

Significance
The duplication of the genetic information (DNA) requires high accuracy to prevent potentially deleterious genetic alterations (mutations). The fidelity of this reaction depends on DNA polymerase selectivity and proofreading functions, postreplicative mismatch repair (MMR), and the abundance of dNTPs, the building blocks of DNA. Here, in a genome-wide screen in budding yeast, we uncovered a group of genes required for high-fidelity DNA replication. When these genes are absent, cells are prone to incorporate incorrect nucleotides, and consequently they heavily rely on DNA polymerase functions and MMR to prevent severe hypermutability. These findings suggest that similar genetic interactions could play a role in human cancer, where inactivation of these genes might act as “minidrivers” that facilitate tumor evolution.
Keywords: DNA replication fidelity, mismatch repair, CTP biosynthesis, DNA polymerases, dNTP pool imbalance
Abstract
Eukaryotic DNA replication fidelity relies on the concerted action of DNA polymerase nucleotide selectivity, proofreading activity, and DNA mismatch repair (MMR). Nucleotide selectivity and proofreading are affected by the balance and concentration of deoxyribonucleotide (dNTP) pools, which are strictly regulated by ribonucleotide reductase (RNR). Mutations preventing DNA polymerase proofreading activity or MMR function cause mutator phenotypes and consequently increased cancer susceptibility. To identify genes not previously linked to high-fidelity DNA replication, we conducted a genome-wide screen in Saccharomyces cerevisiae using DNA polymerase active-site mutants as a “sensitized mutator background.” Among the genes identified in our screen, three metabolism-related genes (GLN3, URA7, and SHM2) have not been previously associated to the suppression of mutations. Loss of either the transcription factor Gln3 or inactivation of the CTP synthetase Ura7 both resulted in the activation of the DNA damage response and imbalanced dNTP pools. Importantly, these dNTP imbalances are strongly mutagenic in genetic backgrounds where DNA polymerase function or MMR activity is partially compromised. Previous reports have shown that dNTP pool imbalances can be caused by mutations altering the allosteric regulation of enzymes involved in dNTP biosynthesis (e.g., RNR or dCMP deaminase). Here, we provide evidence that mutations affecting genes involved in RNR substrate production can cause dNTP imbalances, which cannot be compensated by RNR or other enzymatic activities. Moreover, Gln3 inactivation links nutrient deprivation to increased mutagenesis. Our results suggest that similar genetic interactions could drive mutator phenotypes in cancer cells.
The fidelity of DNA replication is strongly influenced by three processes (1–3): (i) nucleotide selectivity, wherein replicative DNA polymerases select the correct dNTP to be incorporated; (ii) DNA polymerase proofreading activity, which excises wrongly incorporated nucleotides by using the DNA polymerase 3′-to-5′ exonuclease activity; and (iii) mismatch repair (MMR) (4, 5), a DNA replication-coupled repair mechanism (6, 7), which corrects errors that escaped proofreading. Furthermore, the balance and overall concentration of dNTPs not only affect nucleotide selection but also influence DNA polymerase proofreading activity (8). A central player in the biosynthesis of dNTPs is the ribonucleotide reductase (RNR) holoenzyme, which catalyzes the reduction of NDPs to dNDPs (9, 10). In Saccharomyces cerevisiae, RNR is composed of two identical Rnr1 large subunits that associate with two smaller subunits represented by Rnr2 and Rnr4 (11, 12). In addition, a second large subunit has been identified (Rnr3), which is induced upon DNA damage and when overexpressed can rescue the rnr1 lethal phenotype (13).
The interplay between nucleotide selectivity, DNA polymerase proofreading activity, and MMR guarantees the high accuracy of DNA replication, resulting in less than one mutation per genome duplication in S. cerevisiae (14–16). Perturbations in any of these processes are linked to increased mutation rates and, in higher eukaryotes, to increased cancer susceptibility. Accordingly, mutations inactivating DNA polymerase proofreading function or MMR genes are associated with familial colorectal/ovarian cancer (17, 18) and Lynch syndrome (19), respectively.
Eukaryotic DNA synthesis is accomplished by three essential DNA polymerases: Polα, Polε, and Polδ (called in S. cerevisiae Pol1, Pol2, and Pol3, respectively). Polα initiates DNA synthesis at replication origins and at every Okazaki fragment, albeit with low processivity and lack of proofreading activity. Subsequently, the synthesis is taken over by one of the two high-fidelity DNA polymerases, Polε or Polδ. Polε replicates the leading strand, whereas Polδ synthesizes the lagging strand. This “division of labor during eukaryotic DNA replication” model (20) was initially proposed based on the characterization of S. cerevisiae strains carrying active-site mutations in DNA polymerases (e.g., pol2-M644G and pol3-L612M), which confer a weak mutator phenotype with a characteristic mutator signature, without compromising DNA polymerase proofreading activity (21, 22).
Given the intrinsic mechanistic differences between leading- and lagging-strand DNA synthesis, it has been proposed that the two strands may also differ in terms of repair efficiencies. Supporting this idea, reports have shown that errors made in the lagging strand (especially those generated by Pol1) are more efficiently repaired by MMR than errors made in the leading strand (23, 24). In addition, inactivation of yeast Exo1, a 5′–3′ exonuclease that is involved in MMR (25), increased the mutation rates to a greater extent when combined with mutant variants of the lagging-strand polymerases (pol1-L868M or pol3-L612M) than when combined with a mutator variant of the leading-strand polymerase (pol2-M644G) (26, 27). This suggests that lagging-strand MMR might be more dependent on Exo1 function than leading-strand MMR.
In this study, we conducted a genome-wide screen in S. cerevisiae in which we used three active-site DNA polymerase mutants to identify genes that prevent the accumulation of mutations. We uncovered a group of genes that are important for ensuring the fidelity of DNA replication, especially when DNA polymerase or MMR function is compromised. We discovered that inactivation of either the transcription factor Gln3 or the CTP synthetase Ura7 results in reduced dCTP concentrations and DNA damage checkpoint activation, with concomitant up-regulation of the other three dNTPs. Moreover, we showed that glutamine supplementation suppresses mutagenesis in gln3Δ mutants, providing evidence for a link between nutrient deprivation and mutator phenotypes. Mutation spectra analysis in ura7Δ and gln3Δ mutants revealed a mutation signature dominated by C-to-T transitions, which is likely driven by an increased dTTP:dCTP ratio observed in the absence of either of these two genes. Overall, we have found an additional requirement for dNTP pool homeostasis, defined by genes that affect the production of one of the substrates used by RNR. We demonstrated that inactivation of these genes creates a dNTP pool imbalance with high mutagenic potential that, in combination with genetic alterations affecting DNA polymerase nucleotide selectivity, proofreading activity, or MMR, causes a strong mutator phenotype.
Results
A Genome-Wide Screen Uncovers Genes Required for DNA Replication Fidelity.
Numerous studies in S. cerevisiae have shown that DNA polymerase mutations, in combination with MMR mutant alleles, lead to synergistic mutator interactions (28–31). We rationalized that, by using DNA polymerase mutants as a “sensitized mutator background,” we may identify previously unrecognized genes that contribute to DNA replication fidelity. For this purpose, we performed a genome-wide screen where we crossed a nonessential yeast deletion collection (∼4,800 strains) with either a wild-type (WT) strain, or one of three DNA polymerase active-site mutants (pol1-L868M, pol2-M644G, and pol3-L612M), followed by mutator phenotype evaluation. These active-site mutations cause a mild mutator phenotype, allowing us to screen for mutational enhancers. We engineered a modified version of the synthetic genetic array (SGA) protocol (32) to select for haploid cells that simultaneously carry the DNA polymerase mutation, a nonessential gene deletion, and two mutator reporters, one frameshift reporter (lys2-10A), and one forward inactivation reporter (CAN1) (Fig. 1A). To increase the robustness of the screen, we crossed in quadruplicate the deletion collection with the four query strains. Due to the large number of strains, we aimed to screen for mutator phenotypes (∼4,800 strains × 4 queries × 4 ∼ 77,000 strains), we set up a “semi–high-throughput” method in 96-well format, in which each plate contained up to 24 different genotypes. Cells were spotted in yeast extract–peptone–dextrose (YPD) agar plates, grown, and replica plated on reporter plates lacking lysine (lys2-10A frameshift assay) or containing canavanine (CAN1 inactivation assay). After 4 d of incubation, plates were scored for increased number of colonies, which is indicative of potential mutator phenotypes. Fig. 1B illustrates two single mutants showing increased mutator phenotypes. The msh6Δ mutant, which shows a partial MMR deficiency (33), resulted in frameshifts (lysine− plate) and increased CAN1 inactivating mutations (+canavanine plate). Moreover, the ubc13Δ mutant that is defective in error-free postreplication repair (34) caused increased CAN1 inactivation but not frameshifts.
Fig. 1.

Genome-wide screen identifies genes that affect DNA replication fidelity in S. cerevisiae. (A) Strategy used to cross the nonessential gene deletion collection with active-site DNA polymerase mutants. (B) To screen for mutator phenotypes in 96-well format, strains were spotted on YPD, grown, and replica plated on mutator reporter plates. Increased number of colonies is indicative of a potential mutator phenotype. On the right side (zoom-in), msh6Δ results in increased frameshifts (lysine−) and CAN1 mutations (+canavanine), whereas ubc13Δ increases CAN1 mutations, exclusively.
A previous screen done in S. cerevisiae identified 33 genes with different roles in DNA replication and repair (among others) that, when inactivated, caused elevated mutator phenotypes (35). In contrast, we concentrated our work on deletion mutants that confer strong mutator phenotypes in the presence of DNA polymerase mutant alleles.
Qualitative mutator analysis of the double mutants uncovered a group of genes (GLN3, SHM2, URA7, RRM3, and EXO1) that, when inactivated in specific DNA polymerase mutant backgrounds, resulted in strong mutator phenotypes, evidenced by an increased abundance of canavanine-resistant (CanR) colonies (three representative examples are shown in Fig. S1). None of these double mutants (with the exception of exo1Δ combinations) showed an increased mutator phenotype in the lys2-10A frameshift assay, suggesting that the CanR mutator phenotypes are likely a consequence of base substitutions (Fig. S1).
Fig. S1.

Representative pictures of reporter plates (zoom-in) illustrating mutator phenotypes in some S. cerevisiae double-mutant strains. Inactivation of Exo1 in lagging-strand DNA polymerase mutant backgrounds (pol1-L868M or pol3-L612M) results in frequent CAN1 inactivating mutations and frameshifts in lys2-10A allele, indicated by the higher abundance of CanR and lysine+ colonies, respectively. Similarly, inactivation of Gln3 or Shm2 in lagging-strand DNA polymerase mutant backgrounds, results in increased CAN1 inactivation, but not frameshifts.
Besides EXO1, all other identified genes have not been previously linked to an increased mutator phenotype. Intriguingly, most of these gene deletions (GLN3, SHM2, URA7, and EXO1) caused strong mutator phenotypes exclusively in the lagging-strand DNA polymerase mutant backgrounds (pol1-L868M and pol3-L612M). Moreover, gln3Δ, shm2Δ, ura7Δ, and rrm3Δ did not cause a mutator phenotype in the presence of WT DNA polymerases, suggesting that DNA polymerases buffer against mutations in the absence of these genes.
Gln3, Shm2, and Ura7 regulate genes or metabolic reactions that are linked to the synthesis of purines and pyrimidines. Specifically, Ura7 converts UTP into CTP, which is then used as substrate for the production of dCTP and dTTP (36, 37); Gln3 is a transcription factor that controls nitrogen metabolism (38, 39); and last, Shm2 is a serine hydroxymethyltransferase part of the one-carbon (C1) metabolism (40, 41). On the other hand, Exo1 and the helicase Rrm3 belong to the group of proteins implicated in DNA repair and genome stability (42–45).
To validate our initial findings, we first generated de novo single and double mutants and determined their mutation rates by fluctuation analysis (Table 1). In agreement with initial findings, gln3Δ, shm2Δ, ura7Δ, and rrm3Δ single mutants showed mutation rates that were indistinguishable from WT strain. Notably, we found that inactivation of GLN3, SHM2, URA7, or EXO1 resulted in a synergistic increase in the mutation rates when combined with mutator variants of the lagging-strand DNA polymerases (pol1-L868M or pol3-L612M), but not when combined with a leading-strand polymerase mutant (pol2-M644G) (Table 1). The double-mutants pol1-L868M ura7Δ and pol1-L868M gln3Δ showed the highest CAN1 mutation rates, which were 323- and 293-fold higher than WT (or 65- and 59-fold higher than pol1-L868M mutant), respectively. A similar synergistic increase was observed in ura7Δ or gln3Δ mutants in combination with pol1-L868M or pol3-L612M (but not with pol2-M644G) in an alternative forward mutation assay based on the inactivation of the URA3 gene (Table S1). Thus, these results further demonstrate that these double-mutant combinations result in an overall increased mutator phenotype. To test whether the mutator phenotype observed in pol3-L612M ura7Δ (or pol3-L612M gln3Δ) double mutant depends on error-prone translesion synthesis (TLS) DNA polymerases (Polζ, Polη, or Rev1) (46), we measured the mutation rates in pol3-L612M gln3Δ and pol3-L612M ura7Δ strains lacking Polζ (rev3Δ), Polη (rad30Δ), or Rev1 (rev1Δ) polymerases. We found that, in the absence of TLS polymerases, the mutation rates were not reduced (Table S2); therefore, these genes are not responsible for the mutator phenotype.
Table 1.
Mutation rate analysis of the mutants identified in this screen in combination with DNA polymerase active-site mutant alleles
| Mutation rate (fold increase)* CanR | ||||
| Relevant genotype | WT | pol1-L868M | pol2-M644G | pol3-L612M |
| WT | 7.2 [5.7–9.0] × 10−8 (1) | 3.9 [3.3–4.9] × 10−7 (5) | 8.4 [7.3–10.6] × 10−7 (12) | 9.3 [7.7–11.6] × 10−7 (13) |
| exo1Δ | 7.4 [6.3–9.8] × 10−7 (10) | 5.7 [3.1–8.1] × 10−6 (80) | 1.9 [1.1–2.9] × 10−6 (26) | 6.5 [3.6–10.8] × 10−6 (91) |
| gln3Δ | 1.0 [0.8–1.2] × 10−7 (1) | 2.1 [1.4–4.5] × 10−5 (293) | 3.3 [2.6–6.0] × 10−7 (5) | 9.1 [7.3–18.2] × 10−6 (127) |
| shm2Δ | 1.2 [1.1–1.7] × 10−7 (2) | 1.7 [1.0–2.0] × 10−6 (23) | 5.5 [3.9–7.3] × 10−7 (8) | 3.6 [2.1–4.7] × 10−6 (50) |
| ura7Δ | 1.0 [0.9–1.5] × 10−7 (1) | 2.3 [1.3–4.1] × 10−5 (323) | 1.1 [0.7–1.5] × 10−6 (15) | 1.6 [1.1–2.6] × 10−5 (218) |
| rrm3Δ | 1.1 [0.8–1.5] × 10−7 (2) | 3.5 [2.1–4.4] × 10−7 (5) | 2.8 [1.9–4.8] × 10−6 (40) | 3.6 [2.6–6.0] × 10−6 (50) |
Median rates of inactivation of CAN1 gene (CanR) with 95% confidence interval in square brackets and fold increase relative to the WT in parentheses.
