

Summary
Matrix metalloproteinases (MMPs) represent more than 20 zinc-containing endopeptidases that cleave internal peptide bonds, leading to protein degradation. They play a critical role in many physiological cell functions, including tissue remodeling, embryogenesis and angiogenesis. They are also involved in the pathogenesis of a vast array of diseases, including but not limited to systemic inflammation, various cancers, and cardiovascular, neurological, and autoimmune diseases. Here, we describe gel zymography to detect MMPs in cell and tissue samples and in cell culture supernatants.
Keywords: zymography, zymogram, protease, detection, semi-quantitative
1. Introduction
Matrix metalloproteinases (MMPs) are zinc-containing endopeptidases responsible for extracellular matrix degradation via cleavage of internal peptide bonds of specific proteins. More than 20 MMPs have been identified. MMPs can be either membrane-bound or secreted; they can be segregated into six groups based on their substrate specificity and structure: collagenases, gelatinases, stromelysins, matrilysins, membrane type, and others.
MMPs are crucial to development and tissue repair and remodeling but are also important players in many disease processes. They have been suggested as either diagnosis tools or therapeutic targets in a broad range of conditions such as multiple sclerosis [2,3], periodontal disease [4,5], cancer [7], rheumatoid arthritis and osteoarthritis [8,9], intestinal disease [10,11], or cardiovascular diseases [12]. The accurate and reproducible detection of different MMPs is therefore critical for both basic and translational research.
A number of techniques have been developed to detect and identify MMPs in various samples. Enzyme linked immunosorbent assays (ELISA) and Western blots require the use of antibodies targeted to the MMP of interest. Here, we describe the procedure for gel zymography, which was first described in 1980 by C. Heussen and E.B. Dowdle [13], and continues to be extensively used to detect MMPs in many cell types and tissues and in most bodily fluids [4,8,3,5].
2. Materials
Prepare all solutions using analytical grade reagents and ultrapure water. All solutions should be stored at room temperature in closed bottles unless otherwise specified and must be discarded after use following all waste disposal regulations in place.
2.1. Buffers for sample preparation
Phosphate-buffered saline (PBS), pH 7.2 (1X): Add 800 mL water to a 1 L graduated cylinder. Weigh 8 g NaCl, 0.2 g KCl, 1.44 g Na2HPO4 (dibasic anhydrous) (see Note 1), and 0.24 g KH2PO4 (monobasic anhydrous) and add to the graduated cylinder. Mix and adjust pH with HCl. Add water to 1 L (see Note 2).
Protease inhibitor cocktail (100X): Add 16 mL water to a 20 mL graduated cylinder. Weigh 20 mg aprotinin, 4 mg leupeptin, 961 mg benzamidine and add to the graduated cylinder. Add water to 20 mL and mix. Aliquot into microcentrifuge tubes and store at −20°C.
NP-40 lysis buffer, pH 7.5 (1X) (see Note 3). Lysis buffer: Add 80 mL water to a 100 mL graduated cylinder. Weigh 394 mg Tris-HCl and 584 mg NaCl, and add to the graduated cylinder. Add 1 mL Nonidet P-40 (NP-40) to the graduated cylinder and mix. Add water to 100 mL. Store at 4°C. Immediately before use, add 10 μL of protease inhibitor cocktail (100X) (step 2.1.2 of the materials) to 1 mL of NP-40 lysis buffer.
Triton X-100 lysis buffer, pH 8.5 (1X) (see Note 3): Add 80 mL water to a 100 mL graduated cylinder. Weigh 315 mg Tris-HCl, 730 mg NaCl, 1 mL Triton X-100 (see Note 4) and add to the graduated cylinder. Add water to 100 mL and mix. Store at 4°C. Immediately before use, add 10 μL of protease inhibitor cocktail (100X) to 1 mL of Triton X-100 lysis buffer.
2.2. Zymogram gel components
Separating gel buffer: Add 80 mL water to a 100 mL glass cylinder. Weigh 4.63 g ammediol.HCl and 0.02 g sodium azide and transfer to the cylinder. Mix and adjust pH to 8.96 with HCl. Add water to 100 mL. Store at 4°C for up to a month.
