | <1.5 × 102 | ||||
| LCMV-WE | 16 | Not done | Not done | 2.6 × 105 | |
| 24 | 1.1 × 106 | 1.6 × 106 | 4.2 × 105 | ||
| 48 | 2.6 × 106 | 1.8 × 106 | 1.1 × 106 | ||
| 72 | 2.7 × 107 | 7.2 × 105 | Not done | ||
| Day 11 or 12 | 1.0 × 106 | ||||
HepG2 and Huh7 are hepatoma cell lines. NHep are normal human liver cells (cadaver derived). The last data column is for liver tissue taken at necropsy (days 11 and 12 after infection) from monkeys infected with LCMV-ARM or LCMV-WE. All data represent averages of at least two independent titration experiments, and the variation was within 1 order of magnitude.
The numbers of PFU per milliliter were determined from the medium surrounding hepatic cell cultures. The amount of cell-associated virus was <10%, so only the amount of released virus in the medium is shown. Virus-containing medium was titrated on Vero cell monolayers at six different dilutions, as described in Materials and Methods.
Viruses in liver tissue were titrated on Vero cell monolayers.
Primary hepatocytes, isolated by proteolytic digestion of cadaver tissues, were too traumatized to be used for most cell culture studies. The virus titration experiments were replicated several times to confirm that the replication of LCMV-WE was slightly higher in primary liver cells than the replication of LCMV-ARM. The phenomenon we described for liver cell lines (cell cycle perturbation by the virus) was overwhelmed in primary cells by efforts to regain homeostasis. The primary cells did not last long in culture, and their dying also confused any results. Therefore, most of our conclusions were limited to what could be observed in liver tissue and liver cell lines. The numbers supplied in Table 1 to compare titers of WE and Armstrong in monkey liver tissues were derived from matched animals.
Infection with LCMV-WE causes PRH to disappear from hepatic nuclei.
PRH functions in the nucleus as a transcription factor and a suppressor of eIF4E-mediated mRNA transport (21, 61, 62). To determine whether virus infection affects PRH, we infected human hepatic cell lines with virulent LCMV-WE or nonvirulent LCMV-ARM for 24, 48, and 72 h. Confocal microscopy was used to differentiate between nuclear and cytoplasmic localizations of PRH. The virulent LCMV-WE infection altered PRH distribution and reduced its signal similarly in two human hepatic cell lines, HepG2 and Huh7 cells (Fig. 1). Both cell lines were stained with an affinity-purified antibody to monitor endogenous human PRH bodies. Staining for PML and eIF4E served as a contrast for PRH staining in our confocal studies, as PML and/or eIF4E sometimes colocalized with PRH. Uninfected cells showed the punctate nuclear pattern that is characteristic of PML staining and had punctate and diffuse nuclear and cytoplasmic staining, respectively, for PRH. As reported previously (62), most of the PRH nuclear structures colocalized with PML/eIF4E bodies. For PML staining, there was a dramatic difference between the punctate (nuclear) appearance in the top three panels (Fig. 1A) and the more diffuse (cytoplasmic) staining in the bottom panel, showing that both viral infections equally affected PML bodies in HepG2 and Huh7 hepatic cell lines, as previously observed for infected fibroblasts (10). Neither infection with LCMV-WE nor infection with LCMV-ARM affected the distribution of eIF4E bodies, which is consistent with our previous findings (12).
FIG. 1.


Confocal images of intracellular localization of PRH. Hepatic HepG2 or Huh7 cells were grown on coverslips and infected with LCMV as described in Materials and Methods. (A) Infected or uninfected HepG2 cells were stained with anti-PRH (red), anti-PML (blue), or anti-eIF4E (green) to determine how infection affects the localization of PML, PRH, and eIF4E in hepatic cells. Huh7 cells (B) or HepG2 cells (C) were stained with anti-PRH (red), anti-PML (blue), or anti-LCMV Z (green). All cells were counterstained for DNA with DAPI (gray). The overlay is shown in yellow (OV), and the overlay of DAPI staining is designated OV+DAPI. These confocal images represent single optical slices through the cells. Magnification, ×300. FITC and Texas Red channels were recorded independently.
LCMV-WE reduced the expression of nuclear PRH and dispersed PRH bodies at the same rate in HepG2 and Huh7 cells. There were no differences in PRH staining between uninfected cells and cells infected with LCMV-ARM for 24, 48, or 72 h. After 72 h of infection, <10% of LCMV ARM-infected cells had lost PRH staining in the nucleus. In contrast, 100% of WE-infected cells had lost nuclear PRH by 72 h (Fig. 1). Both LCMV-WE and LCMV-ARM caused a redistribution of PML from nuclear bodies to diffuse/punctate cytoplasmic staining, as previously reported for fibroblasts (10, 29). Thus, PRH redistribution and PML redistribution were unlinked and were affected by different mechanisms.
The Z protein is associated with PRH bodies in cells infected with LCMV.
We demonstrated previously by yeast two-hybrid analysis and immunoprecipitation that PRH can bind the arenavirus Z protein (58). In order to demonstrate this association in situ, we grew hepatic cells on coverslips and infected them as described in Materials and Methods. Infected cells were stained with an affinity-purified LCMV-Z antibody, giving a staining pattern similar to that for PML, with predominant punctate cytoplasmic staining and some nuclear staining (Fig. 1B and C and Fig. 2). Double-staining experiments indicated that the PRH and Z bodies were colocalized. After 48 h of infection with LCMV-WE, almost all of the PRH was found in the cytoplasm and colocalized with the Z protein. LCMV-ARM-infected cells were indistinguishable from uninfected controls (Fig. 1 and 2). In addition to the diffuse cytoplasmic and nuclear distribution, there were distinct PRH bodies that remained associated with eIF4E bodies in the nuclei of control cells. Some of these structures were colocalized with Z (yellow in the overlay) throughout the time course. Yeast two-hybrid (59), coprecipitation (59; M. Djavani, unpublished), and now microscopy analyses have all indicated that PRH interacts with the Z proteins of both LCMV-WE and LCMV-ARM, yet PRH was disrupted only in cells infected with LCMV-WE.