Table S1.
Mutation rate analysis (URA3 inactivation) for several mutant strains
| Mutation rate (fold increase)* 5-FOAR | ||||
| Relevant genotype | wt-POL | pol1-L868M | pol2-M644G | pol3-L612M |
| WT | 9.5 [8.0–14.0] × 10−8 (1) | 4.8 [3.6–6.7] × 10−7 (5) | 1.4 [1.0–2.4] × 10−6 (15) | 6.6 [5.2–10.8] × 10−7 (7) |
| gln3Δ | nd | 1.9 [1.3–2.2] × 10−5 (199) | 2.5 [1.3–3.9] × 10−6 (26) | 6.3 [4.1–15.1] × 10−6 (66) |
| ura7Δ | nd | 1.1 [0.8–1.3] × 10−5 (111) | 2.1 [1.8–3.2] × 10−6 (22) | 1.8 [0.7–3.8] × 10−5 (192) |
Table S2.
Mutation rate analysis (CAN1 inactivation) in pol3-L612M gln3Δ or pol3-L612M ura7Δ strains lacking TLS DNA polymerases
| Relevant genotype | Mutation rate (fold increase)* CanR |
| pol3-L612M gln3Δ rev1Δ | 2.9 [2.3–3.4] × 10−5 (399) |
| pol3-L612M gln3Δ rev3Δ | 2.3 [1.3–3.3] × 10−5 (327) |
| pol3-L612M gln3Δ rad30Δ | 1.8 [0.9–2.8] × 10−5 (247) |
| pol3-L612M ura7Δ rev1Δ | 3.7 [2.7–4.9] × 10−5 (521) |
| pol3-L612M ura7Δ rev3Δ | 1.9 [1.3–4.0] × 10−5 (264) |
| pol3-L612M ura7Δ rad30Δ | 4.3 [3.4–6.2] × 10−5 (597) |
Unlike gln3Δ, ura7Δ, exo1Δ, and shm2Δ mutants that predominantly interacted with pol1-L868M and pol3-L612M, the rrm3Δ mutant mainly interacted with pol2-M644G and pol3-L612M, resulting in mutation rates 40-fold and 50-fold higher than WT, respectively (or threefold and fourfold higher than the polymerase mutants) (Table 1).
Inactivation of Gln3 or Ura7 Results in a Mutagenic Potential That Is Counteracted by DNA Polymerase Proofreading Function and MMR Activity.
Because DNA replication fidelity depends not only on nucleotide selectivity but also on DNA polymerase proofreading activity and MMR function, we tested the consequences of the loss of Gln3, Shm2, Ura7, and Rrm3 in three genetic backgrounds with partially compromised MMR function (exo1Δ, msh3Δ, and msh6Δ) (33, 47), complete lack of MMR (msh2Δ) (33), or in the absence of Polε proofreading activity (pol2-04 mutant allele) (48). Inactivation of GLN3 or URA7 in all tested backgrounds, with the exception of msh3Δ, resulted in strong synergies in the CAN1 inactivation assay (Table 2 and Table S3). For example, exo1Δ gln3Δ resulted in a 15-fold higher mutation rate than exo1Δ, and msh6Δ ura7Δ resulted in 40-fold increase over msh6Δ strain. Inactivation of RRM3 or SHM2 in an msh6Δ background caused a smaller increase in CAN1 inactivation (5.1- and 2.2-fold over msh6Δ strain, respectively) and had no effect in the CAN1 inactivation rate in an exo1Δ background.
Table 2.
Mutation rate analysis of the mutants identified in this screen in combination with alleles causing reduced DNA replication fidelity
| Mutation rate (fold increase)* CanR | ||||
| Relevant genotype | WT | exo1Δ | msh6Δ | pol2-04 |
| WT | 7.2 [5.7–9.0] × 10−8 (1) | 7.4 [6.3–9.8] × 10−7 (10) | 9.6 [7.8–11.7] × 10−7 (13) | 6.2 [4.3–7.6] × 10−7 (6) |
| gln3Δ | 1.0 [0.8–1.2] × 10−7 (1) | 1.1 [0.8–1.4] × 10−5 (146) | 2.4 [1.7–3.4] × 10−5 (334) | 1.1 [0.9–1.6] × 10−5 (154) |
| shm2Δ | 1.2 [0.8–2.8] × 10−7 (2) | 8.4 [7.1–10.5] × 10−7 (12) | 2.1 [1.3–2.6] × 10−6 (30) | 1.5 [1.1–2.3] × 10−6 (22) |
| ura7Δ | 1.0 [0.9–1.5] × 10−7 (1) | 1.9 [0.8–2.4] × 10−5 (261) | 3.8 [3.2–8.5] × 10−5 (524) | 2.5 [1.8–5.2] × 10−5 (354) |
| rrm3Δ | 1.1 [0.8–1.5] × 10−7 (2) | 6.3 [4.3–7.6] × 10−7 (9) | 4.9 [3.6–7.3] × 10−6 (68) | 1.4 [0.9–1.8] × 10−6 (19) |
Median rates of inactivation of CAN1 gene (CanR) with 95% confidence interval in square brackets and fold increase relative to the WT in parentheses.
Table S3.
Mutation rate analysis for several mutants
| Mutation rate (fold increase)* | |||
| Relevant genotype | CanR | Lys+ | Thr+ |
| WT | 7.2 [5.7–9.0] × 10−8 (1) | 1.5 [0.8–2.2] × 10−8 (1) | 2.1 [1.4–3.2] × 10−9 (1) |
| gln3Δ | 1.0 [0.8–1.2] × 10−7 (1) | 1.6 [1.1–3.7] × 10−8 (1) | 2.4 [1.7–3.7] × 10−9 (1) |
| shm2Δ | 1.2 [1.1–1.7] × 10−7 (2) | 3.1 [1.2–5.0] × 10−8 (2) | 2.6 [1.7–5.6] × 10−9 (1) |
| ura7Δ | 1.0 [0.9–1.5] × 10−7 (1) | 1.4 [1.0–2.5] × 10−8 (1) | 1.9 [1.2–5.6] × 10−9 (1) |
| rrm3Δ | 1.1 [0.8–1.5] × 10−7 (2) | 2.4 [1.3–3.0] × 10−8 (2) | 4.6 [2.6–7.9] × 10−9 (2) |
| exo1Δ | 7.4 [6.3–9.8] × 10−7 (10) | 1.4 [0.9–1.8] × 10−7 (10) | 8.7 [6.1–15.0] × 10−9 (4) |
| exo1Δ gln3Δ | 1.1 [0.8–1.4] × 10−5 (146) | 1.2 [0.7–1.6] × 10−6 (83) | 3.5 [2.7–5.0] × 10−7 (170) |
| exo1Δ shm2Δ | 8.4 [7.1–10.1] × 10−7 (12) | 3.5 [2.4–5.1] × 10−7 (24) | 1.8 [1.1–2.5] × 10−8 (9) |
| exo1Δ ura7Δ | 1.9 [0.8–2.4] × 10−5 (261) | 1.3 [0.7–1.9] × 10−6 (89) | 6.6 [4.9–8.3] × 10−7 (319) |
| exo1Δ rrm3Δ | 6.3 [4.3–7.6] × 10−7 (9) | 1.3 [1.0–1.8] × 10−7 (9) | 2.5 [2.0–3.1] × 10−8 (12) |
| msh2Δ | 5.4 [4.4–7.2] × 10−6 (75) | 9.9 [8.1–10.8] × 10−5 (6,771) | 6.3 [5.2–12.8] × 10−6 (3,053) |
| msh2Δ gln3Δ | 1.3 [0.8–2.1] × 10−5 (177) | 8.7 [6.9–14.9] × 10−5 (5,972) | 4.5 [3.1–6.5] × 10−6 (2,149) |
| msh2Δ shm2Δ | 7.4 [4.8–8.6] × 10−6 (104) | 1.4 [1.1–2.1] × 10−4 (9,737) | 6.1 [4.4–8.2] × 10−6 (2,918) |
| msh2Δ ura7Δ | 3.5 [2.6–4.2] × 10−5 (492) | 6.1 [4.7–8.8] × 10−5 (4,161) | 5.7 [4.1–8.5] × 10−6 (2,738) |
| msh2Δ rrm3Δ | 1.7 [1.2–2.6] × 10−5 (234) | 1.1 [0.9–1.2] × 10−4 (7,198) | 1.6 [1.1–2.4] × 10−5 (7,491) |
| msh3Δ | 1.1 [0.8–1.2] × 10−7 (1) | 2.5 [2.0–3.0] × 10−7 (17) | 2.7 [2.0–4.2] × 10−8 (13) |
| msh3Δ gln3Δ | 1.6 [1.1–2.6] × 10−7 (2) | 1.9 [1.4–2.3] × 10−7 (13) | 1.8 [1.5–1.9] × 10−8 (9) |
| msh3Δ shm2Δ | 1.5 [1.3–2.9] × 10−7 (2) | 1.2 [1.3–2.6] × 10−7 (12) | 2.7 [1.7–3.7] × 10−8 (13) |
| msh3Δ ura7Δ | 1.5 [1.3–2.0] × 10−7 (2) | 1.2 [0.8–2.1] × 10−7 (8) | 1.7 [1.1–3.0] × 10−8 (8) |
| msh3Δ rrm3Δ | 2.6 [1.9–3.4] × 10−7 (4) | 3.7 [3.5–4.3] × 10−7 (25) | 6.1 [4.9–8.3] × 10−8 (30) |
| msh6Δ | 9.6 [7.8–11.7] × 10−7 (13) | 1.3 [0.9–1.6] × 10−6 (86) | 1.3 [0.9–1.6] × 10−8 (6) |
| msh6Δ gln3Δ | 2.4 [1.7–3.4] × 10−5 (334) | 1.2 [0.7–4.0] × 10−6 (80) | 1.0 [0.6–1.6] × 10−7 (48) |
| msh6Δ shm2Δ | 2.1 [1.3–2.6] × 10−6 (30) | 1.0 [0.9–1.3] × 10−6 (71) | 3.5 [2.7–5.4] × 10−8 (17) |
| msh6Δ ura7Δ | 3.8 [3.2–8.5] × 10−5 (524) | 8.6 [6.6–20.6] × 10−7 (59) | 9.2 [4.5–26.2] × 10−8 (44) |
| msh6Δ rrm3Δ | 4.9 [3.6–7.3] × 10−6 (68) | 9.1 [6.1–13.8] × 10−7 (62) | 5.5 [3.9–6.8] × 10−8 (26) |
| dun1Δ | 5.6 [4.2–9.1] × 10−8 (1) | 2.1 [0.9–3.5] × 10−8 (1) | 2.4 [1.7–5.2] × 10−9 (1) |
| pol1-L868M dun1Δ | 9.6 [5.4–15.0] × 10−8 (1) | nd | nd |
| pol2-04 gln3Δ dun1Δ | 8.6 [6.4–16.5] × 10−8 (1) | nd | nd |
| pol2-04 dun1Δ | 9.4 [6.5–17.4] × 10−8 (1) | nd | nd |
| pol3-L612M gln3Δ dun1Δ | 3.1 [1.8–4.0] × 10−6 (43) | nd | nd |
| exo1Δ ura7Δ dun1Δ | 8.5 [5.4–11.3] × 10−7 (12) | 2.6 [1.8–3.6] × 10–7 (18) | 8.3 [4.7–10.7] × 10−9 (4) |
Exo1Δ gln3Δ and exo1Δ ura7Δ double mutants also showed increased levels of frameshift mutations (Table S3). We confirmed these frameshifts by sequencing 50 independent hom3-10 revertants (Thr+) from exo1Δ gln3Δ and exo1Δ ura7Δ double mutants. We found that all Thr+ revertants contained the same single-nucleotide deletion (−1 T) in a run of 7 Ts (starting at nucleotide 646), a hotspot for frameshift mutations previously identified in MMR-deficient mutants (33).
Mutational analysis in msh2Δ shm2Δ, msh2Δ gln3Δ, msh2Δ ura7Δ, and msh2Δ rrm3Δ double mutants revealed 1.4-, 2.4-, 6.6-, and 3.1-fold increase in the CAN1 inactivation rate (compared with msh2Δ strain), respectively, without a significant impact on frameshift mutations (Table S3). With the exception of msh2Δ shm2Δ mutant, all other double mutants showed CAN1 inactivation rates significantly higher than msh2Δ strain (according to 95% confidence intervals).
Inactivation of MSH3 in gln3Δ, shm2Δ, ura7Δ, or rrm3Δ mutant backgrounds revealed no major changes in the mutation rates, with the exception of msh3Δ rrm3Δ, which showed a small increase in all three assays (Table S3). These observations are in agreement with the predominant role of Msh6 over Msh3 in MMR, specifically in the repair of base substitutions (33).
Double-mutant combinations of gln3Δ or ura7Δ with pol2-04 revealed synergistic increases in the CAN1 mutation rates (Table 2). For example, pol2-04 gln3Δ and pol2-04 ura7Δ double mutants resulted in 26- and 59-fold increase over pol2-04, respectively. Together, these findings indicate that loss of Gln3 or Ura7 predisposes to base substitutions that are prevented by DNA polymerases or corrected by MMR. On the other hand, inactivation of Shm2 or Rrm3 in msh6Δ, msh2Δ, or pol2-04 mutant backgrounds resulted in a less pronounced increase in the mutation rates (Table 2 and Table S3). These results may reflect a smaller contribution of Shm2 and Rrm3 in DNA replication fidelity, compared with either Ura7 or Gln3.
