

Abstract
Crouzon syndrome craniofacial dysostosis type I [OMIM 123500] is caused by mutations in the gene encoding fibroblast growth factor receptor-2 (FGFR2). An overlapping phenotype with Muenke and Crouzon syndrome with acanthosis nigricans (FGFR3 mutations) is known. The clinical diagnosis can be corroborated by molecular studies in about 80–90% of the cases. No clear genotype/phenotype correlation has been identified yet. Here, we describe a second family with a mild phenotype in which the FGFR2 mutation c.943G>T leading to the amino acid substitution p.Ala315Ser was detected. Five affected family members showed craniofacial dysostosis without overt craniosynostosis. They all had midface hypoplasia. Crouzonoid appearance with mild protrusion of bulbi was only apparent in our index patient as well as obstructive sleep apnea episodes leading to reduced oxygen saturation; therefore, surgical intervention was suggested. One other affected family member additionally had iris coloboma.
Key Words: Craniosynostosis, FGFR2, Midface hypoplasia, Obstructive sleep apnea
Mutations in the fibroblast growth factor receptor 2 (FGFR2) gene are responsible for several severe craniosynostosis syndromes such as Crouzon, Apert, Pfeiffer, Jackson Weiss, and Beare-Stevenson syndrome [Bonaventure and El Ghouzzi, 2003; Cunningham et al., 2007]. FGFR2 belongs to a family of 4 fibroblast growth factor receptors; 3 play an important role in embryogenesis and have a special signaling function in cranial sutures and embryonic development of the limbs [Lajeunie, 2006].
The Crouzon phenotype is characterized by major dysmorphisms: craniosynostosis, proptosis, midface hypoplasia, and, in some cases, a beaked nose. However, the clinical presentation shows variability even within families.
We present a family with 5 affected and genetically tested family members. Only in the index patient we suspected the diagnosis Crouzon syndrome; the other family members had snoring since birth, but no apparent Crouzonoid appearance. All members of our family lack overt craniosynostosis. Because of the facial features, we suggested the diagnosis Crouzon syndrome in the index patient and performed FGFR2 testing. After revealing an FGFR2 mutation c.943G>T (p.Ala315Ser), further family members were surprisingly found to also be mutation carriers.
Case Report
Our 27-year-old proband (Fig. 1, III-1) was born to Caucasian nonconsanguineous parents. He had difficulties breathing through his nose. Due to right-sided choanal stenosis, he had nasal speech. Therefore, he underwent speech and language therapy in infancy (pre-speech and pre-language skills).
Fig. 1.

A Pedigree showing our proband (arrow) and his family. Black symbols: affected family members molecularly confirmed. Gray symbols: members said to be clinically affected with mild midface hypoplasia and snoring, not available for testing. B Sanger sequencing of the FGFR2 gene showing a heterozygous change c.943G>T in exon IIIc (RefSeq NG_012449.1), following the nomenclature proposed by Ingersoll et al. [2001], leading to a missense mutation p.Ala315Ser. The index patient's sequence is depicted.
He actually presented with obstructive sleep apnea leading to significant lack of sleep. In the polysomnography, a desaturation of oxygen was verified. Continual positive airway pressure was recommended. He had a Crouzonoid facial appearance, i.e., mild proptosis, hypertelorism, and midface hypoplasia. He had no beaked nose (Fig. 2A, B). Eye examination as well as hearing was normal. Midface hypoplasia with corresponding class III malocclusion (“pseudo-prognathism”) and dental malposition were present (Fig. 3).
Fig. 2.

Clinical photographs of proband (A, B) his mother (C, D), sister (E, F), nephew 1 (G, H), and nephew 2 (I, J). Note the left-sided iris coloboma of nephew 2 (K).
Fig. 3.