FIG. 2.

The L segment of LCMV-WE controls the disappearance of PRH from liver cell nuclei. HepG2 cells were infected with LCMV reassortants (ARM/WE or WE/ARM) as described in Materials and Methods. The cells were stained with anti-Z (green) and anti-PRH (red) or anti-PML (blue). All cells were counterstained for DNA with DAPI. The effects of reassortant viruses on PRH expression and localization were determined by confocal microscopy. Hepatic cells infected with LCMV carrying the L RNA segment of WE had the same disruptive effects on PRH distribution as those infected with LCMV-WE (WE/WE) (similar to Fig. 1A and B). However, the hepatic cells infected with LCMV carrying the L RNA segment of ARM had no effect on the subcellular distribution of PRH (similar to Fig. 1). Magnification, ×300. The overlay is shown in yellow (OV), and the overlay of DAPI staining is designated OV+DAPI. FITC and Texas Red channels were recorded independently.
The ability to disrupt nuclear PRH bodies maps to the large (L) segment of LCMV-WE.
Arenaviruses have two genome segments, the large (L) and small (S) segments, that can be interchanged between strains to form genetic reassortants. To determine which viral genome segment is responsible for the PRH-disrupting phenotype, we infected hepatic cells with reassortants containing the L segment of ARM and the S segment of WE (ARM/WE) or the L segment of WE and the S segment of ARM (WE/ARM). Cells were grown on coverslips and infected as described in Materials and Methods. The effect of reassortant viruses on PRH expression was determined by confocal microscopy (Fig. 2). HepG2 cells infected with LCMV carrying the L RNA segment of WE had the same disruptive effects on PRH distribution as the parental virus (LCMV-WE) (Fig. 1). However, cells infected with LCMV carrying the L RNA segment of ARM had no effect on the subcellular distribution of PRH (Fig. 2). Therefore, genes carried on the LCMV L RNA segment of WE (and not that of ARM) were responsible for PRH suppression and redistribution.
Hepatic cell lines transfected with the Z genes of Lassa virus, LCMV-WE, and LCMV-ARM were not depleted of PRH.
To determine whether expression of the Z gene alone could suppress the expression of PRH, we transfected HepG2 cells with plasmids expressing the Lassa Z protein, the LCMV-WE Z protein, or the LCMV-ARM Z protein. All transfected cells expressed PRH equally well and retained PRH bodies in their nuclei (Fig. 3).
FIG. 3.

Cells transfected with the Z gene alone do not down-regulate or relocate PRH but do relocate PML. As described previously for infected fibroblasts, the Z protein alone, even if it is derived from a nonvirulent virus, can relocate PML (9). Here we show that the same phenomenon occurs in liver cells. Nevertheless, Z alone cannot affect PRH.
PRH mRNA and protein as well as PRH function are diminished in hepatic cells infected by LCMV-WE.
Quantitative real-time PCRs were done with cDNAs from uninfected cells, LCMV-ARM-infected cells, and LCMV-WE-infected cells to determine whether PRH mRNA or protein levels were changed by infection. The expression of PRH mRNA relative to that of ribosomal 18S rRNA or GAPDH mRNA was assessed at various times after the infection of cultured hepatic cells. PRH mRNA was down-regulated after LCMV-WE infection, while it remained unchanged after infection with LCMV-ARM (Fig. 4).
FIG. 4.

PRH transcription in human liver HepG2 cell line. Total RNAs from HepG2 cells infected for 24, 48, or 72 h were subjected to real-time RT-PCR analysis, and the levels of PRH mRNA relative to those of GAPDH were determined and graphed as fold mRNA changes, or relative quantitation (RQ) values. RQ values were calculated from the cycles (CT) needed to see threshold levels of PCR products such that 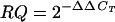 . RNAs were extracted from triplicate wells of cultured cells. PCR products are displayed below the graph in an ethidium bromide-stained agarose gel of PRH-specific cDNAs. Similar results are shown in Table 2, describing PRH mRNA levels in infected monkeys.
. RNAs were extracted from triplicate wells of cultured cells. PCR products are displayed below the graph in an ethidium bromide-stained agarose gel of PRH-specific cDNAs. Similar results are shown in Table 2, describing PRH mRNA levels in infected monkeys.
To determine whether a virus infection of hepatic cells modulates the expression of the PRH protein, we grew cells and infected them with one of the two viruses. Protein extracts from equal numbers of uninfected and infected cells were compared by Western blotting to measure the steady-state levels of PRH. PRH was down-regulated during LCMV-WE infection and remained unchanged after LCMV-ARM infection (Fig. 5A). Chemiluminescence was detected by scanning with a phosphorimager (Molecular Dynamics) and was analyzed with ImageQuant software (Molecular Dynamics) to compare band intensities for host proteins and human β-actin standards (Sigma). The area quantitation report derived from these scans was used to compare the relative amounts of the proteins. To determine whether virus infection of the monkey liver modulates the activity of eIF4E, we probed for VEGF, a protein controlled by eIF4E at the level of translation (31). VEGF was expected to have increased expression when eIF4E was released from PRH suppression. As expected, and in contrast to PRH, VEGF was upregulated in LCMV-WE-infected livers but not in LCMV-ARM-infected livers (Fig. 5B).
FIG. 5.