10X casein substrate solution (see Note 5): Add 100 mL 0.1 N NaOH to a glass beaker. Weigh 800 mg casein and transfer to the beaker. Heat the solution to 37°C with occasional vortexing until full dissolution (see Note 6). Store at −20°C for up to 6 months.
10X gelatin substrate solution (see Note 5): Add 100 mL water to a glass beaker. Weigh 800 mg porcine skin gelatin. Heat in the microwave until the solution just boils (see Note 7). Swirl thoroughly to ensure homogeneous distribution (see Note 8). Store at 4°C for one week or at −20°C for up to 6 months.
Sucrose solution: Add 50 mL water to a 100 mL glass cylinder. Weigh 50 g sucrose and 0.02 g sodium azide and transfer to the cylinder. Add 30 μL toluene and make up to 100 mL with water. Store at 4°C for up to a month.
Separating gel (see Note 9): Mix the components in the appropriate proportions (Table 1) for the choice of gel (see Note 10).
Stacking gel buffer: Add 80 mL water to a 100 mL glass cylinder. Weigh 3.51 g ammediol · HCl and 0.02 g sodium azide and transfer to the cylinder. Mix and adjust pH to 8.37 with HCl. Add water to 100 mL. Store at 4°C for up to a month.
Stacking gel (see Note 9): Mix 142 μL acrylamide/bisacrylamide solution (37.5:1), 285 μL stacking buffer, 285 μL sucrose solution, 428 μL water, 14 μL 10% (weight/volume) ammonium persulfate, and 3 μL TEMED (N,N,N′,N′-tetramethylethylenediamine) (see Note 11).
Tris-HCl, 0.5 M, pH 6.8: Add 400 mL water to a 1 L glass cylinder (see Note 12). Weigh 39.39 g Tris and transfer to the cylinder. Mix and adjust pH with HCl. Add water to 500 mL.
Sample buffer (2X): In a 10 mL glass cylinder, add 2.5 ml of a 0.5 M solution of Tris-HCl, pH 6.8, 2.0 ml of glycerol, 10% (weight/volume) sodium dodecyl sulfate (SDS), and 0.1% (weight/volume) bromophenol blue. Add water to 10 mL.
Running buffer (10X): To prepare 1 L of running buffer, add 100 mL water to a 1 L graduated cylinder. Weigh 29 g of Tris Base, 144 g of glycine, and 10 g of SDS. Add all three to the graduated cylinder and add water to 1 L. Mix with a magnetic stir-bar until fully dissolved.
Table 1.
Recipes for preparing zymogram separating gels with different percentages of acrylamide. The volumes are sufficient for a single mini-gel (9 cm × 6 cm × 0.75 cm).
| Solution | Final acrylamide concentration in gel | |||
|---|---|---|---|---|
| 6% | 7.5% | 10% | 12% | |
| Acrylamide/bisacrylamide solution 37.5:1 | 0.8 mL | 1.0 mL | 1.32 mL | 1.61 mL |
| Separating gel buffer | 1.0 mL | 1.0 mL | 1.0 mL | 1.0 mL |
| 10X casein or gelatin substrate solution | 0.4 mL | 0.4 mL | 0.4 mL | 0.4 mL |
| Sucrose solution | 0.86 mL | 0.86 mL | 0.86 mL | 0.86 mL |
| Water | 0.94 mL | 0.74 mL | 0.42 mL | 0.13 mL |
| 10% (w/v) ammonium persulfate | 14 μL | 14 μL | 14 μL | 14 μL |
| TEMED* | 1.5 μL | 1.5 μL | 1.5 μL | 1.5 μL |
TEMED: N,N,N′,N′-tetramethylethylenediamine.
2.3. Buffers for protease detection
Renaturing buffer (1X) (see Note 13): Add 50 mL of water to a 100 mL graduated cylinder. Add 2.5 ml of Triton X-100 to the cylinder (see Note 4). Add water to 100 mL and mix thoroughly. Do not store for more than a day.
Developing buffer (10X): To prepare 1 L of developing buffer, add 100 mL water to a 1 L graduated cylinder. Weigh 12.1 g Tris Base, 63 g Tris-HCl, 117 g NaCl, 7.4 g CaCl2 and add all four to the cylinder. Add 2 mL Brij 35 to the cylinder (see Note 14). Add water to 1 L and mix thoroughly.
