

Abstract
We used next generation sequencing (NGS) of the immunoglobulin genes to evaluate residual disease in 153 specimens from 32 patients with adult B cell ALL enrolled in a single, multi-center study. The sequencing results were compared to multi-parameter flow cytometry (MFC) data in 66 specimens (25 patients) analyzed by both methods. There was a strong concordance (82%) between the methods in the qualitative determination of the presence of disease. However, in 17% of cases leukemia was detected by sequencing, but not by MFC. In 54 bone marrow (BM) and peripheral blood (PB) paired specimens, the burden of leukemia detected by NGS was lower in PB than BM, although still detectable in 68% of the 28 paired specimens with positive BM. Lastly, patients without disease detected by NGS or MFC had a 5-year relapse free survival (RFS) of > 80%. The results suggest that residual disease detection by immunoglobulin gene sequencing is an extremely sensitive technique, and may identify patients that might benefit from transplant. Moreover, the increased sensitivity of the method may allow frequent peripheral blood testing to supplement marrow sampling to measure disease response.
Keywords: Acute lymphoblastic leukemia, minimal residual disease
Introduction
The detection of measurable residual disease (MRD) is an important marker of an increased risk of relapse in pediatric and adult acute lymphoblastic leukemia (ALL). MRD is associated with higher relapse rate, and worse event- or relapse-free survival, after conventional therapy or allogeneic transplantation 1–7. U.S. and European pediatric studies perform risk stratification based on MRD level, both to modify therapy in cases with elevated MRD, and/or decrease therapy in the absence of MRD 2–4. The use of MRD in adult ALL has followed pediatric studies and similar clinical studies are being conducted in adult patients with ALL 1, 4, 8, 9.
MRD in ALL is generally measured either by multi-parametric flow cytometry (MFC), polymerase chain reaction (PCR) of the IgH VDJ and/or TCR gene rearrangements, or leukemia-specific fusion transcripts (e.g. BCR-ABL in Ph+ ALL). MFC assesses the expression of multiple antigens at a single cell level using the enumeration of a discrete population with an aberrant immunophenotype as the readout of the assay. The sensitivity of MFC is approximately 10−4 (that is, one ALL blast in 104 normal cells). The main advantage of MFC is speed, but also its relative ease and cost-effectiveness of the procedure, and its broad applicability. Disadvantages of MFC include a current lack of standardization across testing sites and different inter-sample sensitivity depending on the specific abnormal immunophenotype of the leukemic population and the number of normal cells of similar type in the sample. Quantitative PCR of allele-specific IgH VDJ rearrangements is a highly sensitive method of detection of MRD (~ 10−4 – 10−5). It is, however, labor intensive and costly, as the MRD assay requires the characterization of leukemia-specific Ig/TCR gene rearrangement for each patient, as well as specific assay design and optimization for each patient. MRD levels between molecular and immuno-phenotypic approaches are highly correlated 10–14.
In chronic myeloid leukemia, standardized molecular monitoring of BCR-ABL chimeric mRNA has revolutionized study design as several clinical trials use MRD as a surrogate endpoint of outcome in pivotal drug trials15, 16. In ALL, MRD could play a similar role in radically changing treatment strategies and allowing for rapid drug development. This will require reproducible, sensitive, and scalable (i.e., available in many settings) MRD detection methods. Recently, methods that combine multiplex PCR of the IgH VDJ sequences with “next generation” sequencing (NGS) to quantitatively and sensitively measure MRD in lymphoid malignancies have been developed 17–20. In this study we compare MFC and NGS in adult ALL patients studied in a single clinical trial.
Methods
Patients and specimens
Cryopreserved diagnostic and follow-up peripheral blood (PB, N=108) and bone marrow samples (BM, N = 89) from 32 adult patients with ALL treated on SWOG S0333 were used for this study. Forty-four specimens were collected at study registration, and 153 were collected for MRD analysis per protocol schedule or suspicion of relapse. The median number of follow-up specimen time points per patient was 3 (range: 1 – 5). Flow cytometry data was available for 66 of the follow-up specimens.
S0333 was a phase II study of double induction combination chemotherapy to treat newly diagnosed adult ALL. Total accrual for the study was 78 patients, and the median age 42 y/o (range: 18 – 64y/o). The median age of the patients included here was 34 y/o (range: 18–64 y/o), and the median WBC 22.6 × 109/L (range: 1.1 × 109/L – 219.1 × 109/L). All patients gave written informed consent to the banking of specimens and their use in research protocols.