Despite several attempts, we could not obtain a ura7Δ mutant in a Polδ proofreading-defective background (pol3-01) (28) by yeast matings. This observation suggested that the pol3-01 ura7Δ double-mutant combination might result in synthetic lethality or severe growth defect. Similar genetic interactions have been previously described for pol3-01 in combination with mutations that abolish MMR function (e.g., msh2Δ combined with pol3-01) (29), supporting a model in which the extreme mutator phenotype results in “error-induced extinction” (28, 31). Indeed, plasmid-shuffling experiments in a haploid pol3Δ ura7Δ strain complemented by a pol3-01 plasmid (“ura7Δ + pol3-01”) revealed a strong growth defect (Fig. 2A), which was less severe in a diploid homozygous pol3Δ ura7Δ strain complemented with the same plasmid (Fig. 2B). This observation is in agreement with the ∼10-fold higher error extinction threshold that a diploid strain (compared with haploids) can tolerate (49). Moreover, even a diploid ura7Δ + pol3-01 strain showed reduced proliferation rates compared with isogenic strains transformed with WT POL3 plasmid (Fig. 2C). As ura7Δ + pol3-01 haploid mutants showed severe growth defects, we determined CAN1 mutation rates in ura7Δ + pol3-01 diploid strains hemizygous for the CAN1 locus (CAN1/can1Δ) (Fig. 2D). Remarkably, we found that ura7Δ + pol3-01 diploid strain has a mutation rate of 1.6 × 10−3 (6.482-fold higher than a WT diploid strain). This mutation rate is at the error extinction threshold (1 × 10−3 mutations per cell division) reported for haploids (49) but is below the one for diploids (1 × 10−2 mutations per cell division), arguing that the severe growth defect observed in haploids is a consequence of error-induced extinction. These findings demonstrate that Ura7 inactivation leads to an extreme mutator phenotype, which in the absence of Pol3 proofreading activity is almost incompatible with cell viability.
Fig. 2.

Inactivation of URA7 in Pol3 proofreading-defective background (pol3-01) results in severe growth defects and synergistic increases in mutation rates. (A) Plasmid shuffling strains pol3Δ, pol3Δ ura7Δ, and pol3Δ msh2Δ [all haploids (n) complemented with a WT POL3-URA3 plasmid] were transformed with either WT POL3 or pol3-01 LEU2-plasmids. Transformants were grown on Ura− Leu− SD plates or 5-FOA–containing plates to select against WT POL3-URA3 plasmid. Double-mutant combination msh2Δ + pol3-01 serves as positive control for a synthetic lethal interaction. (B) Haploid (n) or diploid homozygous (2n) pol3Δ ura7Δ mutants expressing either WT POL3 or mutant pol3-01, were grown as in A. (C) Proliferation curves. Diploid homozygous pol3Δ or pol3Δ ura7Δ strains were transformed with either WT POL3 or pol3-01 plasmids. Three independent isogenic strains for each genotype were grown overnight in YPD and diluted next day to OD600 = 0.1 in fresh YPD. Proliferation was followed by OD600 measurements, and the values were plotted as mean ± SD on log2 scale. (D) Quantification of CAN1 inactivation rates in diploid strains hemizygous for CAN1 locus (SI Materials and Methods for additional details) and homozygous for pol3Δ or pol3Δ ura7Δ mutations complemented with POL3 or pol3-01 plasmids. Error bars represent the 95% confidence intervals (CIs) and numbers on top indicate the fold increase in the mutation rate over the WT diploid strain (2.4 × 10−7 CanR mutants per cell division).
Loss of Gln3 or Ura7 Causes Activation of the DNA Damage Response.
Three of the mutants identified here (gln3Δ, ura7Δ, and rrm3Δ) were previously shown to have an extended S-phase (50). Because a prolonged S-phase could indicate DNA damage or replication stress, we investigated whether gln3Δ or ura7Δ mutants show activation of the DNA damage response (DDR) (Fig. 3A), similar as described for rrm3Δ mutant (51). DNA damage or replication stress triggers phosphorylation of checkpoint kinase Rad53 (52, 53), which in turns activates checkpoint kinase Dun1. Dun1 phosphorylation results in the inhibition of three repressors of RNR: Sml1, which binds and inhibits RNR1 (54, 55); Crt1, which acts as a transcriptional repressor of RNR2–4 (12, 13, 56); and Dif1, which prevents RNR cytoplasmic holoenzyme assembly by sequestering Rnr2–Rnr4 into the nucleus (57, 58). Thus, phosphorylation of Dun1 releases the negative regulation on RNR, promoting high expression levels of RNR subunits (RNR1–4) and RNR holoenzyme assembly, consequently resulting in increased dNTP production (59) (Fig. 3A).
Fig. 3.

Inactivation of Ura7 or Gln3 results in DDR checkpoint activation. (A) Simplified diagram depicting DDR response in S. cerevisiae. (B) Whole-cell lysates of logarithmically growing cells were analyzed by Western blotting with Rad53 and RNR1-4 antibodies. WT cells treated with 200 mM hydroxyurea (HU) were used as control for activation of DDR. (C) DNA content profiles of the indicated strains. (D) Mutation rates in mutant strains in the presence or absence of DUN1. See also Table S3. (E) Mutation rates in the indicated strains grown in YPD media supplemented or not with 5 mM glutamine (Gln). Error bars represent the 95% CI, and numbers on top indicate the fold increase in the mutation rate over WT.
Analysis of whole-cell lysates in ura7Δ and gln3Δ mutants by Western blotting revealed that both deletions cause constitutive DDR activation, characterized by Rad53 phosphorylation (as evidenced by a retarded Rad53 electrophoretic mobility) and elevated Rnr2 and Rnr4 levels (and increased Rnr3 levels in ura7Δ mutant) (Fig. 3B). Loss of Shm2 did not affect Rad53 phosphorylation or RNRs levels with the exception of Rnr4, which was slightly elevated. Accordingly, mutants with constitutive activation of DDR, like pol2-M644G (60) or as shown here gln3Δ and ura7Δ deletion mutants, all presented an accumulation of cells in S-phase (Fig. 3C). Unlike pol2-M644G, other active-site DNA polymerase mutants (pol1-L868M and pol3-L612M) did not cause activation of DDR (Fig. 3 B and C).
Previous reports have shown that mutator phenotypes observed in strains carrying DNA polymerase mutations preventing proofreading (pol2-04 or pol3-01) (31, 60, 61) or altering nucleotide selectivity function (pol3-L612M or pol3-R696W) (49, 62) can be suppressed by deletion of DUN1, which leads to reduced dNTP pools. To test whether the strong mutator phenotypes identified here can be modulated by dNTP levels, we introduced the dun1Δ mutation in several double mutants. Notably, deletion of DUN1 almost completely suppressed the CAN1 mutator phenotype in pol1-L868M gln3Δ from 293-fold to 4-fold over the WT rate (Fig. 3D). Similar results were obtained when the dun1Δ mutation was introduced in pol3-L612M ura7Δ, pol2-04 gln3Δ, and pol3-L612M gln3Δ double mutants (Fig. 3D and Table S3). Interestingly, we found that deletion of DUN1 in exo1Δ ura7Δ double mutant also reduced the CAN1 mutation rate up to exo1Δ levels (from 261-fold to 12-fold over WT) (Fig. 3D). Thus, the gain in DNA replication fidelity observed in the absence of DUN1 is not restricted to strains carrying DNA polymerase mutant alleles, and it is likely a consequence of reduced dNTP concentrations. On the other hand, Dun1 inactivation enhanced the S-phase delay observed in ura7Δ (and had no effect in gln3Δ mutant) as indicated by DNA content analysis (Fig. 3C).
Gln3, a member of the GATA transcription factor family negatively regulated by target of rapamycin (TOR), activates genes subject to nitrogen catabolite repression, particularly during glutamine limitation (63). Glutamine is an energy source and a substrate for the synthesis of purines and pyrimidines, among other nitrogen-containing molecules (39, 64). Thus, we tested whether supplementation of the culture media (YPD) with 5 mM glutamine can suppress the mutator phenotype of pol3-L612M gln3Δ, pol2-04 gln3Δ, and exo1Δ gln3Δ double mutants. Remarkably, glutamine supplementation resulted in a partial 3.3-, 3.3-, and 4-fold reduction in the CAN1 mutation rates, respectively (Fig. 3E). These observations suggest that, although gln3Δ cells were grown in rich media, at later stages during the growth of the culture, glutamine becomes limited resulting in increased mutagenesis.
Gln3 and Ura7 Are Both Required for Maintenance of Normal dNTP Pools.
Because deletion of GLN3 and URA7 both resulted in activation of DDR and both genes have metabolism-related functions, we hypothesized that their loss influences the balance of nucleotide pools. Quantification of ribonucleoside 5′-triphosphate (NTP) concentrations in these mutants (Fig. 4A and Table S4) revealed a 66% reduction in CTP levels in ura7Δ, which is in agreement with a previous report (37). Remarkably, gln3Δ also presented 46% reduction in CTP levels and a 1.7-fold increase in UTP pools. Because CTP is converted into CDP and then reduced by RNR into dCDP, which can be used for either dCTP or dTTP biosynthesis (Fig. 5A) (9), we tested whether lower CTP levels might affect dNTP pools. Strikingly, we found that gln3Δ and ura7Δ mutations both resulted in ∼50% reduction in dCTP levels and a concomitant increase in dTTP, dATP, and dGTP ranging from 2.4- up to 4-fold over their respective WT dNTP concentrations (Fig. 4B). In contrast, NTP and dNTP levels in shm2Δ, pol1-L868M, or pol3-L612M mutants were indistinguishable from WT strain (Fig. 4 A and B and Tables S4 and S5). In agreement with previous reports, we found that pol2-M644G mutant presents an overall increase in dNTP pools (60, 65).
Fig. 4.

Inactivation of Gln3 or Ura7 results in NTP and dNTP imbalance causing increased G:C-to-A:T transitions. (A) NTP and (B) dNTP pool measurements in the indicated strains (Tables S4 and S5). Error bars represent SD. (C) Independent CanR clones (n ≥ 91 per genotype) were sequenced for CAN1 mutations. Graphs indicate the type of identified mutations, in percentages (see also Tables S6 and S7). (D) The G-to-A mutational hotspot at nucleotide 788 was frequently found in msh6Δ gln3Δ, msh6Δ shm2Δ, and msh6Δ ura7Δ strains. Predicted mutation is noted in red. Nucleotides marked in green are facilitating mispair rapid extension prior proofreading due to their higher abundance in gln3Δ or ura7Δ mutants, compared with the WT strain. (E) The G-to-A mutational hotspot at position 497 was frequently found in msh6Δ and msh6Δ shm2Δ, but not in msh6Δ gln3Δ and msh6Δ ura7Δ. Here, the predicted G-dT mispair is immediately followed by incorporation of dCTP (in black), which is less abundant in gln3Δ and ura7Δ strains and, therefore, unlikely to support mispair rapid extension.
Table S4.
NTP concentrations for several mutants
| Relevant genotype | CTP | UTP | ATP | GTP |
| WT | 2,360 ± 532 (1.0) | 5,384 ± 1,406 (1.0) | 12,088 ± 2,351 (1.0) | 3,705 ± 912 (1.0) |
| pol1-L868M | 2,440 ± 483 (1.0) | 5,704 ± 942 (1.1) | 12,310 ± 2,029 (1.0) | 3,719 ± 669 (1.0) |
| pol2-M644G | 2,825 ± 1,171 (1.2) | 5,959 ± 2,511 (1.1) | 15,418 ± 6,078 (1.3) | 4,443 ± 1,902 (1.2) |
| pol3-L612M | 2,870 ± 1,085 (1.2) | 6,654 ± 2,626 (1.2) | 15,524 ± 5,465 (1.3) | 4,529 ± 1,765 (1.2) |
| gln3Δ | 1,267 ± 443 (0.5). | 8,957 ± 2,458 (1.7) | 13,167 ± 3,592 (1.1) | 2,929 ± 1,130 (0.8) |
| shm2Δ | 3,411 ± 1,485 (1.4) | 7,302 ± 3,243 (1.4) | 17,439 ± 7,467 (1.4) | 5,243 ± 2,429 (1.4) |
| ura7Δ | 808 ± 288 (0.3) | 6,575 ± 1,225 (1.2) | 13,080 ± 1,958 (1.1) | 3,587 ± 745 (1.0) |
| dun1Δ gln3Δ* | 1,645 ± 172 (0.7) | 7,533 ± 2,824 (1.4) | 12,246 ± 1,371 (1.0) | 3,272 ± 246 (0.9) |
| dun1Δ ura7Δ* | 1,160 ± 81 (0.5) | 8,338 ± 874 (1.5) | 14,723 ± 502 (1.2) | 4,151 ± 119 (1.1) |
Relates to Fig. 4. NTP concentrations (in picomoles per 108 cells) are the average of two biological replicates ± SD with the fold increase over WT in parentheses.
NTP concentrations were measured at a different time point and normalized according to a WT strain included in the same run.
Fig. 5.

(A) Pathways of de novo dNTP biosynthesis in S. cerevisiae (adapted from ref. 9). Essential genes are shown in bold (RNR1, 2, and 4 are nonessential in certain yeast genetic backgrounds). Metabolism-related genes (GLN3, URA7, and SHM2) identified in this screen were encircled in red. (B) Gln3 and Ura7 promote DNA replication fidelity by preventing dNTP pool imbalances. Inactivation of Gln3 or Ura7 results in low CTP/dCTP levels, triggering DNA damage checkpoint activation. Up-regulation of RNR subunits, instead of compensating low dCTP pools, creates a severe dNTP pool imbalance that, in combination with altered DNA polymerase functions or partial MMR defects, causes severe mutator phenotypes.
Table S5.
dNTP concentrations for several mutants
| Relevant genotype | dCTP | dTTP | dATP | dGTP |
| WT | 75 ± 4 (1.0) | 159 ± 14 (1.0) | 102 ± 3 (1.0) | 59 ± 0 (1.0) |
| pol1-L868M | 70 ± 1 (0.9) | 214 ± 11 (1.3) | 114 ± 1 (1.1) | 63 ± 3 (1.1) |
| pol2-M644G | 292 ± 20 (3.9) | 629 ± 37 (4.0) | 450 ± 20 (4.4) | 205 ± 9 (3.5) |
| pol3-L612M | 85 ± 25 (1.1) | 149 ± 12 (0.9) | 91 ± 4 (0.9) | 54 ± 2 (0.9) |
| gln3Δ | 43 ± 3 (0.6) | 641 ± 76 (4.0) | 293 ± 20 (2.9) | 141 ± 6 (2.4) |
| shm2Δ | 75 ± 2 (1.0) | 178 ± 18 (1.1) | 115 ± 12 (1.1) | 62 ± 3 (1.1) |
| ura7Δ | 35 ± 6 (0.5) | 517 ± 17 (3.3) | 386 ± 3 (3.8) | 158 ± 7 (2.7) |
| dun1Δ gln3Δ* | 27 ± 3 (0.4) | 134 ± 65 (0.8) | 71 ± 34 (0.7) | 47 ± 20 (0.8) |
| dun1Δ ura7Δ* | 27 ± 14 (0.4) | 134 ± 2 (0.8) | 98 ± 2 (1.0) | 42 ± 5 (0.7) |
Relates to Fig. 4. dNTP concentrations (in picomoles per 108 cells) are the average of two biological replicates ± SD with the fold increase over WT in parentheses.
dNTP concentrations were measured at a different time point and normalized according to a WT strain included in the same run.