Coomassie Blue staining solution (1X) (see Note 15): Add 800 mL water to a graduated cylinder. Add 50 mL methanol and 100 mL acetic acid (see Note 16). Weigh 5 g Coomassie Blue R-250 and add to the graduated cylinder. Add water to 1 L.
Coomassie Blue destaining solution (1X) (see Note 15): Add 800 mL water to a 1 L beaker. Add 100 mL methanol and 50 ml acetic acid (see Note 16). Add water to 1 L.
3. Methods
Perform all procedures at room temperature unless otherwise specified and wear personal protective equipment.
3.1. Sample Preparation
3.1.1. Preparation of Cell Culture Supernatants
If the cells to be tested are adherent, plate in complete growth media and allow time to adhere (see Note 17).
Wash the cells 3 times with sterile PBS or serum-free culture media.
Incubate the cells at 37°C with serum-free culture media (see Note 18) for the optimal duration dependent on the cells used (see Note 19).
Collect the culture supernatants.
Proceed to measuring protein concentration (step 3.2 of the methods) or freeze at −80°C until use (see Note 20).
3.1.2. Preparation of Cell Lysates
If the cells to be tested are adherent, plate in complete growth media and allow time to adhere (see Note 21).
Wash the cells 3 times with sterile PBS or culture media.
Incubate the cells at 37°C with culture media for the optimal duration dependent on the cells used (see Note 19).
Place a bottle of PBS and a bottle of NP-40 lysis buffer on ice for a minimum of 20 min to prepare ice-cold buffers.
Wash the cells twice with ice-cold PBS.
Add cold NP-40 lysis buffer at a volume of 2 mL per 150 mm dish.
If the cells to be tested are adherent, scrape them using a cell lifter.
Collect the cell lysate and incubate on ice for 15 min.
Centrifuge the cell lysate 16,000 g for 20 min at 4°C in a microcentrifuge.
Collect the supernatant.
Proceed to measuring protein concentration (step 3.2 of the methods) or freeze at −80°C until use (see Note 20).
3.1.3. Preparation of Tissue Extracts – Technique 1: NP-40 lysis Buffer
Place a bottle of NP-40 lysis buffer on ice for a minimum of 20 min to prepare ice-cold buffers.
Collect the tissue and process immediately.
Cut approximately 50 mg of tissue into small pieces into a 1.5 mL microcentrifuge tube.
Add 0.5 mL of cold NP-40 lysis buffer (see Note 22).
Homogenize the tissue with a pestle over ice.
Centrifuge the homogenate 16,000 g for 20 min at 4°C in a microcentrifuge.
Collect the supernatant.
Proceed to measuring protein concentration (step 3.2 of the methods) or freeze at −80°C until use (see Note 20).
3.1.4. Preparation of Tissue Extracts – Technique 2: Pulverization
Collect the tissue and immediately freeze with dry ice.
Pulverize the frozen tissue with a micro-dismembrator.
Weigh 50 mg of pulverized tissue and resuspend in 0.15 mL Triton X-100 lysis buffer under gentle rotation at 4°C.
Centrifuge the homogenate 16,000 g for 20 min at 4°C in a microcentrifuge.
Collect the supernatant.
Proceed to measuring protein concentration (step 3.2 of the methods) or freeze at −80°C until use (see Note 20).
3.2. Measuring Protein Concentration
Remove the bovine serum albumin (BSA) standard reagent (see Note 23) from 4°C storage and let it warm to room temperature (see Note 24). Invert a few times before use.
Prepare serial dilutions of 2 mg/mL BSA to create a calibration curve (linear) in the range of 200 μg/mL to 1000 μg/mL.
Pipet each dilution of standard and the samples into separate clean cuvettes filled with 500 μL dH2O and 500 μL Bradford reagent and vortex. Prepare a blank sample in a clean cuvette by mixing 500 μL dH2O and 500 μL Bradford reagent.
Incubate at room temperature (see Note 24) for at least 5 min. Do not incubate longer than 1 hr at room temperature (see Note 25).