In hepatic cells, virulent LCMV down-regulates PRH (A) and up-regulates VEGF (B). (A) Cells were infected with virulent or nonvirulent LCMV, and Western blot analyses of the cell lysates were done as described in Materials and Methods. Blots were probed with a rabbit anti-PRH antibody or an anti-β-actin antibody and were revealed by enhanced chemiluminescence. The relative amounts of PRH protein are shown above the Western blot, with the amount of uninfected hepatic cells grown for 72 h set to 1.0. (B) Infected monkey liver sections were subjected to immunohistochemistry to detect the expression of VEGF, which is controlled at the level of translation by eIF4E (30). The liver section on the left expressed little VEGF and was from a monkey infected with LCMV-ARM, whereas the section on the right expressed higher steady-state levels of VEGF and was from a monkey with LCMV-WE-mediated disease. Real-time quantitative PCRs of liver mRNAs from these monkey tissues revealed a three- to sevenfold increase in VEGF mRNA in diseased tissue.
To investigate whether transcriptional regulation by PRH was altered, we used the reporter plasmid p4×HRE-βGAL containing the HRE derived from a bile transporter gene (nctp). HepG2 cells were transfected with reporter plasmids for 24 h and then mock infected (incubated with medium) or infected with virulent LCMV-WE or nonvirulent LCMV-ARM for 24, 48, or 72 h. LCMV-WE infection had decreased β-Gal activity compared to uninfected controls or LCMV-ARM infection (P < 0.05). The parent vector, pGL3, has a simian virus 40 promoter driving the expression of a β-galactosidase reporter gene and was used to transfect control cells. The β-Gal stimulation paralleled endogenous PRH expression in HepG2 cells. An inhibition of β-Gal activity was expected because LCMV-WE down-regulated PRH expression after 48 h of infection. Indeed, LCMV-WE decreased β-Gal expression approximately fivefold in HepG2 cells, suggesting a suppression of PRH function. In contrast, there was a two- to threefold stimulation of β-Gal expression in cells infected with LCMV-ARM compared to control transfected cells (Fig. 6). These results indicate that a nuclear function of PRH, i.e., transcription activity, was diminished by LCMV-WE infection of hepatic cells.
FIG. 6.

Virulent LCMV inhibits reporter protein activity. p4×HRE-βGAL was transfected into HepG2 cells as described in Materials and Methods. Plasmids pGL-3 and pGFP were cotransfected to control for β-Gal activity and transfection success, respectively. Twenty-four hours after transfection, the cells were infected with LCMV-ARM or LCMV-WE for 24, 48, or 72 h. The cells were harvested, and β-Gal activity was measured spectrophotometrically. Each bar represents the mean value + the standard deviation of triplicate measurements from two independent experiments.
PRH RNA and protein were reduced in LCMV-WE-infected rhesus macaque livers.
To determine the effects of LMCV-WE on PRH levels in vivo, we examined PRH mRNA levels in liver samples from monkeys sacrificed during the first few days of an acute infection. PRH mRNA was measured in relation to the internal standards GAPDH and 18S rRNA (Table 2). PRH mRNA levels in livers dropped approximately fourfold during the disease.
TABLE 2.
LCMV and PRH expression in lethally infected monkey livers
| Monkeya | Presence of viral RNA in liverb | Fold inhibition of PRH mRNAc |
|---|---|---|
| Rh-1a-Ctrl | − | 1 |
| Rh-1b-Ctrl | − | 1 |
| Rh.iv3-d1a | − | 2.0 |
| Rh.iv3-d1b | − | 2.3 |
| Rh.iv3-d2a | − | 3.3 |
| Rh.iv3-d2b | − | 2.5 |
| Rh.iv3-d3a | − | 1.6 |
| Rh.iv3-d4a | − | 3.0 |
| Rh.iv3-d4b | + | 2.5 |
| Rh.iv3-d6a | + | 2.7 |
| Rh.iv3-d6b | + | 3.9 |
| Rh.iv3-d7a | + | 4.2 |
| Rh.iv3-d11 | + | 3.5 |
| Rh.iv6-d12 | + | 4.3 |
Ctrl is a mock-infected control animal; d1 is 1 day after infection. Rh.iv3-d11 and Rh.iv6-d12 were two monkeys that were lethally infected with 103 or 106 PFU of virus and then euthanized on day 11 or 12, respectively. Although these two animals were previously described (37, 38), the remaining animals designated Rh.iv3 have not been described. They were also healthy rhesus macaques that were intravenously infected with 103 PFU of virus and then sacrificed on different days after infection (d2, d3, d4, d6, or d7), as indicated by their names.
+, positive by RT-PCR for viral RNA; −, negative by RT-PCR for viral RNA.
Fold inhibition, or relative quantitation (RQ = 2−ΔΔCT), is a quantitative measurement of real-time PCR mRNA levels in comparison with a standard. We used GAPDH and 18S RNA standards and gave the values here for GAPDH standardization, which was less dramatic and more reliable than 18S standardization.
We examined PRH protein expression in livers from healthy and virus-infected rhesus macaques. In uninfected or LCMV-ARM-infected rhesus livers, the PRH protein was expressed abundantly, but in LCMV-WE-infected tissues, positive immunostaining was reduced (Fig. 7, top row). Liver slices from diseased and nondiseased monkeys showed that a high level of Ki-67 staining, which is indicative of cellular proliferation, coincided with low PRH levels (Fig. 7, bottom row). These results were consistent with data obtained by Western blotting of infected hepatic cell lines (Fig. 5A), indicating that LCMV-WE infection lowered the steady-state levels of PRH.
FIG. 7.

During liver disease caused by a hemorrhagic fever virus, PRH decreases and cell division increases. Sections of monkey liver tissue were stained with anti-PRH (top) or an antibody to the proliferation antigen Ki-67 (bottom). The photomicrographs of an LCMV-ARM-infected liver, shown here in the left panels, are identical to pictures of an uninfected liver (not shown) and differ from the diseased liver shown in the right panels. Brown staining indicates the abundant presence of the PRH protein (arrowheads) in the healthy liver and decreased PRH during disease. The Ki-67 nuclear antigen is expressed in cycling cells and was highly expressed in the diseased liver. Ki-67 is seen in hepatocytes and not infiltrating immune cells. In a monkey that recovered from disease, we observed a decrease in Ki-67 staining upon recovery (38). Magnification, ×200 (top panels) and ×400 (bottom panels).