Next, we measured NTP/dNTP levels in dun1Δ gln3Δ and dun1Δ ura7Δ double mutants (Fig. 4 A and B). Importantly, we found that, in the absence of Dun1, nucleotide pools in gln3Δ and ura7Δ strains were almost identical to WT, with exception of CTP and dCTP, which remained 30–50% and 64% lower, respectively. Therefore, Dun1 inactivation partially suppresses the dNTP imbalance in ura7Δ and gln3Δ mutants and consequently its mutagenic potential.
Inactivation of Gln3, Ura7, and Shm2 Causes an Increase in Mutations Dominated by C-to-T Transitions.
To further characterize ura7Δ, gln3Δ, and shm2Δ mutants, we carried out mutational spectra analysis at the CAN1 locus (in an msh6Δ background to prevent the correction of mispairs) aiming to correlate this information with dNTP levels. According to previous findings (33), we observed in msh6Δ mutant a higher proportion of base substitutions compared with the WT strain (92% versus 75%, respectively) (Table S6). Moreover, the percentage of base substitutions increased up to 99% in msh6Δ gln3Δ, msh6Δ ura7Δ, or msh6Δ shm2Δ double mutants. Noteworthy, ∼95% of the mutations in msh6Δ gln3Δ and msh6Δ ura7Δ were G:C-to-A:T transitions (compared with 54% observed in msh6Δ) (Fig. 4C and Table S6). A similar trend was also found in msh6Δ shm2Δ in which G:C-to-A:T transitions represented 80% of CanR events.
Table S6.
CAN1 mutation spectra
| Mutations observed | WT | msh6Δ | msh6Δ gln3∆ | msh6Δ shm2∆ | msh6Δ ura7∆ |
| CanR clones sequenced | 91 | 110 | 94 | 95 | 110 |
| Mutations overall* | 92 (100) | 111 (100) | 96 (100) | 96 (100) | 110 (100) |
| Base substitutions | 69 (75) | 102 (92) | 95 (99) | 95 (99) | 109 (99) |
| A-T → G-C | 6 (7) | 9 (8) | 1 (1) | 1 (1) | 0 (0) |
| G-C → A-T | 18 (20) | 60 (54) | 92 (96) | 77 (80) | 104 (95) |
| G-C → T-A | 29 (32) | 27 (24) | 0 (0) | 14 (15) | 5 (5) |
| A-T → C-G | 3 (3) | 2 (2) | 0 (0) | 3 (3) | 0 (0) |
| A-T → T-A | 7 (8) | 1 (1) | 0 (0) | 0 (0) | 0 (0) |
| C-G → G-C | 6 (7) | 3 (3) | 2 (2) | 0 (0) | 0 (0) |
| Transitions | 24 (26) | 69 (62) | 93 (97) | 78 (81) | 104 (95) |
| Transversions | 45 (49) | 33 (30) | 2 (2) | 17 (18) | 5 (5) |
| One-base pair frameshifts | 15 (16) | 7 (6) | 1 (1) | 1 (1) | 1 (1) |
| Complex mutations† | 8 (9) | 2 (2) | 0 (0) | 0 (0) | 0 (0) |
Relates to Fig. 4. Mutation spectra analysis based on DNA sequencing of the CAN1 gene in independent CanR mutants, shown as the number of clones containing the indicated mutations, and in parentheses as the percentage relative to the total.
In few cases (about 1–2% of the sequenced clones), two simultaneous CAN1 mutations (more than 100 bp apart) were found. These mutations were included in the analysis and considered as independent mutational events.
Include multiple mutations within 10 nt, insertions or deletions of more than 1 nt, and duplication events.
Analysis of the mutation spectra in msh6Δ gln3Δ identified G788A, G806A, G980A, G1018A, and G1622A as mutational hotspots occurring at a frequency at least 3.5 times higher than in the msh6Δ strain and two other weaker hotspots (G584A and C1426T) (Fig. 4D and Table S7). Comparing the mutation spectra in msh6Δ gln3Δ with msh6Δ ura7Δ revealed interesting similarities including hotspots G584A, G788A, G980A, G1018A, and C1426T. However, in msh6Δ ura7Δ, only G670A and G788A hotspots are significantly different from msh6Δ. Interestingly, some of these mutations, including G670A, G788A, and G980A, were also found in msh6Δ shm2Δ (Table S7), although this mutant in general had a broader distribution of the G:C-to-A:T transitions across the CAN1 gene. The increased frequency of G:C-to-A:T transitions in msh6Δ gln3Δ and msh6Δ ura7Δ strains is in agreement with the reduced dCTP and elevated dTTP levels, resulting in a dCTP:dTTP ratio of 1:15 compared with the 1:2 ratio existing in WT cells. We also identified one mutational hotspot (G497A) preferentially found in strains with normal dCTP levels (msh6Δ and msh6Δ shm2Δ) (Fig. 4E). Thus, this mutation appears to be counterselected in strains with reduced dCTP levels, given the sequence context that demands dCTP, immediately after the predicted dTTP misincorporation.
Table S7.
Mispair base substitution hotspots identified in gln3Δ, ura7Δ, and shm2Δ mutants
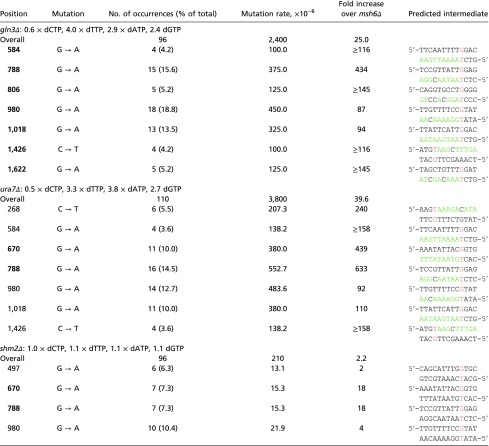 |
Relates to Fig. 4. Mutations are shown relative to the coding strand. The predicted mutation is noted in red. Nucleotides following the mutation and which dNTP pools are increased in the mutants in comparison with WT are noted in green. dNTP levels are shown as fold over the WT (Fig. 4 and Table S5). The mutation spectra analysis was done in an msh6Δ background. Mutation hotspots that are significantly different to the msh6Δ control (Fisher’s exact test, value of P ≤ 0.05) are shown in bold.
Discussion
Gln3, Shm2, Ura7, and Exo1 Increase Lagging-Strand DNA Replication Fidelity.
We discovered that, like EXO1 deletion, the inactivation of GLN3, URA7, and SHM2 caused synergistic increases in mutation rates exclusively in combination with lagging-strand DNA polymerase active-site mutant alleles (pol1-L868M or pol3-L612M). These findings contrast with the synergistic increase in mutation rates reported after introducing an msh2Δ mutation in either leading (pol2-M644G)- or lagging (pol3-L612M)-strand DNA polymerase mutant backgrounds (26). There are several possible explanations for this Polδ/lagging-strand bias: (i) a higher DNA replication fidelity during leading-strand synthesis facilitated by Polε checkpoint activation (66–68), which might provide additional time for proofreading or MMR; (ii) an increased base selectivity of Polε (by a factor of 10) compared with Polδ (2); (iii) intrinsic differences of how these active-site mutations interfere with Polε and Polδ replication fidelity; or (iv) leading- and lagging-strand DNA synthesis being mainly done by Polδ. This fourth hypothesis, supported by a recent study (69), opened up a strong controversy about the assignment of replicative DNA polymerases to the leading- or lagging-strand synthesis (70). At this point, we cannot distinguish between these different possibilities. However, it is interesting that ura7Δ and gln3Δ mutants showed remarkable similarities to a previously reported rnr1 mutant (rnr1-Q288A), which causes altered dNTP pools and increased mutagenesis (68). Similar to ura7Δ or gln3Δ, rnr1-Q288A shows reduced dCTP pools, resulting in the same dCTP:dTTP = 1:15 ratio, and activation of the DDR. Moreover, rnr1-Q288A CAN1 mutation spectra contained several hotspots (G670A, G788A, and G1018A) frequently found in msh6Δ ura7Δ (some of them also present in msh6Δ gln3Δ). Notably, mutational hotspots identified in the rnr1-Q288A strain were predicted as a consequence of lagging-strand replication. Thus, the bias for lagging-strand mutagenesis, observed in gln3Δ, ura7Δ, and rnr1-Q288A mutant strains, might be explained by inadequate dNTP pools causing S-phase checkpoint activation during Polε’s leading-strand synthesis. In this way, checkpoint activation might provide additional time for polymerase proofreading and/or MMR. Future studies will be required to investigate why most of the mutations identified here show a mutator bias for active-site mutants of lagging-strand DNA polymerases.
Rrm3 and Shm2 Contribute to DNA Replication Fidelity.
The Rrm3 helicase facilitates the passage through natural pausing sites of genomic regions containing replication fork barriers (42, 43, 51). Similarly, Rrm3 could facilitate the passage through DNA pausing sites resulting from low-fidelity DNA replication conditions, and therefore be preferentially required for polymerases with high processivity (Polε and Polδ) (71). Alternatively, the elevated dNTP pools in rrm3Δ mutant (72, 73) might promote mutagenesis, as it has been described for other mutations causing increased dNTP pools (74, 75). Moreover, a recent publication described an alternative helicase-independent Rrm3 function required for restricting DNA replication under replication stress (76), which could potentially enhance DNA replication fidelity. Future studies will address whether the effects described here are dependent on Rrm3’s helicase activity or perhaps related to this alternative role of Rrm3.
Shm2 is a cytosolic serine hydroxymethyltransferase that synthesizes 5,10-methylene-tetrahydrofolate (5,10-CH2-THF) (40, 41), an intermediate in the one-carbon (C1) cycle and precursor in de novo synthesis of purines and pyrimidines (Fig. 5A). In mammals, the same reaction is catalyzed by the serine hydroxymethyltransferase 1 (Shmt1) (Shmt1 is the mammalian homolog of yeast Shm2). Mammalian Shmt1 also serves as a scaffold protein that facilitates the interaction with DHFR and thymidylate synthase at the nuclear lamina (77). Remarkably, inactivation of Shmt1 in mice (78, 79) or its down-regulation in human lung cells (80) results in increased uracil incorporation into DNA as result of impaired de novo dTMP biosynthesis.
Here, we found that inactivation of Shm2 results in increased mutation rates in DNA polymerase mutant backgrounds (pol1-L868M, pol3-L612M, or pol2-04) (Tables 1 and 2) but not when MMR has been inactivated (msh2Δ strain) (Table S3). These observations indicate that the type of damage generated in the absence Shm2 is not repaired by MMR and does not causes mutagenesis unless DNA polymerase function is affected.
Because Shm2 generates 5,10-CH2-THF, which is used as cofactor during nucleotide biosynthesis, we hypothesized that loss of Shm2 might cause dNTP pool imbalances and consequently DNA replication infidelity. Interestingly, inactivation of Shm2 in S. cerevisiae neither caused alterations in NTP or dNTP pools (Fig. 4 A and B and Tables S4 and S5) nor resulted in activation of the DDR (Fig. 3 B and C), which otherwise is a common feature described for mutants with dNTP pools below their normal level (81). One possible explanation is that loss of Shm2 results in increased oxidative damage, leading to modified cytosine or uracil bases, which upon incorporation into DNA might be susceptible to deamination events frequently associated with C-to-T transitions. Supporting this hypothesis, quantitative metabolic flux analysis done in mammalian cells revealed that the C1 cycle, through oxidation of 5,10-CH2-THF, contributes to the production ∼40% of NADPH (82), a central cofactor in redox homeostasis. Alternatively, loss of Shm2 in S. cerevisiae, similar to inactivation of mammalian Shmt1 (78, 80), might cause uracil accumulation and increased mutagenesis. However, our analysis in shm2Δ mutants did not reveal major differences in NTP/dNTP pools or signs of DDR activation. Thus, if loss of Shm2 causes dUTP accumulation in S. cerevisiae, this increase might have been below our limit of detection, but nevertheless sufficient to cause genome instability.
Low dCTP Levels as an Achilles’ Heel of High-Fidelity DNA Replication.
Among the mutants identified here, both ura7Δ and gln3Δ showed the strongest mutator synergies with DNA polymerases or MMR mutant alleles (Tables 1 and 2 and Table S3). Although inactivation of Gln3 or Ura7 (in an msh6Δ background) mainly resulted in base substitutions (Fig. 4C and Table S6), we also found a small increase in frameshift mutations in combination with msh6Δ or exo1Δ, but not with msh3Δ or msh2Δ backgrounds (Table S3). We propose that this increase in frameshifts is not directly associated to the dNTP imbalance caused by gln3Δ and ura7Δ mutations, but rather a consequence of an overload of MMR capacity due to a large quantity of base substitutions, thus preventing the recognition of frameshifts.
Previous reports demonstrated that Ura7 contributes to a great extent (∼80%) to the production of CTP (a minor isoform called Ura8 gives account for the remaining CTP production) (37). However, the potential consequences of reduced CTP levels on dNTP pools remained unknown. We found that loss of Ura7 not only affects CTP pools but also results in a 50% reduction in dCTP pools with a concomitant increase in the concentration of the other three dNTPs (Fig. 4 A and B and Tables S4 and S5).
The role of the transcription factor Gln3 in NTP/dNTP pool maintenance has not been previously investigated. Here, we provide evidence that, although glutamine is used for de novo synthesis of both purines and pyrimidines, loss of Gln3 under our experimental conditions preferentially affects CTP and dCTP concentrations. Similar to ura7Δ mutant, the gln3Δ strain had a 50% reduction in CTP and dCTP levels as well as higher levels on the other dNTPs (Fig. 4 A and B and Tables S4 and S5). Importantly, we demonstrated that the strong mutator phenotype of gln3Δ double mutants was largely suppressed by glutamine supplementation (Fig. 3E). Glutamine is used as nitrogen source for protein and nucleotide biosynthesis and is considered one important “fuel” for cancer cells. This is illustrated by the fact that some cancer cell lines are strongly dependent on external glutamine for survival (“glutamine addiction”) (64, 83). Consequently, glutamine analogs inhibit cell proliferation, in part by inactivating glutamine-requiring enzymes involved in purine and pyrimidine biosynthesis (including CTP synthetase). Moreover, the glutamine analog Acivicin, which strongly inhibits CTP synthetase activity (84), has been shown to induce a dNTP imbalance characterized by reduced CTP/dCTP levels and increased UTP levels (85, 86), which is reminiscent of the gln3Δ mutant phenotype. In light of our findings, it would be interesting to investigate whether a low glutamine environment, as recently described for the core region in solid tumors (87) or due to glutamine analog treatment, causes increased mutagenesis, accelerating tumor evolution and the acquisition of cancer drug resistance.