Warm up the spectrophotometer for 10 min. Zero the instrument with the blank sample. Measure the absorbance of the standards and the samples at 280 nm.
Create a standard curve by plotting 280 nm readouts on the y-axis and concentration of the serial standard dilutions on the x-axis. Determine the slope of the standard curve.
Using the slope and 280 nm readouts, calculate the concentration of the samples (see Note 26).
3.3. Gel Preparation
Assemble the electrophoresis plates (see Note 27).
Prepare the separating gel and immediately pour (see Note 28) between the electrophoresis plates up to 2 cm from the top.
Overlay with water-saturated n-butanol and allow the gel to polymerize approximately 30 min at room temperature (see Note 29).
Prepare the stacking gel (see Note 28).
Decant the n-butanol from the gel and gently rinse with water (see Note 30).
Pour the stacking gel on top of the separating gel.
Immediately insert the well comb (see Note 31).
Allow the stacking gel to polymerize approximately 20 min at room temperature.
3.4. Loading and Electrophoresis
Remove the well comb from the gel and rinse gently with 1X running buffer.
Install the zymography gel in the electrophoresis setup.
Fill the inside chamber with 1X running buffer and the outside chamber with 500 mL 1X running buffer (see Note 32).
Place 10 μL of 2X sample buffer on a Parafilm membrane and add 10 μL of sample. Pipet a few times to mix and load the mixture into each well using gel loading pipet tips (see Note 33).
Load a protein molecular weight standard in at least one well per gel.
Load appropriate recombinant MMP or a control known to contain the MMP of interest to identify the sample MMPs (optional) (see Note 34).
Run the gel at 125 V for 90 min or until the loading dye reaches the bottom of the gel.
3.5. Gel Renaturation
Place 100 mL of 1X renaturing buffer in a container large enough to fit the whole gel flat.
Open the gel plates (or cassette in the case of a commercial pre-cast gel).
Make a diagonal cut in a corner of the gel to mark its orientation.
Remove the gel from the plate or cassette (see Note 35).
Place the gel in the container with renaturing buffer.
Incubate for 30 min at room temperature with gentle agitation on a bench rocker.
3.6. Gel Development
Remove the renaturing buffer by gently pouring it into a waste container (see Note 36).
Add 100 mL of 1X developing buffer to the container with the gel.
Incubate for 30 min at room temperature with gentle agitation on a bench rocker.
Remove the developing buffer by gently pouring it into a waste container
Add 100 mL of 1X developing buffer to the container with the gel. Close the lid tightly.
Incubate overnight (16–18 hrs) at 37°C (see Note 37).
3.7. Gel Staining and Destaining
Remove the developing buffer by gently pouring it into a waste container.
Add 100 mL of water to the container with the gel.
Incubate for 5 min at room temperature with gentle agitation on a bench rocker.
Remove the water by gently pouring it into a waste container.
Repeat steps 2–4 twice more.
Scan the gel to record the exact position of the protein standard bands as they will become difficult to see after gel staining and destaining.
3.7.1. Staining with Coomassie Blue R-250
Add 20 mL of Coomassie blue staining solution (see Note 15) to the container with the gel.
Incubate for 1 hr at room temperature with gentle agitation on a bench rocker (see Note 38).
Remove the staining buffer by gently pouring it into a waste container.
Add 100 ml of 1X destaining solution to the container with the gel.
Incubate at room temperature with gentle agitation on a bench rocker until areas of proteolytic activity are clearly visible (see Note 39).
3.7.2. Staining with colloidal Coomassie Blue (G-250)
Add 20 mL of ready-to-use commercial buffer with colloidal Coomassie Blue to the container with the gel.
Incubate for 1 hr at room temperature with gentle agitation on a bench rocker (see Note 38).
Remove the staining buffer by gently pouring it into a waste container.
Add 100 mL of water to the container with the gel.
Incubate for 1 hr at room temperature with gentle agitation on a bench rocker (see Note 40).
3.8. Data Acquisition and Analysis
Remove the gel from the water, place in a plastic sheet protector, and gently remove air bubbles.
Scan the gel with a digital scanner (see Note 41).
The integrative intensity of the MMP bands can be quantified with ImageJ (freely available from the National Institutes of Health) or similar software.
Figure 1.