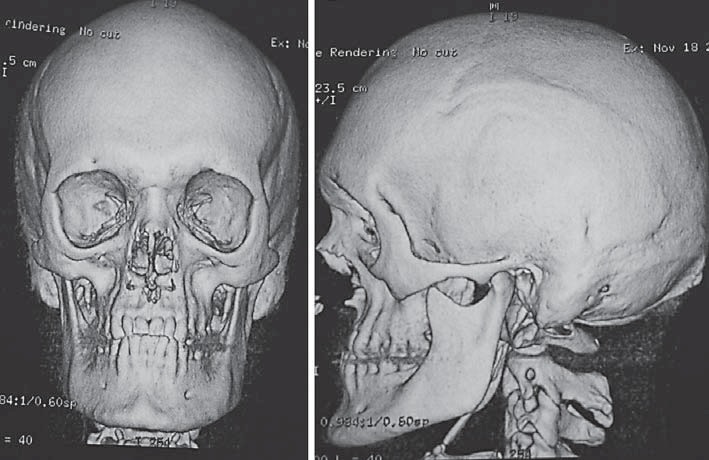
3D reconstruction of the craniofacial skeleton of the index patient showing maxillary and midfacial hypoplasia corresponding in class III malocclusion.
After verifying the mutation in our proband we clinically and genetically analyzed further family members possibly carrying the mutation (pedigree in Fig. 1).
The proband's 64-year-old mother (Fig. 1, II-2) had mild midface hypoplasia (Fig. 2C, D) and has primarily been a mouth breather since birth. The wisdom teeth led to compression of adjacent teeth.
The proband's 36-year-old sister (Fig. 1, III-2) had a right-sided narrow nasal passage, broad forehead, mild facial asymmetry, and midface hypoplasia (Fig. 2E, F). Malposition of teeth is noted and a medical history of 5 persistent primary teeth, which were removed at the age of 14, was reported.
The proband's older nephew (Fig. 1, IV-1) aged 12 years showed mild facial asymmetry (Fig. 2G, H) and malocclusion. Therefore, he got a clip which pulls the upper jaw forward as well as braces correcting the malposition of teeth.
The proband's younger nephew (Fig. 1, IV-3) aged 2 years presented with a large fontanel at birth measuring 4 × 4 cm, and a narrow nasal passage on the right side. He showed a high, broad forehead, mild midface hypoplasia, left-sided iris coloboma (Fig. 2I,2J,2K), no vision defect, mild malposition of teeth, and mild prognathism.
All affected family members have been suffering from snoring since birth. None of them had limb abnormalities. Clinical features of the described patients are summarized in Table 1. Five other family members were said to have severe snoring since birth. They are possibly affected but not available for genetic testing (gray symbols in Fig. 1).
Table 1.
Comparison of clinical features of FGFR2 Ala315Ser carriers
| Our family |
Johnson et al. [2000] |
|||||||
|---|---|---|---|---|---|---|---|---|
| proband 27 years | mother 64 years | sister 36 years | nephew 1 13 years | nephew 2 2 years | proband 6 months | mother | maternal Grandfather | |
| Head | ||||||||
| Craniosynostosis | – | – | – | – | – | + | – | – |
| Face | ||||||||
| Broad forehead | + | – | + | + | + | + | + | + |
| Ocular hypertelorism | + | – | – | + | + | + | + | + |
| Protrusio bulbi | + | – | – | – | – | – | – | – |
| Choanal stenosis/atresia | + | – | – | – | – | n.a. | n.a. | n.a. |
| Facial asymmetry | + | – | – | + | – | + | + | + |
| Midface hypoplasia | + | + | + | + | + | – | – | – |
| Malocclusion | + | – | – | + | – | n.a. | n.a. | n.a. |
| Prognathism | + | + | + | + | + | + | + | + |
| Eyes | ||||||||
| Iris coloboma | – | – | – | – | + | n.a. | n.a. | n.a. |
| Teeth | ||||||||
| Dental malposition | + | + | + | + | + | n.a. | n.a. | n.a. |
| Respiratory | ||||||||
| Apnea | + | – | – | – | – | n.a. | n.a. | n.a. |
| Snoring since birth | + | + | + | + | + | n.a. | n.a. | n.a. |
+, present; –, absent; n.a., not available.
Methods and Results
Sanger sequencing of the mutation hot spot regions in FGFR2 and FGFR3 was performed using genomic DNA extracted from blood samples.
A heterozygous base exchange c.943G>T was detected in the exon referred to as exon IIIc of FGFR2 (FGFR2c spliceform, i.e., exon 8 in RefSeq NM_000141.4), following the nomenclature of Ingersoll et al. [2001]. This change led to a missense mutation p.Ala315Ser in the index patient. Sequence analysis was done using Alamut (http://www.interactive-biosoftware.com/alamut-visual/).
Segregation analysis also revealed this heterozygous change in his mother, his sister, and 2 of his nephews (Fig. 1).