The finding that deletion of DUN1 suppresses the strong mutator phenotype of double mutants carrying ura7Δ or gln3Δ mutations in DNA replication fidelity-compromised backgrounds (Fig. 3D), suggests that this phenotype is in part caused by DDR activation. This hypothesis is further supported by the finding that suppression of the mutator phenotype upon DUN1 deletion correlates with a reduction in dATP, dTTP, and dGTP levels (Fig. 4B), resulting in a dNTP pool with less mutagenic potential given the lower dTTP:dCTP ratio. The differences between dun1Δ ura7Δ and dun1Δ gln3Δ DNA content profiles (Fig. 3C) are consistent with a major role of Ura7 during CTP synthesis, whereas Gln3 is required under special circumstances (e.g., glutamine limitation). We speculate that conditions restricting dNTPs biosynthesis (e.g., dun1Δ) will prevent depletion of glutamine pools, and consequently cells might not heavily rely on Gln3 functions.
RNR catalytic activity responds to sophisticated allosteric regulation that “senses” three out of the four dNTPs (RNR is refractory to dCTP levels) and can “fine-tune” them, according to the cellular demands (Fig. 5A) (88). Excess in dCDP pools can be redirected to the synthesis of dTTPs by the action of dCMP deaminase. However, cells are not able to compensate for reductions in dCTP pools. As illustrated in Fig. 5B, ura7Δ or gln3Δ mutation interferes with the production of CTP, which is used as substrate for dCTP biosynthesis. Reduced dCTP levels likely cause stalled replication forks and activation of the DDR, which, instead of compensating for low dCTP levels, creates a severe dNTP imbalance with high mutagenic potential.
Mutation spectra analysis in gln3Δ or ura7Δ mutants revealed a strong increase in G:C-to-A:T transitions, representing about 95% of the identified mutations (Fig. 4C and Table S7). The reduced dCTP and high dTTP levels observed in gln3Δ or ura7Δ mutants are likely driving the misincorporation of dTTP at positions where dCTP would be required.
Without any exception, all mutational hotspots identified in this study can be explained by the “next-nucleotide effect” (8, 89). According to this model, after the misincorporation of a nucleotide, the high concentration of the next nucleotide (given by the sequence context) favors its incorporation (rapid extension) before proofreading of the previously misincorporated nucleotide (Fig. 4 D and E and Table S7). For example, at G788A transition, which occurs 16 times more frequently in msh6Δ ura7Δ compared with msh6Δ, the predicted T misincorporation (resulting in a G-dT mispair) is followed by the correct incorporation of 5 nt, in which none of them is C, and all nucleotides are at least 2.7-fold more abundant than in the WT strain (Fig. 4D). The same holds true for other hotspots, with some variations in the number of nucleotides that are introduced after the mispaired base, until the next C is required.
The characterization of mammalian cell lines resistant to inhibitory concentrations of specific nucleosides (or their analogs) (90, 91) revealed that mutations affecting the allosteric regulation of key enzymes involved in dNTP biosynthesis (RNR, CTP synthetase, and dCMP deaminase) can lead to dNTP imbalances, and in some cases, increased mutator phenotypes. Consequently, the identification and characterization of genes affecting dNTP pools might provide insights into mutagenesis and cancer susceptibility. Unfortunately, quantification of dNTP pools is laborious and not well suited for high-content screening. Therefore, this type of analysis has been limited to a relatively small number of gene mutations. In addition, not all dNTP imbalances correlate with increased mutation rates. For example, inactivation of dCMP deaminase (dcd1Δ) in S. cerevisiae resulted in 3-fold reduction in dTTP pool and about 30-fold increased in dCTP levels, without any consequences on mutation rates at the URA4 reporter (92).
Future studies will be required to understand in more detail the consequences of dNTP imbalances and S-phase checkpoint activation on DNA replication fidelity. As it has been suggested by Kumar et al. (81), a collection of strains with diverse well-defined dNTP pool imbalances would be extremely useful to expand our understanding how dNTP imbalances and checkpoint activation affect DNA replication fidelity.
In summary, we uncovered a group of previously unrecognized genes (GLN3, SHM2, RRM3, and URA7) that contribute to DNA replication fidelity. Two of these mutations (gln3Δ and ura7Δ) caused imbalanced dNTP pools by preventing the production of substrates used by RNR for dNTP biosynthesis. Importantly, these dNTP imbalances, when combined with partial defects on DNA polymerase functions or MMR activity, cause severe mutator phenotypes. In light of these observations, it is likely that still-unrecognized mutations (or environmental conditions) might influence the balance of dNTPs without immediate consequences on mutation rates. Such mutations or conditions can have dramatic effects on mutation rates when combined with defects in other DNA replication fidelity determinants often found in cancer cells. Therefore, our results not only highlight the superb buffer capabilities of the eukaryotic DNA replication and MMR system, but they also open up avenues to investigate genetic interactions that might drive mutator phenotypes and potentially tumor evolution. Thus, the human counterparts of the genes identified here could represent potential “minidrivers” for cancer development, which might participate in polygenic interactions resulting in increased mutagenesis.
Materials and Methods
All S. cerevisiae strains used in this study (Table S8) were derivatives of the S288C strains RDKY3686 (MATα ura3-52 leu2Δ1 trp1Δ63 hom3-10 his3Δ200 lys2-10A) (93) or RDKY5964 (a MATa version of RDKY3686) (26). Strains were cultivated at 30 °C according to standard protocols. Gene deletions and gene tagging were performed using standard PCR-based recombination methods (94), followed by confirmation by PCR. Correct insertion of tags or point mutations, as well as absence of additional unwanted mutations, were confirmed by sequencing. Specific mutations (pol1-L868M, pol2-M644G, pol3-L612M, pol2-04, cyh2-Q38K) were introduced at the chromosomal locus by pop-in/pop-out or PCR-based recombination methods and the presence of the desired mutations, as well as the absence of additional mutations, was confirmed by sequencing (for details, see SI Materials and Methods).
Table S8.
S. cerevisiae strains used in this study
| Name | Relevant genotype* | Reference |
| RDKY3686 | MATα ura3-52 leu2∆1 trp1∆63 hom3-10 his3∆200 lys2-10A | Ref. 93 |
| RDKY5964 | MATa ura3-52 leu2∆1 trp1∆63 hom3-10 his3∆200 lys2-10A | Ref. 26 |
| HHY6443 | MATa ura3-52 leu2∆1 trp1∆63 hom3-10 his3∆200 lys2-10A iYEL072W::hph can1::hisG yel072w::CAN1/URA3 bar1::loxP.klLEU2.loxP | This study |
| HHY6537 | MATα ura3-52 leu2∆1 trp1∆63 hom3-10 his3∆200 lys2-10A 10A iYEL072W::hph can1::hisG yel072w::CAN1/URA3 bar1::loxP.klLEU2.loxP | This study |
| HHY5298 | RDKY5964 cyh2 Q38K hom3-10.HIS3 pMFA1-klLEU2.hphNT1.lys2-10A MLH2.klURA3 POL1.natNT2 | This study |
| HHY5292 | RDKY5964 cyh2 Q38K hom3-10.HIS3 pMFA1-klLEU2.hphNT1.lys2-10A MLH2.klURA3 pol1-L868M.natNT2 | This study |
| HHY5284 | RDKY5964 cyh2 Q38K hom3-10.HIS3 pMFA1-klLEU2.hphNT1.lys2-10A MLH2.klURA3 pol2-M644G.natNT2 | This study |
| HHY5289 | RDKY5964 cyh2 Q38K hom3-10.HIS3 pMFA1-klLEU2.hphNT1.lys2-10A MLH2.klURA3 pol3-L612M.natNT2 | This study |
| HHY1794 | RDKY5964 exo1::hphNT1 | This study |
| HHY6372 | RDKY5964 gln3::HIS3 | This study |
| HHY6378 | RDKY5964 rrm3::kanMX4 | This study |
| HHY6374 | RDKY5964 shm2::kanMX4 | This study |
| HHY6376 | RDKY5964 ura7::kanMX4 | This study |
| HHY6425 | RDKY5964 dun1::hphNT1 | This study |
| HHY6517 | RDKY5964 dun1::hphNT1 gln3::HIS3 | This study |
| HHY6519 | RDKY5964 dun1::hphNT1 ura7::kanMX4 | This study |
| HHY5746 | RDKY5964 exo1::hphNT1 gln3::HIS3 | This study |
| HHY5752 | RDKY5964 exo1::hphNT1 rrm3::kanMX4 | This study |
| HHY6415 | RDKY5964 exo1::hphNT1 shm2::kanMX4 | This study |
| HHY5743 | RDKY5964 exo1::hphNT1 ura7::kanMX4 | This study |
| HHY6505 | RDKY5964 msh2::HIS3 | This study |
| HHY6507 | RDKY5964 msh2::natNT2 gln3::HIS3 | This study |
| HHY5596 | RDKY5964 msh2::natNT2 rrm3::kanMX4 | This study |
| HHY6509 | RDKY5964 msh2::natNT2 shm2::kanMX4 | This study |
| HHY5749 | RDKY5964 msh2::natNT2 ura7::kanMX4 | This study |
| HHY5195 | RDKY5964 msh3::HIS3 | This study |
| HHY6511 | RDKY5964 msh3::hphNT1 gln3::HIS3 | This study |
| HHY2248 | RDKY5964 msh3::HIS3 rrm3::kanMX4 | This study |
| HHY6513 | RDKY5964 msh3::hphNT1 shm2::kanMX4 | This study |
| HHY6515 | RDKY5964 msh3::HIS3 ura7::kanMX4 | This study |
| HHY780 | RDKY5964 msh6::hphNT1 | This study |
| HHY6419 | RDKY5964 msh6::hphNT1 gln3::HIS3 | This study |
| HHY2246 | RDKY5964 msh6::hphNT1 rrm3::kanMX4 | This study |
| HHY6421 | RDKY5964 msh6::hphNT1 shm2::kanMX4 | This study |
| HHY6423 | RDKY5964 msh6::hphNT1 ura7::kanMX4 | This study |
| HHY6252 | RDKY5964 pol1-L868M.natNT2 | This study |
| HHY6428 | RDKY5964 pol1-L868M.natNT2 dun1::hphNT1 | This study |
| HHY6379 | RDKY5964 pol1-L868M.natNT2 exo1::hphNT1 | This study |
| HHY6381 | RDKY5964 pol1-L868M.natNT2 gln3::HIS3 | This study |
| HHY6431 | RDKY5964 pol1-L868M.natNT2 gln3::HIS3 dun1::hphNT1 | This study |
| HHY6399 | RDKY5964 pol1-L868M.natNT2 rrm3::kanMX4 | This study |
| HHY6387 | RDKY5964 pol1-L868M.natNT2 shm2::kanMX4 | This study |
| HHY6393 | RDKY5964 pol1-L868M.natNT2 ura7::kanMX4 | This study |
| HHY6405 | RDKY5964 pol2-04.natNT2 | This study |
| HHY6429 | RDKY5964 pol2-04.natNT2 dun1::hphNT1 | This study |
| HHY6407 | RDKY5964 pol2-04.natNT2 gln3::HIS3 | This study |
| HHY6433 | RDKY5964 pol2-04.natNT2 gln3::HIS3 dun1::hphNT1 | This study |
| HHY6413 | RDKY5964 pol2-04.natNT2 rrm3::kanMX4 | This study |
| HHY6409 | RDKY5964 pol2-04.natNT2 shm2::kanMX4 | This study |
| HHY6411 | RDKY5964 pol2-04.natNT2 ura7::kanMX4 | This study |
| HHY1993 | RDKY5964 pol2-M644G.natNT2 | This study |
| HHY1947 | RDKY5964 pol2-M644G.natNT2 exo1::hphNT1 | This study |
| HHY6383 | RDKY5964 pol2-M644G.natNT2 gln3::HIS3 | This study |
| HHY6401 | RDKY5964 pol2-M644G.natNT2 rrm3::kanMX4 | This study |
| HHY6389 | RDKY5964 pol2-M644G.natNT2 shm2::kanMX4 | This study |
| HHY6395 | RDKY5964 pol2-M644G.natNT2 ura7::kanMX4 | This study |
| HHY1996 | RDKY5964 pol3-L612M.natNT2 | This study |
| HHY1943 | RDKY5964 pol3-L612M.natNT2 exo1::hphNT1 | This study |
| HHY6385 | RDKY5964 pol3-L612M.natNT2 gln3::HIS3 | This study |
| HHY6435 | RDKY5964 pol3-L612M.natNT2 gln3::HIS3 dun1::hphNT1 | This study |
| HHY6497 | RDKY5964 pol3-L612M.natNT2 gln3::HIS3 rad30::hphNT1 | This study |
| HHY6501 | RDKY5964 pol3-L612M.natNT2 gln3::HIS3 rev1::klTRP1 | This study |
| HHY6163 | RDKY5964 pol3-L612M.natNT2 gln3::HIS3 rev3::kanMX4 | This study |
| HHY6403 | RDKY5964 pol3-L612M.natNT2 rrm3::kanMX4 | This study |
| HHY6391 | RDKY5964 pol3-L612M.natNT2 shm2::kanMX4 | This study |
| HHY6397 | RDKY5964 pol3-L612M.natNT2 ura7::kanMX4 | This study |
| HHY6437 | RDKY5964 pol3-L612M.natNT2 ura7::kanMX4 dun1::hphNT1 | This study |
| HHY6495 | RDKY5964 pol3-L612M.natNT2 ura7::kanMX4 rad30::hphNT1 | This study |
| HHY6503 | RDKY5964 pol3-L612M.natNT2 ura7::kanMX4 rev1::klTRP1 | This study |
| HHY6499 | RDKY5964 pol3-L612M.natNT2 ura7::kanMX4 rev3::hphNT1 | This study |
| HHY6453 | HHY6537 pol1-L868M.natNT2 | This study |
| HHY6471 | HHY6443 pol1-L868M.natNT2 gln3::HIS3 | This study |
| HHY6459 | HHY6443 pol1-L868M.natNT2 ura7::kanMX4 | This study |
| HHY6455 | HHY6537 pol2-M644G.natNT2 | This study |
| HHY6473 | HHY6443 pol2-M644G.natNT2 gln3::HIS3 | This study |
| HHY6461 | HHY6443 pol2-M644G.natNT2 ura7::kanMX4 | This study |
| HHY6457 | HHY6537 pol3-L612M.natNT2 | This study |
| HHY6475 | HHY6443 pol3-L612M.natNT2 gln3::HIS3 | This study |
| HHY6463 | HHY6443 pol3-L612M.natNT2 ura7::kanMX4 | This study |
| HHY6481 | MATα ura3-52, leu2∆1, trp1∆63, hom3-10, his3∆200, lys2-10A, pol3::hphNT1 + pHHB388 (pRS316-POL3) | This study |
| HHY6482 | HHY6481 ura7::kanMX4 | This study |
| HHY6483 | HHY6481 msh2::HIS3 | This study |
| HHY6521 | MATa/α ura3-52/ura3-52, leu2∆1/leu2∆1, trp1∆63/trp1∆63, hom3-10/hom3-10, his3∆200/his3∆200, lys2-10A/lys2-10A, pol3::hphNT1/pol3::hphNT1 + pHHB388 (pRS316-POL3) | This study |
| HHY6523 | HHY6521 ura7::kanMX4/ura7::kanMX4 | This study |
| HHY6533 | HHY6521 can1::klTRP1/CAN1.natNT2 | This study |
| HHY6535 | HHY6521 can1::klTRP1/CAN1.natNT2 ura7::kanMX4/ura7::kanMX4 | This study |
Relates to Materials and Methods.