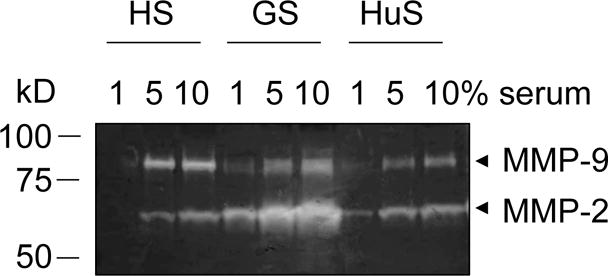
Gelatin zymography gel loaded with different dilutions of horse serum (HS), goat serum (GS), and human serum (HuS) (1, 5, 10%). Stronger bands of MMP-2 (~ 65 kD) and MMP-9 (~ 85 kD) are visible with increasing concentrations of the different sera.
Figure 2.
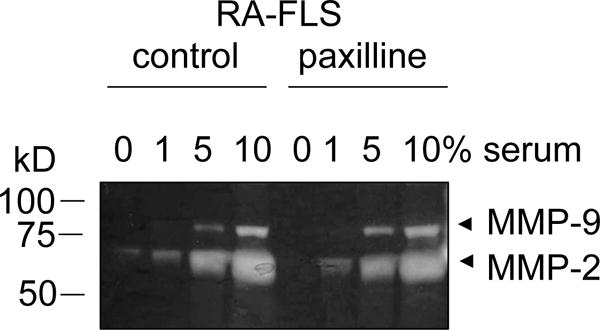
Gelatin zymography gel comparing MMP-2 and MMP-9 production in the supernatant of untreated fibroblast-like synoviocytes isolated from a patient with rheumatoid arthritis (RA-FLS; control) and paxilline-treated RA-FLS incubated in the presence of 0, 1, 5, or 10% of fetal bovine serum. Paxilline-treated RA-FLS exhibit a decrease in MMP-2 production when incubated without serum. This effect is masked by incubation with 1, 5, 10% serum.
Acknowledgments
This work was supported in part by funding from Baylor College of Medicine and National Institutes of Health grants AR059838 and NS073712 to CB.
Footnotes
The 1.44 g of Na2HPO4 (dibasic anhydrous) can be replaced by 1.81 g Na2HPO4 · 2 H2O (dibasic dihydrate) or by 2.72 g Na2HPO4 · 7H2O (dibasic septahydrate) in the preparation of PBS.
If PBS is to be kept more than a day, it can be sterilized to prevent bacterial growth by either autoclaving or sterile filtration (0.22 μm filter pores).
Choice of the lysis buffer for protein extraction will depend on the specimen but also on the target MMP in the sample. For example, MMP-23 can only be efficiently extracted using the non-ionic detergent Triton X-100 [1].
Triton X-100 is very viscous. When pipetting Triton X-100, only place the very tip of the pipet into the liquid to avoid coating the outside of the pipet with the detergent. Apply suction very slowly to allow the liquid to reach the correct volume in the pipet. If Triton X-100 is visible on the outside of the pipet, remove it with a clean tissue. Empty the pipet very slowly into the receiving container already containing water and wait until the pipet has completely emptied (can take a few minutes).
The choice of substrate in the gel will depend on the MMPs of interest. For example, MMP-2 and MMP-9 can be detected on gelatin gels, as can also be MMP-1, MMP-8, and MMP-13 although gelatin is not their preferred substrate. MMP-1 and MMP-13 are best detected on collagen gels while casein is the preferred substrate for MMP-11 and also allows for the detection of MMP-1, MMP-3, MMP-7, MMP-12, and MMP-13 [6]
Full dissolution of casein is very important to ensure homogeneity of the separating gel during zymography. Lack of homogeneity will prevent data interpretation.
Do not allow the solution to boil over as that would change its concentration.
Full dissolution of the gelatin is very important to ensure homogeneity of the separating gel during zymography. Lack of homogeneity will prevent data interpretation.
Pre-cast gels can be purchased from various vendors. These gels are provided in sealed pouches and have strict expiration dates; expired gels will not provide expected results as the substrate will have started degrading in a heterogeneous manner.