Next generation sequencing and data analysis
Pre-treatment specimens were analyzed to identify the clonal leukemic sequence of the VDJ or DJ fragment(s). Clonal sequences representing more than 5% of the total reads were considered leukemic (following guidelines for conventional diagnostic techniques), also considering the distribution of sequence frequencies across the entire repertoire of IgH molecules profiled in the sample. Follow-up specimens were subsequently sequenced, and the presence of the leukemic marker sequence(s) previously identified in the diagnostic sample was searched for explicitly.
DNA was extracted from cryopreserved white blood cells, using the Qiagen DNeasy kit (Hilden, Germany). Amplification and sequencing of the IgH Complementarity Determining Region 3 (CDR3) region was performed using the clonoSEQ™ assay (Adaptive Biotechnologies, Seattle, WA, www.clonoSeq.com), starting with 6–7 μg of input DNA, yielding limit of detection of approximately 1 leukemic cell in a background of 1 million nucleated cells. Calculation of the frequency of clonal sequences and the level of MRD was as previously described 17, 21, 22. Sequencing analysis was performed blinded to the MFC result.
Multi-parametric flow cytometry
MFC was performed on fresh specimens as described previously, using the antibody combination of CD20 FITC, CD10 PE, CD34 PerCP-Cy5.5, CD38 A594, CD19 PE-Cy7, CD58 APC, and CD45 APC-H7. Cells with surface marker patterns that differed from normal B-cell maturation were defined and quantified as MRD as previously described23.
Statistical methods
Overall survival (OS) and relapse-free survival (RFS) were measured from the date of registration for second induction until death from any cause or date of first relapse or death respectively, with patients last known to be alive (and in complete remission for RFS) censored at the date of last contact. Concordance between MRD positive/negative status was tabulated, and between quantitative measurements was visually summarized with scatterplots. OS and RFS were estimated using the Kaplan-Meier method and P values were calculated using log-rank tests.
Results
NGS marker identification
At least one Ig clonal sequence was identified in pre-treatment specimens from 29/32 (91%) of cases analyzed. The three patients lacking a detectable clonal sequence in pre-treatment material were excluded from further analysis (in all three cases only PB specimen at diagnostic was available). In the remaining 29 cases, the leukemic clonal sequence was a complete VDJ rearrangement in 17/29 patients (59%), an incomplete DJ rearrangement in 8/29 patients (28%), and in 3/29 (10%) cases both VDJ and DJ sequences were present. One patient had a light chain rearrangement (kappa). 17/29 (59%) cases contained more than one IgH clonal rearrangement at diagnosis, median = 2 (range: 1 – 4)
Comparison of disease detection determined by sequencing and flow cytometry
A total of 66 follow-up specimens (61 BM and 5 PB) had data from both flow cytometry and sequencing. The determination of the presence (or absence) of leukemia was concordant in 54/66 (82%) of samples. MRD was detected by sequencing in 11 specimens that were negative by MFC and one sample was positive by MFC, but no MRD was detected by NGS (flow level of MRD 0.002%). Figure 1A shows the correlation of leukemic burden detected by NGS and MFC in BM specimens.
Figure 1.

A. Comparison of tumor burden measurements obtained by NGS (Y axis) and MFC (X axis) in 61 BM specimens. Diamonds represent the 20 specimens positive both by NGS and MFC. The triangle denotes the single specimen negative by NGS and positive by MFC. Asterisks show 9 samples positive by HTS and negative by MFC. Circles show 31 specimens negative by both methods. NGS is presented as a fraction of the total nucleated cells calculated using the % B cells determined by MFC, and MFC as a fraction of total white blood cells. NGS and MFC values of disease burden are highly correlated (R2 = 0.91, P < 0.001). B. A comparison of disease burden in BM and PB. This figure shows 24 cases with a BM positive for ALL, with a time point paired sample from PB. The X-axis displays disease burden in BM as a fraction of nucleated cells, calculated from MFC data. Triangles represent specimens for which NGS analysis of the equivalent PB specimen detected disease, and circles, those BM specimens for which NGS analysis of the concomitant PB specimen was negative for MRD. Of the nine samples with ALL detected in BM but not in PB, the levels of MRD are very low (~ 1:104) in eight cases, suggesting that PB may be able to supplement BM testing in patients with MRD.