All strains derived from S288C. The genotype corresponds to the listed strain with the indicated modifications.
Genome-Wide Screen in S. cerevisiae.
Here, we engineered a modified version of the SGA protocol (32) to cross (using a RoToR robot; Singer Instruments) the nonessential gene deletion collection (BY4742) (MATα his3Δ1 leu2Δ0 ura3Δ lys2Δ yfg::kanMX4) with four queries (HHY5298, HHY5292, HHY5984, and HHY5289), carrying the wt-POL1, pol1-L868M, pol2-M644G, or pol3-L612M alleles, respectively [marked with a nourseothricin (nat) cassette at the 3′-UTR]. Otherwise, all four queries shared the following genotype: MATa ura3-52 leu2Δ1 trp1Δ63 his3Δ200 cyh2-Q38K hom3-10.HIS3 pMFA1-klLEU2.hphNT1.lys2-10A MLH2.klURA3, which allowed the systematic mating, sporulation, and selection procedure (for details about query strain construction and SGA modifications, see SI Materials and Methods).
For qualitative mutator analysis, double-mutant cells were spotted on YPD-agar using Liquidator 96 (Mettler Toledo) and grown for 2 d at 30 °C. Next, plates were imaged using the GelDoc system (Bio-Rad) and replica plated onto two different mutator reporter plates, either lacking lysine (lys2-10A frameshift reversion assay) or containing canavanine (CAN1 inactivation assay), and grown for 4 d at 30 °C. Plates were imaged for documentation and scored visually. Positive hits were rechecked and those double mutants that showed increased mutator phenotype were generated in RDKY5964 (or RDKY3686) for further analysis.
Determination of Mutation Rates.
Mutation rates using frameshift reversion assays (hom3-10 and lys2-10A) and the CAN1 inactivation assay were determined by fluctuation analysis as previously described (33, 93). URA3 forward inactivation rates were similarly determined by fluctuation analysis based on the spontaneous appearance of 5-fluoroorotic acid (5-FOA)-resistant colonies. Each mutation rate was determined by using two biological isolates and at least 14 independent cultures.
Yeast Cell Lysates and Immunoblotting.
S. cerevisiae whole-cell protein extracts were generated as previously described (26) and were analyzed on 7% or 8% SDS/PAGE followed by immunoblotting using anti-Rad53 (EL7.E1; Abcam), anti-Rnr1 (AS09576; Agrisera), anti-Rnr2 (AS09575; Agrisera), and anti-Rnr3 (AS09574; Agrisera). YL1/2 antibody (Sigma) was used to probe for Rnr4 and tubulin (81).
DNA Content Analysis.
Logarithmic S. cerevisiae cultures were processed as described in ref. 6 and analyzed using BD FACSCanto II (BD Biosciences).
Determination of NTP and dNTP Pools.
NTP and dNTPs were measured as described (60).
CAN1 Mutation Spectra Analysis.
The CAN1 gene from individual CanR clones was amplified from genomic DNA by PCR and sequenced (GATC Biotech). Sequences were analyzed with Lasergene 12 (DNASTAR). Mutation spectra distributions and mutational hotspots were compared with Fisher’s exact test in R. Values of P ≤ 0.05 were defined as significantly different.
SI Materials and Methods
Media and Growth Conditions.
S. cerevisiae strains were grown at 30 °C either in yeast extract–peptone–dextrose (YPD) media [1% Bacto yeast extract (Becton Dickinson), 2% Bacto peptone (Becton Dickinson), and 2% glucose] or in synthetic dropout (SD) media [0.67% Difco yeast nitrogen base without amino acids (Becton Dickinson), 2% glucose, and supplemented with the appropriate amino acid dropout mix]. For the CAN1 inactivation assay, plates were prepared in SD media lacking arginine (Arg), supplemented with 60 mg/L canavanine (Sigma). 5-FOA (US Biological) plates were done in SD media lacking Ura supplemented with 1 g/L 5-FOA and 50 mg/L uracil. Antibiotics were used at the following final concentrations (unless otherwise specified): 200 μg/mL geneticin (G418) (Santa Cruz Biotechnology), 300 μg/mL hygromycin B (hph) (Thermo Fisher Scientific), 100 μg/mL nourseothricin (nat) (clonNAT; Werner BioAgents), and 10 μg/mL cycloheximide (Serva Electrophoresis).
Query Strain Constructions.
The selectable markers used in the query of the original SGA protocol (32) are incompatible with the genetic markers required for the mutator assays. Because the CAN1 inactivation assay and the lys2-10 frameshift reversion assay require a functional CAN1 and LYP1 gene, respectively (lys2 strains are dependent on the lysine permease Lyp1 for survival), we could not use canavanine and thialysine to kill diploid cells. Therefore, we introduced in our queries one point mutation in the CYH2 gene (cyh2-Q38K) that results in cycloheximide resistance (95), and an URA3 cassette downstream of MLH2 gene, to kill diploid cells with a combination of cycloheximide and 5-FOA. Second, the LEU2 gene from Kluyveromyces lactis (klLEU2) under the control of a MFA1 promoter was introduced together with an hphNT1 cassette upstream of the lys2-10A allele. This allows the selection of haploid MATa progeny containing the lys2-10A reporter with a SD media lacking Leu supplemented with hph. Third, a natNT2 cassette was introduced downstream of the DNA polymerase mutant alleles (pol1-L868M, pol2-M644G, and pol3-L612M) or the WT POL1 gene to select for the mutant or WT polymerase allele using nat.
In detail, the query strains were constructed as follows. First, we amplified the klLEU2 ORF (including 143 bp of the 3′-UTR) from pOM13 (Euroscarf) with primers 5′-AAC TGT TTC TCG GAT AAA ACC AAA ATA AGT ACA AAG CCA TCG AAT AGA AAT GTC TAA GAA TAT CGT TGT C-3′ and 5′-AAA AGG AAG ATA AAG GAG GGA GAA CAA CGT TTT TGT ACG CAG AAA GAT CCG CAG GCT AAC CGG AA-3′ and used to replace the MFA1 ORF in RDKY5964 (MATa, ura3-52, trp1Δ63, leu2Δ1, his3Δ200, lys2-10A, hom3-10), resulting in HHY6484.
In parallel, a hygromycin cassette (hphNT1) from pFA6a-hphNT1 (94) was amplified with primers 5′-GTC TAT ATT CAT TGA AAC TGA TTA TTC GAT TTT CTT CTT GCT GAC CGT ACG CTG CAG GTC GAC-3′ and 5′-TTG AAG AGT TTT CCT CGC TAA AAC TGT GCG ATG CCT CTA GAA GCG ATC GAT GAA TTC GAG CTC G-3′ and inserted upstream the lys2-10A gene in HHY5218 (MATa ura3-52, leu2Δ1, trp1Δ63, hom3-10, his3Δ200, lys2-10A CAN1::URA3) to generate HHY6485.
Next, we prepared genomic DNA from HHY6484 and used it as template to amplify the MFA1 promoter followed by the klLEU2 gene (pMFA1-klLEU2), with primers 5′-GGC GCG CCT TAA TTA ACC CGG GGA TCC GTC GAC CTG CAG CGT ACG GAT CCG CAG GCT AAC CGG AA-3′ and 5′-GTC TAT ATT CAT TGA AAC TGA TTA TTC GAT TTT CTT CTT GCT GAC CAG GAT AGT GTG CAA CGT GG-3′. The pMFA1-klLEU2 cassette was inserted immediately upstream the hphNT1 cassette in HHY6485 to generate HHY6486.
In parallel, the cyh2-Q38K mutation resulting in cycloheximide resistance was introduced into RDKY3686 (MATα, ura3-52, trp1Δ63, leu2Δ1, his3Δ200, lys2-10A, hom3-10) by transformation with a PCR product generated from genomic DNA from RDKY7593 (which harbors the cyh2-Q38K mutation, generously provided by C. D. Putnam and R. D. Kolodner, Ludwig Institute for Cancer Research, San Diego) with primers 5′-GGC TTC CAG ATG TTA ACT GC-3′ and 5′-GAA CAG TCA TAC TGT CTA CTC-3′ generating HHY6487. Next, a HIS3 cassette was amplified from pRS303 with primers 5′-ATC CAC CTT TCT TCT TCA CTT TAA TGA TAG AAT ATT AAT TTT CCC TTT ATG AGC AGA TTG TAC TGA GAG TGC ACC-3′ and 5′-ATT AAT ATA TAT GTA AAT ATA TGT GCG CGT ATA TAT ATA TAT ATA TAT ATC TCC TTA CGC ATC TGT GCG GTA TTT C-3′ and introduced at the 3′-UTR of hom3-10 gene in strain HHY6487, to generate HHY6488.
HHY6486 and HHY6488 were crossed and sporulated to obtain HHY6489 (MATα ura3-52, trp1Δ63, leu2Δ1, his3Δ200, pMFA1-klLEU2.hphNT1.lys2-10A, hom3-10.HIS3, cyh2-Q38K). klURA3 was amplified from pUG72 (Euroscarf) with primers 5′-CTC TAA TAT TGC ATT GTT ACG ACA TCC TGT TGT CAT GCG ACT AAA CAA TAC AAC AGA TCA CGT G-3′ and 5′-AAC CTC ACA GAA TCA GAT GAA TGA GAT ATA TAC AGT ATA AAT ACC GTT TTA TTT AGG TTC TAT CGA GG-3′ and integrated at the 3′-UTR of MLH2 gene in HHY6489, to generate HHY6490.
DNA polymerase active-site mutations pol2-M644G (22) and pol3-L612M (96) were introduced in RDKY5964 by pop-in/out strategy as previously reported (26). Pol1-L868M mutation was introduced in RDKY5964 following the same strategy, but with plasmid pHHB97, previously linearized with BamHI. pHHB97 was generated by site-directed mutagenesis of pRS306-POL1, which contains the full-length WT POL1 gene, including 1 kb of the 5′-UTR and 738 bp of the 3′-UTR, cloned in between the KpnI and SacII sites of pRS306.
A natNT2 cassette was amplified from pFA6a-natNT2 (94) and introduced at the 3′-UTR of DNA polymerase active-site mutant alleles or WT POL1. HHY6490 was crossed against these strains to generate pol1-L868M, pol2-M644G, pol3-L612M, and wt-POL1 queries (HHY5292, HHY5284, HHY5289, and HHY5298, respectively), which otherwise have in common the following genotype (MATa ura3-52, trp1Δ63, leu2Δ1, his3Δ200, pMFA1-klLEU2.hphNT1.lys2-10A, hom3-10.HIS3, cyh2-Q38K, MLH2.klURA3).
SGA.
The four query strains HHY5298, HHY5292, HHY5984, and HHY5289 were crossed with an array of the quadruplicated BY4742 collection by pinning onto a fresh YPD-agar plate using RoToR robot (Singer Instruments) and grown for 1 d at 30 °C. Cells were then subjected to two rounds of pinning onto diploid selection medium (YPD-agar containing G418 + nat) and grown for 2 and 1 d, respectively, at 30 °C. Next, cells were then pinned onto presporulation medium [15 g of Difco nutrient broth (Becton Dickinson), 5 g of Bacto yeast extract (Becton Dickinson), 10 g of Difco agar (Becton Dickinson), and 62.5 mL of 40% glucose per 500 mL] and grown for 1 d at 30 °C. Then, cells were pinned onto sporulation medium [5 g of potassium acetate (Sigma), 0.5 g of Bacto yeast extract (Becton Dickinson), 0.25 g of glucose, 0.05 g of amino acid supplement powder for sporulation (mix containing 2 g of histidine, 10 g of leucine [Leu], 2 g of lysine, and 2 g of uracil [Ura]; all from Sigma), 10 g of Difco agar (Becton Dickinson) per 500 mL, containing a final concentration of 50 mg/L G418] and incubated for 7 d at 23–25 °C. Spores were pinned onto haploid selection medium [1.7 g of Difco yeast nitrogen base without amino acids and ammonium sulfate (Becton Dickinson), 1 g of glutamic acid monosodium salt (Sigma), 2 g of dropout mix (32) without Leu, 20 g of Difco agar, 50 mL of 40% glucose per liter containing G418] and grown for 5 d at 30 °C. Cells were then subjected to two rounds of pinning and grown on double-mutant selection medium [1.7 g of Difco yeast nitrogen base without amino acids and ammonium sulfate, 1 g of glutamic acid monosodium salt, 2 g of dropout mix (32) without Leu, 1 g of 5-FOA, 20 g of Difco agar, 50 mL of 40% glucose per liter, containing cycloheximide, G418, nat, and additional 50 μg/mL hph in the second round of pinning] for 1 d at 30 °C. Then the cells were decondensed from 1536- to 384-format using RoToR robot by pinning onto SD agar medium without Leu supplemented with G418 + nat [1.7 g of Difco yeast nitrogen base without amino acids and ammonium sulfate, 1 g of glutamic acid monosodium salt, 2 g of dropout mix (32) without Leu, 20 g of Difco agar, 50 mL of 40% glucose per liter, containing G418 + nat] and grown for 2 d at 30 °C. Cells were transferred using the RoToR robot to 96-well plates containing liquid SD medium without Leu [1.7 g of Difco yeast nitrogen base without amino acids and ammonium sulfate, 1 g of glutamic acid monosodium salt, 2 g of dropout mix (32) without Leu, 50 mL of 40% glucose per liter, containing 15% glycerol and supplemented with G418 + nat], grown for 2 d at 30 °C, and finally stored at −80 °C.
Strain Construction URA3 Inactivation Assay.
URA3 inactivation rates were measured in the strain HHY6443 (MATa ura3-52 leu2Δ1 trp1Δ63 his3Δ200 hom3-10 lys2-10A iYEL072::hph can1::hisG yel072w::CAN1/URA3 bar1::loxP.klLEU2.loxP), containing an URA3 cassette at the left arm of chromosome V. HHY6443 was generated as follows. First, RDKY6678 (97) strain containing the URA3 gene was crossed with RDKY3686 (93) and sporulated to generate HHY6491 (MATα ura3-52 leu2Δ1 trp1Δ63 his3Δ200 hom3-10 lys2-10A iYEL072::hph can1::hisG yel072w::CAN1/URA3) and HHY6492 (a MATa version of HHY6491). Next, a TRP1 cassette from Kluyveromyces lactis (klTRP1) was amplified from pYM22 (94) and introduced at the SML1 locus in HHY6491 to generate HHY6493 (MATα ura3-52 leu2Δ1 trp1Δ63 his3Δ200 hom3-10 lys2-10A iYEL072::hph can1::hisG yel072w::CAN1/URA3 sml1::klTRP1). In parallel, a LEU2 cassette from Kluyveromyces lactis (klLEU2) flanked by loxP sites was amplified from pUG73 (Euroscarf) and introduced at the BAR1 locus in HHY6492 to generate HHY6494 (MATa ura3-52 leu2Δ1 trp1Δ63 his3Δ200 hom3-10 lys2-10A iYEL072::hph can1::hisG yel072w::CAN1/URA3 bar1::loxP.klLEU2.loxP). Finally, HHY6493 was crossed with HHY6494 and sporulated to generate HHY6443.