The correct acrylamide percentage to use will depend on the molecular weight of the MMP of interest. Proteins with high molecular masses will resolve better on low percentage gels whereas proteins with low molecular masses will resolve better on high percentage gels. The 10% gels are the most commonly used for zymography as they resolve proteins between 20 and 100 kDa.
The volumes given here are sufficient for a single mini-gel (9 cm × 6 cm × 0.75 cm).
Placing water at the bottom of the cylinder will help dissolve the Tris faster. In addition, warming the water to 37°C will hasten the dissolution of Tris. However, the solution should be brought to room temperature before adjusting pH.
The renaturing buffer should be prepared fresh on the day it will be used.
Brij 35 is very viscous. When pipetting Brij 35, only place the very tip of the pipet into the liquid to avoid coating the outside of the pipet with the detergent. Apply suction very slowly to allow the liquid to reach the correct volume in the pipet. If Brij 35 is visible on the outside of the pipet, remove it with a clean tissue. Empty the pipet very slowly into the receiving container already containing water and wait until the pipet has completely emptied (can take a few minutes).
Coomassie Blue R-250 can be replaced by a ready-to-use commercial solution containing colloidal Coomassie blue G-250. An advantage of the colloidal Coomassie blue is that it only requires water (no methanol or acetic acid) for destaining.
Always add concentrated acid to a large volume of water; never add water to concentrated acid as this would result in a violent exothermic reaction.
The number of cells to use for preparing cell culture supernatants for zymography will vary depending on the type of cells studied and must be tested prior to assays with a novel cell system. As a starting point, MMP-2 was detectable in the culture supernatants of human fibroblast-like synoviocytes isolated from patients with rheumatoid arthritis when cells were plated at a density of 50,000 cells per well with 0.3 ml culture medium per well in a 24-well plate [14].
Serum contains MMPs (Fig. 1); it is therefore crucial to remove all serum for the cell culture and then maintain the cells in serum-free media during MMP production. Indeed, any serum present in the culture may obscure effects from differential treatment of the cells (Fig. 2) [15,14,16,10,4,8]
The duration of incubation will differ with the cell types assessed and must be tested prior to assays with a novel cell system. As examples, when assessing MMP-2 levels in the culture supernatants of human fibroblast-like synoviocytes isolated from patients with rheumatoid arthritis or from rats with a model of rheumatoid arthritis, a 24 hr incubation gave optimal results [14,16,3,4]. In contrast, rabbit corneal fibroblasts were cultured for 72 hr before supernatant collection [17,8] whereas a 1 hr incubation was sufficient to detect pro-MMP-9 in the culture supernatants of human neutrophils [18,3].
It is crucial that MMPs are not denatured to remain functional. If the samples are to be frozen, they should be frozen immediately upon collection. In addition, every effort should be made to avoid repeated cycles of freezing and thawing of the samples.
The number of cells to use for preparing cell culture supernatants for zymography will vary depending on the type of cells studied and must be tested prior to assays with a novel cell system. As a starting point, MMP-2 was detectable in the culture supernatants of human fibroblast-like synoviocytes isolated from patients with rheumatoid arthritis when cells were plated at a density of 50,000 cells per well with 0.3 ml culture medium per well in a 24-well plate [14].
The volume of lysis buffer to add will depend on the type of tissue to be homogenized as protein extractability varies with specimens.
While BSA is commonly used, any known protein standard can be used as a control.
Cold solutions can condense atmospheric moisture on the outside of the cuvette and scatter light giving erroneous readings.
Absorbance of protein-dye conjugate increases over time and affects accuracy of protein concentration measurements.
If cell supernatants or tissue extracts contain low levels of gelatinases, a phase extraction with Triton X-114 is recommended [19].
The electrophoresis plates must be completely clean and dry at this time or the gel could incompletely polymerize or polymerize in a heterogeneous manner.
Once the polymerization agent (TEMED) is added to the gel, immediately mix and pour as the gel will start polymerizing rapidly. If the gel polymerizes too fast, reduce the ambient temperature as this will delay polymerization.
Ensure that the electrophoresis system is perfectly horizontal during gel pouring and polymerization.
It is crucial to remove all n-butanol to ensure full contact between the separating and stacking gels.