DISCUSSION
Previous studies revealed an association between the arenavirus Z protein and the host homeodomain protein PRH: immunoprecipitation studies and yeast two-hybrid analyses using the viral Z protein as bait showed that an intact RING (zinc-binding) domain was needed for Z-PRH interactions (59). Even though Z proteins from both virulent and nonvirulent viruses could bind PRH (59), only virulent virus infections down-regulated PRH. Since cells expressing only Z do not down-regulate PRH, it is likely that viral genes in addition to the Z gene are needed for this effect.
Arenaviruses have a large (L) and a small (S) genome segment. It has long been known that the genes on the L segment control the virulence of the Old World arenaviruses (18, 49). We used reassortant viruses containing the L segment of the virulent virus and the S segment of the nonvirulent virus and visa versa to show that the ability to suppress PRH can be mapped to the large genome segment. Since the L segment encodes both the viral Z protein and the viral polymerase, we checked whether the level of virus replication could contribute to the PRH-suppressing phenotype. Since the virulent and nonvirulent viruses replicated similarly in two hepatic cell lines (HepG2 and Huh7), simple growth kinetics did not account for the effects on PRH. However, in humans and in a monkey model, arenavirus virulence has always been associated with high virus loads in the liver (38, 39, 44). This may reasonably be attributed to the viral polymerase that is also encoded on the L segment, since small changes in its activities could become more apparent in vivo. Furthermore, it is known that the Z protein binds and coprecipitates with the polymerase (25; M. S. Salvato et al., unpublished data), so it would be reasonable to attribute the effects on PRH to a Z protein-polymerase complex.
From previous studies and our observations of 14 intravenously infected monkeys in this study, the liver appears to be the site of the highest rate of virus replication for LCMV. This is not true for all species or inoculation routes. In a previous publication (38), we showed that high virus titers can also occur in the spleen, but we did not elaborate on the point that virus titers failed to correlate with virus replication. Spleen tissue was consistently negative for in situ hybridization and had low levels of infectious centers compared to liver tissue (M. Djavani, unpublished data); thus, the spleen may have trapped infectious particles (most likely on B-cell receptors), but very few of them were replicating. Although the liver shows little morphological damage, the high levels of virus replication in the liver may affect its systemic functions.
Previous studies indicated that virulent arenavirus infections of primates lead to a physiological state resembling a partial hepatectomy, i.e., a high rate of liver cell proliferation and high circulating levels of interleukin-6, tumor necrosis factor receptors, and interleukin-6 receptors (39). Ordinarily, a hepatectomy initiates liver regeneration by activating the transcription factors NF-κB, Stat3, AP-1, and C/EBP beta and by increasing cell cycling (34). PRH acts during hepatectomy to promote cellular differentiation (57). We showed that a virulent arenavirus infection reduces the expression of PRH and presumably blocks the regenerative process.
In myeloid cell lines, PRH has been shown to promote differentiation and to inhibit proliferation by binding to the nuclear fraction of the translation factor eIF4E (61, 62). When PRH is down-regulated, as in several primary leukemia and lymphoma cells, the block of eIF4E function is released, and it resumes the transport of certain mRNAs, such as cyclin D1, that promote cellular proliferation. PRH binds and inhibits eIF4E via a YXXXXLΦ motif, where “X” is any residue and “Φ” is a hydrophobic residue (56). This motif is found in 199 of 803 homeodomain proteins (in the Swiss Prot database) and is thought to control cellular proliferation and differentiation (62). We speculate that the virus-mediated down-regulation of PRH in liver cells is similar to the down-regulation of PRH in certain types of myeloid leukemias in that decreased PRH releases eIF4E-mediated RNA transport and promotes cell proliferation.
We showed previously by using cultured fibroblasts that LCMV infection (by virulent or nonvirulent isolates) redistributes the oncosuppressor protein PML from the nucleus to the cytoplasm (10). PML is found in all cell types and, like PRH, acts to suppress eIF4E (13, 29, 62). Thus, PML is a general regulator and PRH is a tissue-specific regulator of eIF4E. We showed here that both viruses relocate PML but that only the virulent virus relocates PRH. Hence, it is likely that the relocation of PML is not linked to the disease mechanism and that the relocation of PRH is linked to the disease mechanism.
Several observations suggest that the disease mechanism is connected to the effects of PRH on eIF4E. In the LCMV monkey model, peak disease coincided with a drop in steady-state PRH levels (Fig. 7) and a rise in liver cell proliferation, i.e., 25 to 44% of the liver cells were dividing compared with only 2% cell division upon cessation of the disease (Fig. 7) (39). The effects of PRH on eIF4E are similar to the effects of ribavirin, a nucleotide analog used to treat arenavirus disease. Ribavirin binds to the mRNA cap-binding site of eIF4E and simultaneously inhibits the mRNA transport function of eIF4E and cell proliferation (30, 32). It is compelling that ribavirin can inhibit eIF4E at micromolar concentrations, well below the concentrations at which it affects nucleotide pools and well within the range needed to reverse virus-mediated disease. Our model suggests that LCMV-WE infection blocks the eIF4E-suppressing function of PRH and that this blockage leads to liver pathology.