Synthetic Lethal Interactions with DNA Polymerase Mutants by Plasmid Shuffling.
The POL3 gene, including 1 kb upstream and 200 bp downstream of POL3, was amplified from genomic DNA with primers 5′-CTG ACT GCG GCC GCT CTT CGT TCA ACT TGT TTT CCT TG-3′ and 5′-GGT GAC CCC GGG GTT TAC AAA TTA CTG ACA ATA AA-3′, introducing NotI and SmaI sites, which were used to clone the amplified fragment into pRS315 and pRS316 (98) to generate pRS315-POL3 (pHHB351) and pRS316-POL3 (pHHB388), respectively. To generate pRS315-pol3-01, plasmids RDK3097 (61) and pHHB351 were digested with NcoI and BglII. The 2,015-bp POL3 fragment of RDK3097 plasmid containing D321A and E323A mutations, and the 8,479-bp fragment of pHHB351 were gel extracted and ligated to generate pRS315-pol3-01 (pHHB396).
To create the strains used for the DNA polymerase plasmid shuffling experiments, RDKY3686 and RDKY5964 were mated to generate a diploid WT strain. In this strain, an hphNT1 cassette was used to replace one of the two POL3 alleles. Next, strains were transformed with pHHB388 and sporulated to generate HHY6481. In HHY6481, URA7 gene was deleted with a kanMX4 cassette (amplified from pFA6a-kanMX4), and MSH2 gene with a HIS3 cassette, to generate HHY6482 and HHY6483, respectively. HHY6481, HHY6482, and HHY6483 were transformed with pHHB351 and pHHB396. To check for synthetic lethality, transformants (Ura+Leu+) were streaked on 5-FOA plates (to select for the loss of wt-POL3-URA3 plasmid) and in SD media lacking Ura and Leu (as control). The msh2Δ pol3Δ strain (HHY6483) transformed with pol3-01-LEU2 plasmid (pHHB396) was used as a positive control for a synthetic lethal interaction (29). Strains were imaged after 3 d of growth with a GelDoc system (Bio-Rad). Homozygous diploid strains HHY6521 and HHY6523 were used for plasmid shuffling as described above.
Mutation Rates in Diploid Strains.
CAN1 inactivation rates were determined in diploid plasmid shuffling strains, hemizygous for the CAN1 locus (can1::klTRP1/CAN1.natNT2), which contained deletions of both copies of POL3, complemented with pHHB388 (pRS316-POL3), similar as previously described (49). HHY6533 (MATa/α ura3-52/ura3-52, leu2∆1/leu2∆1, trp1∆63/trp1∆63, hom3-10/hom3-10, his3∆200/his3∆200, lys2-10A/lys2-10A, can1::klTRP1/CAN1.natNT2, pol3::hphNT1/pol3::hphNT1 + pHHB388) and HHY6535, which in addition lacks both URA7 copies (MATa/α ura3-52/ura3-52, leu2∆1/leu2∆1, trp1∆63/trp1∆63, hom3-10/hom3-10, his3∆200/his3∆200, lys2-10A/lys2-10A, can1::klTRP1/CAN1.natNT2, ura7::kanMX4/ura7::kanMX4, pol3::hphNT1/pol3::hphNT1, + pHHB388), were used for plasmid shuffling as described in the previous paragraph.
HHY6533 was generated as follows. First, a TRP1 cassette from Kluyveromyces lactis (klTRP1) was amplified from pYM22 (94) and introduced at the CAN1 locus in HHY6481 to generate HHY6526 (MATα ura3-52, leu2∆1, trp1∆63, hom3-10, his3∆200, lys2-10A, pol3::hphNT1 can1::klTRP1 + pHHB388). Second, a natNT2 cassette was amplified from pFA6a-natNT2 (94) using primers 5′-ACC AAA GAC TTT TTG GGA CAA ATT TTG GAA TGT TGT AGC ATA GAT ATG ACC GTA CGC TGC AGG TCG AC-3′ and 5′-ATG AGG GTG AGA ATG CGA AAT GGC GTG GAA ATG TGA TCA AAG GTA ATA AAA CAT CGA TGA ATT CGA GCT CG-3′ and introduced 7 bp downstream the CAN1 STOP codon in HHY6525 (a MATa version of HHY6481) to generate HHY6528 (MATa ura3-52, leu2∆1, trp1∆63, hom3-10, his3∆200, lys2-10A, pol3::hphNT1 CAN1.natNT2 + pHHB388). Third, HHY6526 was crossed with HHY6528 to generate HHY6533 (MATa/α ura3-52/ura3-52, leu2∆1/leu2∆1, trp1∆63/trp1∆63, hom3-10/hom3-10, his3∆200/his3∆200, lys2-10A/lys2-10A, pol3::hphNT1/pol3::hphNT1, can1::klTRP1/CAN1.natNT2 + pHHB388).
HHY6535 was generated as described for HHY6533, with the modification that the initial CAN1 deletion and the introduction of the natNT2 cassette were done in HHY6482 (MATα ura3-52, leu2∆1, trp1∆63, hom3-10, his3∆200, lys2-10A, pol3::hphNT1, ura7::kanMX4 + pHHB388) and HHY6530 (a MATa version of HHY6482), respectively.
Both HHY6533 and HHY6535 were transformed with either pHHB351 or pHHB396 and then grown in Leu− medium containing 5-FOA + nat, to pop-out pHHB388. Mutation rates for the CAN1 inactivation assay were determined by fluctuation analysis as previously described (33), with the modification that liquid cultures were grown in YPD media supplemented with nat and later on plated on YPD or SD-Arg + canavanine plates, both supplemented with nat to select against CanR mitotic recombinants.
Acknowledgments
We thank Adam J. Rose, Hannah Zhao, Alhassan Abdelsamie, and N. Katarina Horvat for critical reading of this manuscript. Thanks to Richard D. Kolodner for helpful discussions, strains, and reagents. Also, we are grateful to Christopher D. Putnam and Linda Reha-Krantz for valuable discussions and to Tobias P. Dick for sharing reagents. Thanks to Annette Kopp-Schneider for advice on statistical analysis and to Frank Exner for technical support. This work was supported by the German Cancer Research Center and the Marie Curie Career Integration Grant “iMMR” (granted to H.H.), and by the Swedish Cancer Society, the Knut and Alice Wallenberg Foundation, and the Swedish Research Council (granted to A.C.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
See Commentary on page 5561.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1618714114/-/DCSupplemental.
References
- 1.Ganai RA, Johansson E. DNA replication—a matter of fidelity. Mol Cell. 2016;62:745–755. doi: 10.1016/j.molcel.2016.05.003. [DOI] [PubMed] [Google Scholar]
- 2.St Charles JA, Liberti SE, Williams JS, Lujan SA, Kunkel TA. Quantifying the contributions of base selectivity, proofreading and mismatch repair to nuclear DNA replication in Saccharomyces cerevisiae. DNA Repair (Amst) 2015;31:41–51. doi: 10.1016/j.dnarep.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kunkel TA. Evolving views of DNA replication (in)fidelity. Cold Spring Harb Symp Quant Biol. 2009;74:91–101. doi: 10.1101/sqb.2009.74.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reyes GX, Schmidt TT, Kolodner RD, Hombauer H. New insights into the mechanism of DNA mismatch repair. Chromosoma. 2015;124:443–462. doi: 10.1007/s00412-015-0514-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kunkel TA, Erie DA. Eukaryotic mismatch repair in relation to DNA replication. Annu Rev Genet. 2015;49:291–313. doi: 10.1146/annurev-genet-112414-054722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hombauer H, Srivatsan A, Putnam CD, Kolodner RD. Mismatch repair, but not heteroduplex rejection, is temporally coupled to DNA replication. Science. 2011;334:1713–1716. doi: 10.1126/science.1210770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aksenova A, et al. Mismatch repair-independent increase in spontaneous mutagenesis in yeast lacking non-essential subunits of DNA polymerase ε. PLoS Genet. 2010;6:e1001209. doi: 10.1371/journal.pgen.1001209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reha-Krantz LJ. DNA polymerase proofreading: Multiple roles maintain genome stability. Biochim Biophys Acta. 2010;1804:1049–1063. doi: 10.1016/j.bbapap.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 9.Mathews CK. Deoxyribonucleotide metabolism, mutagenesis and cancer. Nat Rev Cancer. 2015;15:528–539. doi: 10.1038/nrc3981. [DOI] [PubMed] [Google Scholar]
- 10.Reichard P. Ribonucleotide reductases: Substrate specificity by allostery. Biochem Biophys Res Commun. 2010;396:19–23. doi: 10.1016/j.bbrc.2010.02.108. [DOI] [PubMed] [Google Scholar]
- 11.Elledge SJ, Davis RW. Identification and isolation of the gene encoding the small subunit of ribonucleotide reductase from Saccharomyces cerevisiae: DNA damage-inducible gene required for mitotic viability. Mol Cell Biol. 1987;7:2783–2793. doi: 10.1128/mcb.7.8.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang M, Elledge SJ. Identification of RNR4, encoding a second essential small subunit of ribonucleotide reductase in Saccharomyces cerevisiae. Mol Cell Biol. 1997;17:6105–6113. doi: 10.1128/mcb.17.10.6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elledge SJ, Davis RW. Two genes differentially regulated in the cell cycle and by DNA-damaging agents encode alternative regulatory subunits of ribonucleotide reductase. Genes Dev. 1990;4:740–751. doi: 10.1101/gad.4.5.740. [DOI] [PubMed] [Google Scholar]
- 14.Serero A, Jubin C, Loeillet S, Legoix-Né P, Nicolas AG. Mutational landscape of yeast mutator strains. Proc Natl Acad Sci USA. 2014;111:1897–1902. doi: 10.1073/pnas.1314423111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lang GI, Parsons L, Gammie AE. Mutation rates, spectra, and genome-wide distribution of spontaneous mutations in mismatch repair deficient yeast. G3 (Bethesda) 2013;3:1453–1465. doi: 10.1534/g3.113.006429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lujan SA, et al. Heterogeneous polymerase fidelity and mismatch repair bias genome variation and composition. Genome Res. 2014;24:1751–1764. doi: 10.1101/gr.178335.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palles C, et al. CORGI Consortium; WGS500 Consortium Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet. 2013;45:136–144. doi: 10.1038/ng.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rayner E, et al. A panoply of errors: Polymerase proofreading domain mutations in cancer. Nat Rev Cancer. 2016;16:71–81. doi: 10.1038/nrc.2015.12. [DOI] [PubMed] [Google Scholar]
- 19.Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348:919–932. doi: 10.1056/NEJMra012242. [DOI] [PubMed] [Google Scholar]
- 20.Lujan SA, Williams JS, Kunkel TA. DNA polymerases divide the labor of genome replication. Trends Cell Biol. 2016;26:640–654. doi: 10.1016/j.tcb.2016.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nick McElhinny SA, Gordenin DA, Stith CM, Burgers PM, Kunkel TA. Division of labor at the eukaryotic replication fork. Mol Cell. 2008;30:137–144. doi: 10.1016/j.molcel.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pursell ZF, Isoz I, Lundström EB, Johansson E, Kunkel TA. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science. 2007;317:127–130. doi: 10.1126/science.1144067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pavlov YI, Mian IM, Kunkel TA. Evidence for preferential mismatch repair of lagging strand DNA replication errors in yeast. Curr Biol. 2003;13:744–748. doi: 10.1016/s0960-9822(03)00284-7. [DOI] [PubMed] [Google Scholar]
- 24.Nick McElhinny SA, Kissling GE, Kunkel TA. Differential correction of lagging-strand replication errors made by DNA polymerases alpha and delta. Proc Natl Acad Sci USA. 2010;107:21070–21075. doi: 10.1073/pnas.1013048107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goellner EM, Putnam CD, Kolodner RD. Exonuclease 1-dependent and independent mismatch repair. DNA Repair (Amst) 2015;32:24–32. doi: 10.1016/j.dnarep.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hombauer H, Campbell CS, Smith CE, Desai A, Kolodner RD. Visualization of eukaryotic DNA mismatch repair reveals distinct recognition and repair intermediates. Cell. 2011;147:1040–1053. doi: 10.1016/j.cell.2011.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liberti SE, Larrea AA, Kunkel TA. Exonuclease 1 preferentially repairs mismatches generated by DNA polymerase α. DNA Repair (Amst) 2013;12:92–96. doi: 10.1016/j.dnarep.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morrison A, Johnson AL, Johnston LH, Sugino A. Pathway correcting DNA replication errors in Saccharomyces cerevisiae. EMBO J. 1993;12:1467–1473. doi: 10.1002/j.1460-2075.1993.tb05790.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tran HT, Gordenin DA, Resnick MA. The 3′→5′ exonucleases of DNA polymerases delta and epsilon and the 5′→3′ exonuclease Exo1 have major roles in postreplication mutation avoidance in Saccharomyces cerevisiae. Mol Cell Biol. 1999;19:2000–2007. doi: 10.1128/mcb.19.3.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Williams LN, Herr AJ, Preston BD. Emergence of DNA polymerase ε antimutators that escape error-induced extinction in yeast. Genetics. 2013;193:751–770. doi: 10.1534/genetics.112.146910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herr AJ, et al. Mutator suppression and escape from replication error-induced extinction in yeast. PLoS Genet. 2011;7:e1002282. doi: 10.1371/journal.pgen.1002282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tong AH, Boone C. Synthetic genetic array analysis in Saccharomyces cerevisiae. Methods Mol Biol. 2006;313:171–192. doi: 10.1385/1-59259-958-3:171. [DOI] [PubMed] [Google Scholar]
- 33.Marsischky GT, Filosi N, Kane MF, Kolodner R. Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes Dev. 1996;10:407–420. doi: 10.1101/gad.10.4.407. [DOI] [PubMed] [Google Scholar]
- 34.Brusky J, Zhu Y, Xiao W. UBC13, a DNA-damage-inducible gene, is a member of the error-free postreplication repair pathway in Saccharomyces cerevisiae. Curr Genet. 2000;37:168–174. doi: 10.1007/s002940050515. [DOI] [PubMed] [Google Scholar]
- 35.Huang ME, Rio AG, Nicolas A, Kolodner RD. A genomewide screen in Saccharomyces cerevisiae for genes that suppress the accumulation of mutations. Proc Natl Acad Sci USA. 2003;100:11529–11534. doi: 10.1073/pnas.2035018100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ozier-Kalogeropoulos O, Fasiolo F, Adeline MT, Collin J, Lacroute F. Cloning, sequencing and characterization of the Saccharomyces cerevisiae URA7 gene encoding CTP synthetase. Mol Gen Genet. 1991;231:7–16. doi: 10.1007/BF00293815. [DOI] [PubMed] [Google Scholar]
- 37.Ozier-Kalogeropoulos O, Adeline MT, Yang WL, Carman GM, Lacroute F. Use of synthetic lethal mutants to clone and characterize a novel CTP synthetase gene in Saccharomyces cerevisiae. Mol Gen Genet. 1994;242:431–439. doi: 10.1007/BF00281793. [DOI] [PubMed] [Google Scholar]