Place the comb carefully so as to not trap air bubbles.
Ensure that there is no leaking of the inside chamber buffer. If the inside chamber buffer leaks before sample loading, empty the gel box and unlock the gel. Wet the chamber gasket with running buffer and install the gel.
Volumes given here are for 12-well gels. These volumes can and should be increased when loading a gel with fewer wells.
Using recombinant MMPs as controls can be very useful for semi-quantification of the MMPs in the samples to be tested if various known concentrations are used. In addition, although MMPs are highly conserved between species in terms of molecular weight and protease activity, a positive control containing the MMP of interest in the species of choice would be beneficial.
Removing the gel from the plate or cassette is a tricky step as the gel is very fragile and easy to damage. It is safer to slide the gel off one side of the cassette rather than to try lifting it.
When removing the buffer after incubating the gel, take precautions not to damage the gel. Wearing gloves, use two fingers to gently hold the gel at the bottom of the container while pouring the liquid out from one of the corners of the container.
We recommend an overnight incubation in developing buffer. If, however, the bands are too strong, the developing time can be reduced to 4 hrs. If, in contrast, the bands are very faint, gels can be incubated in the developing buffer for up to 48 hrs.
The gel should be stained in Coomassie Blue staining solution for 1 hour or until the gel is uniformly dark blue.
If no zones of digestion are evident, use a positive control (e.g. recombinant MMP-2 or MMP-9 for gelatin gels and MMP-1 for casein gels). If these controls are visible, the test samples may be devoid of proteases specific to the chosen substrate or the MMP concentration may be lower than expected in which case increase the incubation time in developing buffer or concentrate the test samples. If these controls are not visible, check that (i) the samples were not boiled prior to gel loading as this would denature the proteins, (ii) the sample loading buffer does not contain anything that inhibits the proteases (e.g. EDTA), and (iii) the renaturing buffer contains sufficient amounts of Triton X-100.
Destaining the gel stained with a ready-to-use commercial solution containing colloidal Coomassie Blue for an hour may not be optimal in all situations. More destaining can be obtained with a second incubation of 1 hr in fresh water. The gel can be scanned multiple times during the destaining process to catch the best contrast in the bands.
If the background contains blotches or streaks, this could indicate an uneven distribution of the substrate protein in the gel. Ensure that the gelatin of casein solution used to prepare the separating gel contains no undissolved materials
References
- 1.Pei D, Kang T, Qi H. Cysteine array matrix metalloproteinase (CA-MMP)/MMP-23 is a type II transmembrane matrix metalloproteinase regulated by a single cleavage for both secretion and activation. J Biol Chem. 2000;275(43):33988–33997. doi: 10.1074/jbc.M006493200. [DOI] [PubMed] [Google Scholar]
- 2.Baranger K, Rivera S, Liechti FD, Grandgirard D, Bigas J, Seco J, Tarrago T, Leib SL, Khrestchatisky M. Endogenous and synthetic MMP inhibitors in CNS physiopathology. Prog Brain Res. 2014;214:313–351. doi: 10.1016/B978-0-444-63486-3.00014-1. [DOI] [PubMed] [Google Scholar]
- 3.Fainardi E, Castellazzi M, Bellini T, Manfrinato MC, Baldi E, Casetta I, Paolino E, Granieri E, Dallocchio F. Cerebrospinal fluid and serum levels and intrathecal production of active matrix metalloproteinase-9 (MMP-9) as markers of disease activity in patients with multiple sclerosis. Mult Scler. 2006;12:294–301. doi: 10.1191/135248506ms1274oa. [DOI] [PubMed] [Google Scholar]