Our findings are consistent with those from studies of another virulent-nonvirulent pair of arenaviruses, Pichinde-P18 and Pichinde-P2 (19). The low-passage-number virus P2 is attenuated in guinea pigs, whereas the high-passage-number stock, P18, causes a lethal disease that models Lassa fever (66). P18 activates the transcription factor NF-κB, whereas P2 inhibits NF-κB activation in monocytic cell lines and in guinea pig peritoneal macrophages (19). Since NF-κB is known to activate eIF4E and to promote cell cycling (61), it is likely that P18 (like LCMV-WE) contributes to disease by diverting host cells from differentiation to proliferation. Since both the NF-κB effects and the PRH depletion are detectable days rather than hours after infection, we have still not discovered the primary triggering event. A possible trigger is replication-mediated interferon production.
The arenavirus LCMV has been implicated as a human teratogen (3, 4, 35, 45, 64), and it would be reasonable to look for viral effects on host developmental proteins as the basis for damage to developing embryos. In rats, LCMV spreads from glial to neuronal cells and blocks the development of several brain regions (9). In murine systems, LCMV infection has been shown to reduce differentiated cell functions, e.g., a reduction of growth hormone stunts growth (63) and a reduction of primary B-cell responses (51) or cytotoxic T-cell responses (52) leads to immunosuppression, all of which may be explained by viral suppression of cellular differentiation. Since most of these viral effects occur with both LCMV-WE and LCMV-ARM, it is likely that both viruses affect many host processes equally but that only one affects a repair process drastically enough that it registers as virulent. For example, we showed that both viruses alter the distribution of PML, which has a role in development (62), but that only one destroys PRH and causes disease. Thus, PML is more likely to mediate teratogenic effects and PRH is more likely to contribute to hemorrhagic fever, with both acting through their associations with cell cycle control.
Since most arenavirus studies have used rodent models and since rodents are generally thought to have an immunopathogenic disease mechanism (8), it may be difficult to relate the mechanism for primate hemorrhagic fever to murine disease mechanisms. Nevertheless, it would be worthwhile in the future to explore the effects of LCMV on Hex (murine PRH) and on developmental processes in rodent models.
In summary, our findings demonstrate the effects of LCMV on PRH gene expression in hepatic cells. Immunofluorescence studies indicated that virulent LCMV-WE disrupts PRH bodies but that PRH remains relatively unchanged during infection with a nonvirulent strain, LCMV-ARM. On a biochemical level, the PRH mRNA, protein, and function are down-regulated as LCMV-WE replicates. The LCMV Z protein, encoded by the L RNA segment, can bind PRH but cannot down-regulate PRH outside the context of a viral infection. These experiments provide the first evidence of a virus that disrupts cell cycling through a homeodomain protein and may explain the effects of the virus on embryonic development and on differentiated cell functions. Our results suggest that a virulent arenavirus infection disrupts PRH function and thereby contributes to acute disease.
Acknowledgments
Infection studies were supported by National Institutes of Health (NIH) grants AI53620 and AI53619 (to M.S.S.) and AI-RR13980 (to I.S.L.). Confocal laser scanning microscopy was performed at the MSSM Microscopy Shared Resource Facility, supported with funding from an NIH-NCI shared resources grant (1 R24 CA095823-01) and an NSF Major Research Instrumentation grant (DBI-9724504).
We are grateful to Marvin Reitz and Alex Kentsis for helpful comments and discussions. We thank L. de Jong and I. Van der Kraan for the gift of MAb 5E10.
REFERENCES
- 1.Aldeguer, X., F. Debonera, A. Shaked, A. M. Krasinkas, A. E. Gelman, X. Que, G. A. Zamir, S. Hiroyasu, R. Taub, and K. M. Olthoff. 2002. Interleukin-6 from intrahepatic cells of bone marrow origin is required for normal murine liver regeneration. Hepatology 35:40-48. [DOI] [PubMed] [Google Scholar]
- 2.Allen, J. D., T. Lints, N. A. Jenkins, N. G. Copeland, A. Strasser, R. P. Harvey, and J. M. Adams. 1991. Novel murine homeo box gene on chromosome 1 expressed in specific hematopoietic lineages and during embryogenesis. Genes Dev. 5:509-520. [DOI] [PubMed] [Google Scholar]
- 3.Barton, L. L., S. C. Budd, W. S. Morfitt, C. J. Peters, T. G. Ksiazek, R. F. Schindler, and M. T. Yoshino. 1993. Congenital lymphocytic choriomeningitis virus infection in twins. Pediatr. Infect. Dis. J. 12:942-946. [DOI] [PubMed] [Google Scholar]
- 4.Barton, L. L., C. J. Peters, and T. G. Ksiazek. 1995. Lymphocytic choriomeningitis virus: an unrecognized teratogenic pathogen. Emerg. Infect. Dis. 1:152-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bedford, F. K., A. Ashworth, T. Enver, and L. M. Wiedemann. 1993. HEX: a novel homeobox gene expressed during haematopoiesis and conserved between mouse and human. Nucleic Acids Res. 21:1245-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bogue, C. W., G. R. Ganea, E. Sturm, R. Iannucci, and H. C. Jacobs. 2000. Hex expression suggests a role in the development and function of organs derived from foregut endoderm. Dev. Dyn. 219:84-89. [DOI] [PubMed] [Google Scholar]