Discussion
The verified mutation in our family has been described previously at least 4 times in the literature [Johnson et al., 2000; Wilkie et al., 2002; Roscioli et al., 2013]. Wilkie et al. [2002] described a family in which this variant was present accompanied by a second mutation in cis. However, in contrast to our cases, this family presented with severe syndactyly and no evidence of craniosynostosis.
To our knowledge, the single c.943G>T (p.Ala315Ser) mutation has been rarely reported in the literature so far, i.e., in one familial case of a 3-generation family [Johnson et al., 2000; Kan et al., 2002] and in 2 other unrelated individuals [Roscioli et al., 2013]. The 2 cases of Roscioli et al. [2013] were diagnosed with Crouzon syndrome and craniosynostosis, suggesting that they may have been more severely affected. Note, the c.943G>T (p.Ala315Ser) mutation was not observed in 121,146 alleles listed by the Exome Aggregation Consortium [Lek et al., 2016].
The mutated residue is conserved in vertebrates and located in the alternatively spliced βC′-βE loop (IgIIIc domain), which forms a hydrophobic core in wild type FGFR2c [Plotnikov et al., 2000; Ibrahimi et al., 2004]. This domain is one of the key regions in the ligand-binding site of FGFR2c [Plotnikov et al., 2000], and thus, c.943G>T (p.Ala315Ser) is likely a ligand-dependent mutation. Ibrahimi et al. [2004] showed a reduced ability of the c.943G>T (p.Ala315Ser) FGFR2c to bind FGF2 ligand.
Johnson et al. [2000] described a 3-generation pedigree with nonsyndromic unicoronal craniosynostosis only observed in one individual of the third generation. They hypothesized that the c.943G>T (p.Ala315Ser) FGFR2 mutation confers a predisposition but requires additional environmental factors for craniosynostosis to occur, i.e., fetal head constraint.
Compared to the syndromic craniosynostosis associated with FGFR2 mutations, isolated unilateral or bilateral coronal synostosis present the mild end of the phenotypic spectrum associated with FGFR2 [Helman et al., 2014]. In the family described here, none of the members needed neurosurgical intervention, and there is no morphological hint of a brachy- or plagiocephaly.
While a more or less severe midface hypoplasia is observed in all carriers of the FGFR2 variant in the family described here, craniosynostosis as reported for the index case by Johnson et al. [2000] is not present. Shared clinical features between our family and the family in Johnson et al. [2000] are a broad forehead, hypertelorism, and prognathism (Table 1). The leading features of our patients – snoring since birth in all of our patients and apnea (index patient) – are not reported in any of the patients described by Johnson et al. [2000]. Iris coloboma is only reported in 1 of our patients (IV-3).
Besides the craniofacial abnormalities, additional features such as obstructive sleep apnea and coloboma in 1 patient were observed in the family described here. Sleep-related breathing disorders are a common symptom in children with craniosynostosis syndromes as upper airways may be narrowed by midface hypoplasia [Hein et al., 2011; Driessen et al., 2013]. They are at an increased risk for hyposomnia, sleep structure disturbance, obstructive sleep apnea, and memory deficits [Doerga et al., 2016]. Midface hypoplasia is widely distributed in our family. In general, a clear genotype-phenotype correlation is not possible based on only 2 families. Thus, sequencing of FGFR2 is recommendable also in nonsyndromic patients lacking overt craniosynostosis but presenting with mild Crouzonoid facial features.
In summary, we report a family with atypically mild Crouzon syndrome. In line with the publication of Johnson et al. [2000], we could show that the mild phenotype of our family seems to be consistent with their observation, suggesting that the FGFR2 variant c.943G>T (p.Ala315Ser) may act as a hypomorphic mutation with reduced ligand binding.
Statement of Ethics
Written permission for the reproduction of photographs was obtained.
Disclosure Statement
The authors have no conflicts of interest to declare.
Acknowledgments
We thank the family for their collaboration. We also acknowledge Andreas Eckert and Martina Eßler from the Schlafmedizinisches Zentrum Helios Klinikum Berlin-Buch for providing polysomnography.