- 4.Baeza M, Garrido M, Hernández-Ríos P, Dezerega A, García-Sesnich J, Strauss F, Aitken JP, Lesaffre E, Vanbelle S, Gamonal J, Brignardello-Petersen R, Tervahartiala T, Sorsa T, Hernández M. Diagnostic accuracy for apical and chronic periodontitis biomarkers in gingival crevicular fluid: an exploratory study. J Clin Periodontol. 2015 doi: 10.1111/jcpe.12479. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 5.Thomadaki K, Bosch J, Oppenheim F, Helmerhorst E. The diagnostic potential of salivary protease activities in periodontal health and disease. Oral Dis. 2013;19:781–788. doi: 10.1111/odi.12069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Snoek-van Beurden PA, Von den Hoff JW. Zymographic techniques for the analysis of matrix metalloproteinases and their inhibitors. Biotechniques. 2005;37:73–83. doi: 10.2144/05381RV01. [DOI] [PubMed] [Google Scholar]
- 7.Cathcart J, Pulkoski-Gross A, Cao J. Targeting matrix metalloproteinases in cancer: bringing new life to old ideas. Genes Dis. 2015;2:26–34. doi: 10.1016/j.gendis.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cuéllar VG, Cuéllar JM, Kirsch T, Strauss EJ. Correlation of synovial fluid biomarkers with cartilage pathology and associated outcomes in knee arthroscopy. Arthroscopy. 2015 doi: 10.1016/j.arthro.2015.08.033. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 9.Murphy G, Nagase H. Reappraising metalloproteinases in rheumatoid arthritis and osteoarthritis: destruction or repair? Nat Clin Pract Rheumatol. 2008;4:128–135. doi: 10.1038/ncprheum0727. [DOI] [PubMed] [Google Scholar]
- 10.Chang J, Wehner S, Schäfer N, Sioutis M, Bortscher S, Hirner A, Kalff JC, Bauer AJ, Overhaus M. Iatrogenic extracellular matrix disruption as a local trigger for postoperative ileus. J Surg Res. 2012;178:632–639. doi: 10.1016/j.jss.2012.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Medina C, Radomski MW. Role of matrix metalloproteinases in intestinal inflammation. J Pharmacol Exp Ther. 2006;318:933–938. doi: 10.1124/jpet.106.103465. [DOI] [PubMed] [Google Scholar]
- 12.Hopps E, Caimi G. Matrix metalloproteases as a pharmacological target in cardiovascular diseases. Eur Rev Med Pharmacol Sci. 2015;19:2583–2589. [PubMed] [Google Scholar]
- 13.Heussen C, Dowdle EB. Electrophoretic analysis of plasminogen activators in polyacrylamide gels containing sodium dodecyl sulfate and copolymerized substrates. Analytical Biochem. 1980;102:196–202. doi: 10.1016/0003-2697(80)90338-3. [DOI] [PubMed] [Google Scholar]
- 14.Hu X, Laragione T, Sun L, Koshy S, Jones KR, Ismailov II, Yotnda P, Horrigan FT, Gulko PS, Beeton C. KCa1.1 potassium channels regulate key pro-inflammatory and invasive properties of fibroblast-like synoviocytes in rheumatoid arthritis. J Biol Chem. 2012;287:4014–4022. doi: 10.1074/jbc.M111.312264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu X, Beeton C. Detection of functional matrix metalloproteinases by zymography. J Vis Exp. 2010;45 doi: 10.3791/2445. pii 2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanner MR, Hu X, Huq R, Tajhya RB, Sun L, Khan FS, Laragione T, Horrigan FT, Gulko PS, Beeton C. KCa1.1 inhibition attenuates fibroblast-like synoviocyte invasiveness and ameliorates rat models of rheumatoid arthritis. Arthritis Rheumatol. 2015;67:96–106. doi: 10.1002/art.38883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pan H, Chen J, Xu J, Chen M, Ma R. Antifibrotic effect by activation of peroxisome proliferator-activated receptor-gamma in corneal fibroblasts. Mol Vis. 2009;15:2279–2286. [PMC free article] [PubMed] [Google Scholar]
- 18.Puente J, Jaque M, Carrasco C, Cruz C, Valenzuela M, Wolf M, Mosnaim A. Triptan drugs, natural killer cell cytotoxicity, and neutrophils pro-matrix metalloproteinase-9 secretion. Headache. 2008;48:1482–1489. doi: 10.1111/j.1526-4610.2008.01136.x. [DOI] [PubMed] [Google Scholar]
- 19.Toth M, Sohail A, Fridman R. Assessment of gelatinases (MMP-2 and MMP-9) by gelatin zymography. Methods Mol Biol. 2012;878:121–135. doi: 10.1007/978-1-61779-854-2_8. [DOI] [PubMed] [Google Scholar]