- 7.Bogue, C. W., P. X. Zhang, J. McGrath, H. C. Jacobs, and R. L. Fuleihan. 2003. Impaired B cell development and function in mice with a targeted disruption of the homeobox gene HEX. Proc. Natl. Acad. Sci. USA 100:556-561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonilla, W. V., D. D. Pinschewer, P. Klenerman, V. Rousson, M. Gaboli, P. P. Pandolfi, R. M. Zinkernagel, M. S. Salvato, and H. Hengartner. 2002. Effects of PML protein on virus-host balance. J. Virol. 76:3810-3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonthius, D. J., J. Mahoney, M. J. Buchmeier, B. Karacay, and D. Taggard. 2002. Critical role for glial cells in the propagation and spread of lymphocytic choriomeningitis virus in the developing rat brain. J. Virol. 76:6618-6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borden, K. L. B., E. J. Campbell Dwyer, G. W. Carlile, M. Djavani, and M. S. Salvato. 1998. Two RING finger proteins, the oncoprotein PML and the arenavirus Z protein, colocalize with the nuclear fraction of the ribosomal P proteins. J. Virol. 72:3819-3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brouqui, P., M. C. Rousseau, M. F. Saron, and A. Bourgeade. 1995. Meningitis due to lymphocytic choriomeningitis virus: four cases in France. Clin. Infect. Dis. 20:1082-1083. [DOI] [PubMed] [Google Scholar]
- 12.Campbell Dwyer, E. J., H. Lai, R. C. MacDonald, M. S. Salvato, and K. L. Borden. 2000. The lymphocytic choriomeningitis virus RING protein Z associates with eukaryotic initiation factor 4E and selectively represses translation in a RING-dependent manner. J. Virol. 74:3293-3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cohen, N., M. Sharma, A. Kentsis, J. M. Perez, S. Strudwick, and K. L. B. Borden. 2001. PML RING suppresses oncogenic transformation by reducing the affinity of eIF4E for mRNA. EMBO J. 20:4547-4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crompton, M. R., T. J. Bartlett, A. D. MacGregor, G. Manfioletti, E. Buratti, V. Giancotti, and G. H. Goodwin. 1992. Identification of a novel vertebrate homeobox gene expressed in haematopoietic cells. Nucleic Acids Res. 20:5661-5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.D'Elia, A. V., G. Tell, D. Russo, F. Arturi, F. Puglisi, G. Manfioletti, V. Gattei, D. L. Mack, P. Cataldi, S. Filetti, C. Di Loreto, and G. Damante. 2002. Expression and localization of the homeodomain-containing protein HEX in human thyroid tumors. J. Clin. Endocrinol. Metab. 87:1376-1383. [DOI] [PubMed] [Google Scholar]
- 16.Denson, L. A., S. J. Karpen, C. W. Bogue, and H. C. Jacobs. 2000. Divergent homeobox gene HEX regulates promoter of the Na(+)-dependent bile acid cotransporter. Am. J. Physiol. Gastrointest. Liver Physiol. 279:G347-G355. [DOI] [PubMed] [Google Scholar]
- 17.Djavani, M., I. S. Lukashevich, A. Sanchez, S. T. Nichol, and M. S. Salvato. 1997. Completion of the Lassa fever virus sequence and identification of a RING-finger protein gene at the 5′ end of the L RNA. Virology 235:414-418. [DOI] [PubMed] [Google Scholar]
- 18.Djavani, M., I. S. Lukashevich, and M. S. Salvato. 1998. Sequence comparison of the large genomic RNA segments of two strains of lymphocytic choriomeningitis virus differing in pathogenic potential for guinea pigs. Virus Genes 17:151-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fennewald, S. M., J. F. Aronson, L. Zhang, and N. K. Herzog. 2002. Alterations in NF-κB and RBP-Jκ by arenavirus infection of macrophages in vitro and in vivo. J. Virol. 76:1154-1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garcin, D., S. Rochat, and D. Kolakofsky. 1993. The Tacaribe arenavirus small zinc finger protein is required for both mRNA synthesis and genome replication J. Virol. 67:807-812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghosh, B., G. R. Ganea, L. A. Denson, R. Ianucci, H. C. Jacobs, and C. W. Bogue. 2000. Immunocytochemical characterization of murine HEX, a homeobox containing protein. Pediatr. Res. 48:634-638. [DOI] [PubMed] [Google Scholar]
- 22.Guo, Y., R. Chan, H. Ramsey, W. Li, X. Xie, W. C. Shelley, J. P. Martinez-Barbera, B. Bort, K. Zaret, M. Yoder, and R. Hromas. 2003. The homeoprotein Hex is required for hemangioblast differentiation. Blood 102:2428-2435. [DOI] [PubMed] [Google Scholar]
- 23.Harlow, E., and D. Lane. 1988. Antibodies: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 24.Hromas, R., J. Radich, and S. Collins. 1993. PCR cloning of an orphan homeobox gene (PRH) preferentially expressed in myeloid and liver cells. Biochem. Biophys. Res. Commun. 195:976-983. [DOI] [PubMed] [Google Scholar]
- 25.Jacamo, R., N. Lopez, M. Wilda, and M. T. Franze-Fernandez. 2003. Tacaribe virus Z protein interacts with the L polymerase to inhibit viral RNA synthesis. J. Virol. 77:10383-10393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jahrling, P., R. A. Hesse, G. A. Eddy, K. S. Johnson, R. T. Callis, and E. L. Stephen. 1980. Lassa virus infection of rhesus monkeys: pathogenesis and treatment with ribavirin. J. Infect. Dis. 141:580-589. [DOI] [PubMed] [Google Scholar]