- 38.Cooper TG. Transmitting the signal of excess nitrogen in Saccharomyces cerevisiae from the Tor proteins to the GATA factors: Connecting the dots. FEMS Microbiol Rev. 2002;26:223–238. doi: 10.1111/j.1574-6976.2002.tb00612.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ljungdahl PO, Daignan-Fornier B. Regulation of amino acid, nucleotide, and phosphate metabolism in Saccharomyces cerevisiae. Genetics. 2012;190:885–929. doi: 10.1534/genetics.111.133306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McNeil JB, et al. Cloning and molecular characterization of three genes, including two genes encoding serine hydroxymethyltransferases, whose inactivation is required to render yeast auxotrophic for glycine. J Biol Chem. 1994;269:9155–9165. [PubMed] [Google Scholar]
- 41.Kastanos EK, Woldman YY, Appling DR. Role of mitochondrial and cytoplasmic serine hydroxymethyltransferase isozymes in de novo purine synthesis in Saccharomyces cerevisiae. Biochemistry. 1997;36:14956–14964. doi: 10.1021/bi971610n. [DOI] [PubMed] [Google Scholar]
- 42.Azvolinsky A, Giresi PG, Lieb JD, Zakian VA. Highly transcribed RNA polymerase II genes are impediments to replication fork progression in Saccharomyces cerevisiae. Mol Cell. 2009;34:722–734. doi: 10.1016/j.molcel.2009.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mohanty BK, Bairwa NK, Bastia D. The Tof1p–Csm3p protein complex counteracts the Rrm3p helicase to control replication termination of Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2006;103:897–902. doi: 10.1073/pnas.0506540103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tran PT, Erdeniz N, Symington LS, Liskay RM. EXO1—a multi-tasking eukaryotic nuclease. DNA Repair (Amst) 2004;3:1549–1559. doi: 10.1016/j.dnarep.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 45.Keijzers G, Liu D, Rasmussen LJ. Exonuclease 1 and its versatile roles in DNA repair. Crit Rev Biochem Mol Biol. 2016;51:440–451. doi: 10.1080/10409238.2016.1215407. [DOI] [PubMed] [Google Scholar]
- 46.Goodman MF, Woodgate R. Translesion DNA polymerases. Cold Spring Harb Perspect Biol. 2013;5:a010363. doi: 10.1101/cshperspect.a010363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goellner EM, et al. PCNA and Msh2-Msh6 activate an Mlh1-Pms1 endonuclease pathway required for Exo1-independent mismatch repair. Mol Cell. 2014;55:291–304. doi: 10.1016/j.molcel.2014.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morrison A, Bell JB, Kunkel TA, Sugino A. Eukaryotic DNA polymerase amino acid sequence required for 3′→5′ exonuclease activity. Proc Natl Acad Sci USA. 1991;88:9473–9477. doi: 10.1073/pnas.88.21.9473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Herr AJ, Kennedy SR, Knowels GM, Schultz EM, Preston BD. DNA replication error-induced extinction of diploid yeast. Genetics. 2014;196:677–691. doi: 10.1534/genetics.113.160960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koren A, Soifer I, Barkai N. MRC1-dependent scaling of the budding yeast DNA replication timing program. Genome Res. 2010;20:781–790. doi: 10.1101/gr.102764.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ivessa AS, et al. The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein-DNA complexes. Mol Cell. 2003;12:1525–1536. doi: 10.1016/s1097-2765(03)00456-8. [DOI] [PubMed] [Google Scholar]
- 52.Rouse J, Jackson SP. Interfaces between the detection, signaling, and repair of DNA damage. Science. 2002;297:547–551. doi: 10.1126/science.1074740. [DOI] [PubMed] [Google Scholar]
- 53.Tourrière H, Pasero P. Maintenance of fork integrity at damaged DNA and natural pause sites. DNA Repair (Amst) 2007;6:900–913. doi: 10.1016/j.dnarep.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 54.Zhao X, Muller EG, Rothstein R. A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol Cell. 1998;2:329–340. doi: 10.1016/s1097-2765(00)80277-4. [DOI] [PubMed] [Google Scholar]
- 55.Chabes A, Domkin V, Thelander L. Yeast Sml1, a protein inhibitor of ribonucleotide reductase. J Biol Chem. 1999;274:36679–36683. doi: 10.1074/jbc.274.51.36679. [DOI] [PubMed] [Google Scholar]
- 56.Huang M, Zhou Z, Elledge SJ. The DNA replication and damage checkpoint pathways induce transcription by inhibition of the Crt1 repressor. Cell. 1998;94:595–605. doi: 10.1016/s0092-8674(00)81601-3. [DOI] [PubMed] [Google Scholar]
- 57.Lee YD, Wang J, Stubbe J, Elledge SJ. Dif1 is a DNA-damage-regulated facilitator of nuclear import for ribonucleotide reductase. Mol Cell. 2008;32:70–80. doi: 10.1016/j.molcel.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu X, Huang M. Dif1 controls subcellular localization of ribonucleotide reductase by mediating nuclear import of the R2 subunit. Mol Cell Biol. 2008;28:7156–7167. doi: 10.1128/MCB.01388-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou Z, Elledge SJ. DUN1 encodes a protein kinase that controls the DNA damage response in yeast. Cell. 1993;75:1119–1127. doi: 10.1016/0092-8674(93)90321-g. [DOI] [PubMed] [Google Scholar]
- 60.Williams LN, et al. dNTP pool levels modulate mutator phenotypes of error-prone DNA polymerase ε variants. Proc Natl Acad Sci USA. 2015;112:E2457–E2466. doi: 10.1073/pnas.1422948112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Datta A, et al. Checkpoint-dependent activation of mutagenic repair in Saccharomyces cerevisiae pol3-01 mutants. Mol Cell. 2000;6:593–603. doi: 10.1016/s1097-2765(00)00058-7. [DOI] [PubMed] [Google Scholar]
- 62.Mertz TM, Sharma S, Chabes A, Shcherbakova PV. Colon cancer-associated mutator DNA polymerase δ variant causes expansion of dNTP pools increasing its own infidelity. Proc Natl Acad Sci USA. 2015;112:E2467–E2476. doi: 10.1073/pnas.1422934112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Crespo JL, Powers T, Fowler B, Hall MN. The TOR-controlled transcription activators GLN3, RTG1, and RTG3 are regulated in response to intracellular levels of glutamine. Proc Natl Acad Sci USA. 2002;99:6784–6789. doi: 10.1073/pnas.102687599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wise DR, Thompson CB. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem Sci. 2010;35:427–433. doi: 10.1016/j.tibs.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nick McElhinny SA, et al. Genome instability due to ribonucleotide incorporation into DNA. Nat Chem Biol. 2010;6:774–781. doi: 10.1038/nchembio.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Navas TA, Zhou Z, Elledge SJ. DNA polymerase epsilon links the DNA replication machinery to the S phase checkpoint. Cell. 1995;80:29–39. doi: 10.1016/0092-8674(95)90448-4. [DOI] [PubMed] [Google Scholar]
- 67.Pursell ZF, Kunkel TA. DNA polymerase epsilon: A polymerase of unusual size (and complexity) Prog Nucleic Acid Res Mol Biol. 2008;82:101–145. doi: 10.1016/S0079-6603(08)00004-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kumar D, et al. Mechanisms of mutagenesis in vivo due to imbalanced dNTP pools. Nucleic Acids Res. 2011;39:1360–1371. doi: 10.1093/nar/gkq829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Johnson RE, Klassen R, Prakash L, Prakash S. A major role of DNA polymerase δ in replication of both the leading and lagging DNA strands. Mol Cell. 2015;59:163–175. doi: 10.1016/j.molcel.2015.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Burgers PM, Gordenin D, Kunkel TA. Who is leading the replication fork, Pol ε or Pol δ? Mol Cell. 2016;61:492–493. doi: 10.1016/j.molcel.2016.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reijns MA, et al. Lagging-strand replication shapes the mutational landscape of the genome. Nature. 2015;518:502–506. doi: 10.1038/nature14183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Poli J, et al. dNTP pools determine fork progression and origin usage under replication stress. EMBO J. 2012;31:883–894. doi: 10.1038/emboj.2011.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.O’Rourke TW, et al. Differential involvement of the related DNA helicases Pif1p and Rrm3p in mtDNA point mutagenesis and stability. Gene. 2005;354:86–92. doi: 10.1016/j.gene.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 74.Chabes A, et al. Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase. Cell. 2003;112:391–401. doi: 10.1016/s0092-8674(03)00075-8. [DOI] [PubMed] [Google Scholar]
- 75.Buckland RJ, et al. Increased and imbalanced dNTP pools symmetrically promote both leading and lagging strand replication infidelity. PLoS Genet. 2014;10:e1004846. doi: 10.1371/journal.pgen.1004846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Syed S, Desler C, Rasmussen LJ, Schmidt KH. A novel Rrm3 function in restricting DNA replication via an Orc5-binding domain is genetically separable from Rrm3 function as an ATPase/helicase in facilitating fork progression. PLoS Genet. 2016;12:e1006451. doi: 10.1371/journal.pgen.1006451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Anderson DD, Woeller CF, Chiang EP, Shane B, Stover PJ. Serine hydroxymethyltransferase anchors de novo thymidylate synthesis pathway to nuclear lamina for DNA synthesis. J Biol Chem. 2012;287:7051–7062. doi: 10.1074/jbc.M111.333120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Macfarlane AJ, Perry CA, McEntee MF, Lin DM, Stover PJ. Shmt1 heterozygosity impairs folate-dependent thymidylate synthesis capacity and modifies risk of Apc(min)-mediated intestinal cancer risk. Cancer Res. 2011;71:2098–2107. doi: 10.1158/0008-5472.CAN-10-1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.MacFarlane AJ, et al. Cytoplasmic serine hydroxymethyltransferase regulates the metabolic partitioning of methylenetetrahydrofolate but is not essential in mice. J Biol Chem. 2008;283:25846–25853. doi: 10.1074/jbc.M802671200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Paone A, et al. SHMT1 knockdown induces apoptosis in lung cancer cells by causing uracil misincorporation. Cell Death Dis. 2014;5:e1525. doi: 10.1038/cddis.2014.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kumar D, Viberg J, Nilsson AK, Chabes A. Highly mutagenic and severely imbalanced dNTP pools can escape detection by the S-phase checkpoint. Nucleic Acids Res. 2010;38:3975–3983. doi: 10.1093/nar/gkq128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fan J, et al. Quantitative flux analysis reveals folate-dependent NADPH production. Nature. 2014;510:298–302. doi: 10.1038/nature13236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J Clin Invest. 2013;123:3678–3684. doi: 10.1172/JCI69600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Weber G, et al. Multi-enzyme-targeted chemotherapy by acivicin and actinomycin. Adv Enzyme Regul. 1982;20:75–96. doi: 10.1016/0065-2571(82)90009-7. [DOI] [PubMed] [Google Scholar]
- 85.Denton JE, Lui MS, Aoki T, Sebolt J, Weber G. Rapid in vivo inactivation by acivicin of CTP synthetase, carbamoyl-phosphate synthetase II, and amidophosphoribosyltransferase in hepatoma. Life Sci. 1982;30:1073–1080. doi: 10.1016/0024-3205(82)90527-6. [DOI] [PubMed] [Google Scholar]
- 86.Neil GL, Berger AE, McPartland RP, Grindey GB, Bloch A. Biochemical and pharmacological effects of the fermentation-derived antitumor agent, (alphaS,5S)-alpha-amino-3-chloro-4,5-dihydro-5-isoxazoleacetic acid (AT-125) Cancer Res. 1979;39:852–856. [PubMed] [Google Scholar]
- 87.Pan M, et al. Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat Cell Biol. 2016;18:1090–1101. doi: 10.1038/ncb3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Reichard P. Interactions between deoxyribonucleotide and DNA synthesis. Annu Rev Biochem. 1988;57:349–374. doi: 10.1146/annurev.bi.57.070188.002025. [DOI] [PubMed] [Google Scholar]
- 89.Kunkel TA. DNA replication fidelity. J Biol Chem. 1992;267:18251–18254. [PubMed] [Google Scholar]
- 90.Trudel M, Van Genechten T, Meuth M. Biochemical characterization of the hamster thy mutator gene and its revertants. J Biol Chem. 1984;259:2355–2359. [PubMed] [Google Scholar]
- 91.Weinberg GL, Ullman B, Wright CM, Martin DW., Jr The effects of exogenous thymidine on endogenous deoxynucleotides and mutagenesis in mammalian cells. Somat Cell Mol Genet. 1985;11:413–419. doi: 10.1007/BF01534835. [DOI] [PubMed] [Google Scholar]
- 92.Sánchez A, et al. Replication fork collapse and genome instability in a deoxycytidylate deaminase mutant. Mol Cell Biol. 2012;32:4445–4454. doi: 10.1128/MCB.01062-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Amin NS, Nguyen MN, Oh S, Kolodner RD. exo1-dependent mutator mutations: Model system for studying functional interactions in mismatch repair. Mol Cell Biol. 2001;21:5142–5155. doi: 10.1128/MCB.21.15.5142-5155.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Janke C, et al. A versatile toolbox for PCR-based tagging of yeast genes: New fluorescent proteins, more markers and promoter substitution cassettes. Yeast. 2004;21:947–962. doi: 10.1002/yea.1142. [DOI] [PubMed] [Google Scholar]
- 95.Käufer NF, Fried HM, Schwindinger WF, Jasin M, Warner JR. Cycloheximide resistance in yeast: The gene and its protein. Nucleic Acids Res. 1983;11:3123–3135. doi: 10.1093/nar/11.10.3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li L, Murphy KM, Kanevets U, Reha-Krantz LJ. Sensitivity to phosphonoacetic acid: A new phenotype to probe DNA polymerase delta in Saccharomyces cerevisiae. Genetics. 2005;170:569–580. doi: 10.1534/genetics.104.040295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Putnam CD, Hayes TK, Kolodner RD. Specific pathways prevent duplication-mediated genome rearrangements. Nature. 2009;460:984–989. doi: 10.1038/nature08217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]