References
- Bonaventure J, EL Ghouzzi V. Molecular and cellular bases of syndromic craniosynostoses. Expert Rev Mol Med. 2003;5:1–17. doi: 10.1017/S1462399403005751. [DOI] [PubMed] [Google Scholar]
- Cunningham ML, Seto ML, Ratisoontorn C, Heike CL, Hing AV. Syndromic craniosynostosis: from history to hydrogen bonds. Orthod Cranifac Res. 2007;10:67–81. doi: 10.1111/j.1601-6343.2007.00389.x. [DOI] [PubMed] [Google Scholar]
- Doerga PN, Spruijt B, Mathijssen IM, Wolvius EB, Joosten KF, van der Schroeff MP. Upper airway endoscopy to optimize obstructive sleep apnea treatment in Apert and Crouzon syndromes. J Craniomaxillofac Surg. 2016;44:191–196. doi: 10.1016/j.jcms.2015.11.004. [DOI] [PubMed] [Google Scholar]
- Driessen C, Joosten KF, Bannink N, Bredero-Boelhouwer HH, Hoeve HL, et al. How does obstructive sleep apnoea evolve in syndromic craniosynostosis? A prospective cohort study. Arch Dis Child. 2013;98:538–543. doi: 10.1136/archdischild-2012-302745. [DOI] [PubMed] [Google Scholar]
- Hein A, Schweitzer T, Straßburg HM, Wurm M. Diagnosis and therapy of obstructive sleep apnea syndrome in children with premature craniosynostosis syndromes (in German). Klin Padiatr. 2011;223:424–429. doi: 10.1055/s-0031-1297972. [DOI] [PubMed] [Google Scholar]
- Helman SN, Badhey A, Kadakia S, Myers E. Revisiting Crouzon syndrome: reviewing the background and management of a multifaceted disease. Oral Maxillofac Surg. 2014;18:373–379. doi: 10.1007/s10006-014-0467-0. [DOI] [PubMed] [Google Scholar]
- Ibrahimi OA, Zhang F, Eliseenkova AV, Itoh N, Linhardt RJ, Mohammadi M. Biochemical analysis of pathogenic ligand-dependent FGFR2 mutations suggests distinct pathophysiological mechanisms for craniofacial and limb abnormalities. Hum Mol Genet. 2004;13:2313–2324. doi: 10.1093/hmg/ddh235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingersoll RG, Paznekas WA, Tran AK, Scott AF, Jiang G, Jabs EW. Fibroblast growth factor receptor 2 (FGFR2): genomic sequence and variations. Cytogenet Cell Genet. 2001;94:121–126. doi: 10.1159/000048802. [DOI] [PubMed] [Google Scholar]
- Johnson D, Wall SA, Mann S, Wilkie AO. A novel mutation, Ala315Ser, in FGFR2: a gene-environment interaction leading to craniosynostosis? Europ J Human Genet. 2000;8:571–577. doi: 10.1038/sj.ejhg.5200499. [DOI] [PubMed] [Google Scholar]
- Kan SH, Elanko N, Johnson D, Cornejo-Roldan L, Cook J, et al. Genomic screening of fibroblast growth-factor receptor 2 reveals a wide spectrum of mutations in patients with syndromic craniosynostosis. Am J Hum Genet. 2002;70:472–486. doi: 10.1086/338758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lajeunie E, Heuertz S, El Ghouzzi V, Martinovic J, Renier D, et al. Mutation screening in patients with syndromic craniosynostosis indicates that a limited number of recurrent FGFR2 mutations account for severe forms of Pfeiffer syndrome. Eur J Hum Genet. 2006;14:289–298. doi: 10.1038/sj.ejhg.5201558. [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotnikov AN, Hubbard SR, Schlessinger J, Mohammadi M. Crystal structures of two FGF-FGFR complexes reveal the determinants of ligand-receptor specificity. Cell. 2000;101:413–424. doi: 10.1016/s0092-8674(00)80851-x. [DOI] [PubMed] [Google Scholar]
- Roscioli T, Elakis G, Cox TC, Moon DJ, Venselaar H, et al. Genotype and clinical care correlations in craniosynostosis: findings from a cohort of 630 Australian and New Zealand patients. Am J Med Genet C Semin Med Genet. 2013;163C:259–270. doi: 10.1002/ajmg.c.31378. [DOI] [PubMed] [Google Scholar]
- Wilkie AOM, Patey SJ, Kan S, van den Ouweland AMW, Hamel BCJ. FGFs, their receptors, and human limb malformations: clinical and molecular correlations. Am J Med Genet. 2002;112:266–278. doi: 10.1002/ajmg.10775. [DOI] [PubMed] [Google Scholar]