- 27.Jayaraman, P. S., J. Frampton, and G. Goodwin. 2000. The homeodomain protein PRH influences the differentiation of haematopoietic cells. Leukoc. Res. 24:1023-1031. [DOI] [PubMed] [Google Scholar]
- 28.Keng, V. W., H. Yagi, M. Ikawa, T. Nagano, Z. Myint, K. Yamada, T. Tanaka, A. Sato, I. Muramatsu, M. Okabe, M. Sato, and T. Noguchi. 2000. Homeobox gene Hex is essential for onset of mouse embryonic liver development and differentiation of the monocyte lineage. Biochem. Biophys. Res. Commun. 276:1155-1161. [DOI] [PubMed] [Google Scholar]
- 29.Kentsis, A., E. Campbell Dwyer, J. M. Perez, M. Sharma, A. Chen, Z. Q. Pan, and K. L. B. Borden. 2001. The RING domains of the promyelocytic leukemia protein PML and the arenaviral protein Z repress translation by directly inhibiting translation initiation factor eIF4E. J. Mol. Biol. 312:609-623. [DOI] [PubMed] [Google Scholar]
- 30.Kentsis, A., I. Topisirovic, B. Culjkovic, L. Shao, and K. L. B. Borden. 2004. Ribavirin suppresses eIF4E-mediated oncogenic transformation by physical mimicry of the 7-methyl guanosine mRNA cap. Proc. Natl. Acad. Sci. USA 101:18105-18110. [DOI] [PMC free article] [PubMed]
- 31.Kevil, C., A. De Benedetti, K. D. Payne, L. L. Coe, S. Larouz, and S. Alexander. 1996. Translational regulation of vascular permeability factor by eukaryotic initiation factor 4E: implications for tumor angiogenesis. Int. J. Cancer 65:785-790. [DOI] [PubMed] [Google Scholar]
- 32.Kochhar, D. M., J. D. Penner, and T. B. Knudsen. 1980. Embryotoxic, teratogenic, and metabolic effects of ribavirin in mice. Toxicol. Appl. Pharmacol. 52:99-112. [DOI] [PubMed] [Google Scholar]
- 33.Kongsuwan, K., E. Webb, P. Housiaux, and J. M. Adams. 1988. Expression of multiple homeobox genes within diverse mammalian haemopoietic lineages. EMBO J. 7:2131-2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kountouras, J., P. Boura, and N. J. Lygidakis. 2001. Liver regeneration after hepatectomy. Hepatogastroenterology 48:556-562. [PubMed] [Google Scholar]
- 35.Larsen, P. D., S. A. Chartrand, K. M. Tomashek, L. G. Hauser, and T. G. Ksiazek. 1993. Hydrocephalus complicating lymphocytic choriomeningitis virus infection. Pediatr. Infect. Dis. J. 12:528-531. [DOI] [PubMed] [Google Scholar]
- 36.Lee, K. J., M. Perez, D. D. Pinschewer, and J. C. de la Torre. 2002. Identification of the lymphocytic choriomeningitis virus (LCMV) proteins required to rescue LCMV RNA analogs into LCMV-like particles. J. Virol. 76:6393-6397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lukashevich, I. S., M. Djavani, K. Shapiro, A. Sanchez, E. Ravkov, S. T. Nichol, and M. S. Salvato. 1997. The Lassa fever virus L gene: nucleotide sequence, comparison, and precipitation of a predicted 250 kDa protein with monospecific serum. J. Gen. Virol. 78:547-551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lukashevich, I. S., M. Djavani, J. D. Rodas, J. C. Zapata, A. Usborne, C. Emerson, J. Mitchen, P. B. Jahrling, and M. S. Salvato. 2002. LCMV infection of rhesus macaque causes hemorrhagic fever after intravenous but not after intragastric inoculation. J. Med. Virol. 67:171-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lukashevich, I. S., I. Tikhonov, J. D. Rodas, J. C. Zapata, M. Djavani, and M. S. Salvato. 2003. Arenavirus-mediated liver pathology: acute lymphocytic choriomeningitis virus infection of rhesus macaques is characterized by high interleukin-6 expression and hepatocyte proliferation. J. Virol. 77:1727-1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mack, D. L., D. S. Leibowitz, S. Cooper, H. Ramsey, H. E. Broxmeyer, and R. Hromas. 2002. Down-regulation of the myeloid homeobox protein Hex is essential for normal T-cell development. Immunology 107:444-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Magli, M. C., P. Barba, A. Celetti, G. De Vita, C. Cillo, and E. Boncinelli. 1991. Coordinate regulation of HOX genes in human hematopoietic cells. Proc. Natl. Acad. Sci. USA 88:6348-6352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Manfioletti, G., V. Gattei, E. Buratti, A. Rustighi, A. De Iuliis, D. Aldinucci, G. H. Goodwin, and A. Pinto. 1995. Differential expression of a novel proline-rich homeobox gene (PRH) in human hematolymphopoietic cells. Blood 85:1237-1245. [PubMed] [Google Scholar]
- 43.Martinez Barbera, J. B., M. Clements, P. Thomas, T. Rodriquez, D. Meloy, D. Kioussis, and R. S. Beddington. 2000. The homeobox gene HEX is required in definitive endodermal tissues for normal forebrain, liver and thyroid formation. Development 127:2433-2445. [DOI] [PubMed] [Google Scholar]
- 44.McCormick, J. B., D. H. Walker, I. J. King, P. A. Webb, L. H. Elliott, S. G. Whitfield, and K. M. Johnson. 1986. Lassa virus hepatitis: a study of fatal Lassa fever in humans. Am. J. Trop. Med. Hyg. 35:401-407. [DOI] [PubMed] [Google Scholar]
- 45.Mets, M. B., L. L. Barton, A. Khan, and T. G. Ksiazek. 2000. Lymphocytic choriomeningitis virus: an underdiagnosed cause of congenital chorioretinitis. Am. J. Ophthalmol. 130:209-215. [DOI] [PubMed] [Google Scholar]
- 46.Newman, C. S., F. Chia, and P. A. Krieg. 1997. The Xhex homeobox gene is expressed during development of the vascular endothelium: overexpression leads to an increase in vascular endothelial cell number. Mech. Dev. 66:83-93. [DOI] [PubMed] [Google Scholar]
- 47.Nishina, H., C. Vaz, P. Billa, M. Nghiem, T. Sasaki, J. L. De la Pompa, K. Furlonger, C. Paige, C.-C Hui, K. D. Fischer, H. Kishimoto, H. Iwatsubo, T. Katada, J. R. Woodget, and J. M. Penninger. 1999. Defective liver formation and liver cell apoptosis in mice lacking the stress signaling kinase SEK1/MKK4. Development 126:505-516. [DOI] [PubMed] [Google Scholar]
- 48.Pellizzari, L., A. D'Elia, A. Rustighi, G. Manfioletti, G. Tell, and G. Damante. 2000. Expression and function of the homeodomain-containing protein HEX in thyroid cells. Nucleic Acids Res. 28:2503-2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Riviere, Y., R. Ahmed, P. J. Southern, M. J. Buchmeier, and M. B. A. Oldstone. 1985. Genetic mapping of lymphocytic choriomeningitis virus pathogenicity: virulence in guinea pigs is associated with the L RNA segment. J. Virol. 55:704-708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rodas, J. D., I. S. Lukashevich, J. C. Zapata, C. Cairo, I. Tikhonov, M. Djavani, C. D. Pauza, and M. S. Salvato. 2004. Mucosal arenavirus infection of primates can protect them from lethal hemorrhagic fever. J. Med. Virol. 72:424-435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ruedi, E., H. Hengartner, and R. M. Zinkernagel. 1990. Immunosuppression in mice by lymphocytic choriomeningitis virus infection: time dependence during primary and absence of effects on secondary antibody responses. Cell. Immunol. 130:501-512. [DOI] [PubMed] [Google Scholar]
- 52.Salvato, M., E. Shimomaye, P. Southern, and M. B. Oldstone. 1988. Virus-lymphocyte interactions. IV. Molecular characterization of LCMV Armstrong (CTL+) small genomic segment and that of its variant, clone 13 (CTL−). Virology 164:517-522. [DOI] [PubMed] [Google Scholar]
- 53.Salvato, M. S., and E. M. Shimomaye. 1989. The completed sequence of lymphocytic choriomeningitis virus reveals a unique RNA structure and a gene for a zinc finger protein. Virology 173:1-10. [DOI] [PubMed] [Google Scholar]
- 54.Salvato, M. S., K. J. Schweighofer, J. Burns, and E. M. Shimomaye. 1992. Biochemical and immunological evidence that the 11 kDa zinc-binding protein of lymphocytic choriomeningitis virus is a structural component of the virus. Virus Res. 22:185-198. [DOI] [PubMed] [Google Scholar]
- 55.Salvato, M. S., and J. D. Rodas. 2004. Arenaviruses, p. 629-650. In L. H. Collier and B. W. J. Mahy (ed.), Topley and Wilson's microbiology and microbial infections, 10th ed. Arnold Publishing, London, United Kingdom.
- 56.Sonenberg, N., and A. C. Gingras. 1998. The mRNA 5′ cap-binding protein eIF4E and control of cell growth. Curr. Opin. Cell Biol. 10:268-275. [DOI] [PubMed] [Google Scholar]
- 57.Tanaka, T., T. Inazu, K. Yamada, Z. Myint, V. W. Keng, Y. Inoue, N. Taniguchi, and T. Noguchi. 1999. cDNA cloning and expression of rat homeobox gene, HEX, and functional characterization of the protein. Biochem. J. 339:111-117. [PMC free article] [PubMed] [Google Scholar]
- 58.Thomas, P. Q., A. Brown, and R. S. P. Beddington. 1998. HEX: a homeobox gene revealing peri-implantation asymmetry in the mouse embryo and an early transient marker of endothelial cell precursors. Development 125:85-94. [DOI] [PubMed] [Google Scholar]
- 59.Topcu, Z., D. L. Mack, R. A. Hromas, and K. L. B. Borden. 1999. The promyelocytic leukemia protein PML interacts with the proline-rich homeodomain protein PRH: a RING may link hematopoiesis and growth control. Oncogene 18:7091-7100. [DOI] [PubMed] [Google Scholar]
- 60.Topisirovic, I., A. D. Capili, and K. L. B. Borden. 2002. Gamma interferon and cadmium treatments modulate eukaryotic initiation factor 4E-dependent mRNA transport of cyclin D1 in a PML-dependent manner. Mol. Cell. Biol. 22:6183-6198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Topisirovic, I., M. L. Guzman, M. J. McConnell, J. D. Licht, B. Culjkovic, S. J. Neering, C. T. Jordan, and K. L. B. Borden. 2003. Aberrant eukaryotic initiation factor 4E-dependent mRNA transport impedes hematopoietic differentiation and contributes to leukemogenesis. Mol. Cell. Biol. 23:8992-9002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Topisirovic, I., B. Culjkovic, N. Cohen, J. M. Perez, L. Skrabanek, and K. L. B. Borden. 2003. The proline-rich homeodomain protein, PRH, is a tissue-specific inhibitor of eIF4E-dependent cyclin D1 mRNA transport and growth. EMBO J. 22:689-703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Valsamakis, A., Y. Riviere, and M. B. Oldstone. 1987. Perturbation of differentiated functions in vivo during persistent viral infection. III. Decreased growth hormone mRNA. Virology 156:214-220. [DOI] [PubMed] [Google Scholar]
- 64.Wright, R., D. Johnson, M. Neumann, T. G. Ksiazek, P. Rollin, R. V. Keech, D. J. Bonthius, P. Hitchon, C. F. Grose, W. E. Bell, and J. F. Bale, Jr. 1997. Congenital lymphocytic choriomeningitis virus syndrome: a disease that mimics congenital toxoplasmosis or cytomegalovirus infection. Pediatrics 100:E9. [DOI] [PubMed] [Google Scholar]
- 65.Zaret, K. 1998. Early liver differentiation: genetic potentiation and multilevel growth control. Curr. Opin. Genet. Dev. 8:526-531. [DOI] [PubMed] [Google Scholar]
- 66.Zhang, L., K. Marriott, and J. F. Aronson. 1999. Sequence analysis of the small RNA segment of guinea pig-passaged Pichinde virus variants. Am. J. Trop. Med. Hyg. 61:220-225. [DOI] [PubMed] [Google Scholar]