Comparison of disease detection by NGS in matching BM and PB specimens
Leukemia was detected in 33/68 (49%) of the BM samples and in 30/69 (43%) of the PB samples. There were 54 paired-samples of BM and PB (Figure 2). Twenty-five pairs (46%) showed no detectable MRD in either specimen and 19 (35%) had leukemia detected in both specimens. In the concordant positive pairs, the leukemic clone in BM was 6-fold higher than in PB (range 0.39 to 821-fold, Figure 1B).
Figure 2.
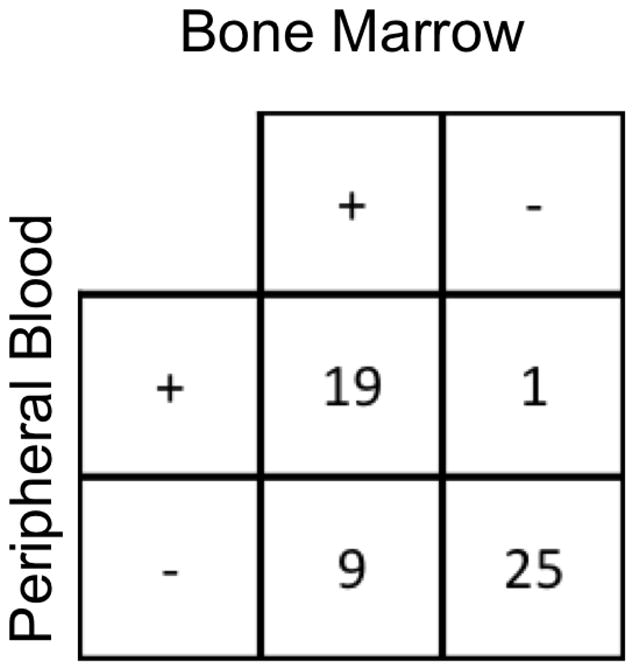
Comparison of MRD detection in BM and PB in 54 samples.
Nine pairs of samples (17%) had disease detectable in BM but not in PB by NGS. In 6, of them the BM MRD by MFC was negative, and for the 3 remaining pairs MRD was detected at low levels by MFC (0.003%, 0.004%, and 0.013%). One pair had disease detectable only in PB. In this case, MRD was not detected by either MFC or NGS of the marrow sample.
A graphic view of the correlation between the levels of disease in BM and PB is presented in Figure 1B, where the subset of 24 BM specimens with positive disease detected by NGS and coexisting MFC data are colored according to the presence or absence of disease in the corresponding PB specimen.
MRD levels and outcome
Overall survival and RFS of patients from second induction therapy (a protocol specified time-point for sample collection) was analyzed (Figure 3A and B). Twenty-one patients had specimens (BM or PB) available for analysis, and all of them had less than 5% blasts by morphology. The median time from registration at first induction to registration at second induction was 41 days (range: 35 – 75 days).
Figure 3. Survival by MRD status.

Panels A and B show OS and RFS according to MRD status at the time of registration to the protocol mandated second induction. The solid line shows survival for patients with MRD negative by MFC and NGS; ticked line shows survival for patients with MRD positive both by NGS an MFC; and dotted line shows survival for patients with MRD positive by NGS and negative by flow. Panels C and D illustrate RFS for patients with MRD negative (solid line) or positive (ticked line) by flow and NGS respectively.
Patients who were MRD negative by NGS and MFC had an excellent OS and RFS of above 80% while patients positive by both NGS and flow cytometry have the poorest outcome (p = 0.0049 and p = 0.003 for OS and RFS, respectively). Patients who were NGS positive, MFC negative had an intermediate outcome (p= 0.028 and p = 0.04 for OS and RFS when compared to patients with MRD positive by both MFC and NGS).
Only 1 of 7 (14%) patients with MRD negative by NGS relapsed, in contrast to 5 of 12 (42%) patients who were MRD negative by MFC. RFS was significantly better for MRD negative patients detected by NGS or MFC compared to patients with detectable residual disease (P = 0.018 for NGS, and P = 0.0032 for MFC, see Figure 3C, D).
Discussion
In this study we found: 1) a strong qualitative and quantitative correlation between values obtained by NGS and MFC in determining leukemic burden in B-ALL patients; 2) when discordance occurred, it was strongly biased towards the detection of MRD by NGS when MRD was negative by MFC. This suggests that NGS is a more sensitive assay, consistent with previous studies with T cell and pediatric B cell ALL 17–19, 21, 22; 3) levels of MRD detected by NGS were higher in BM than in PB, and 4) cases without MRD by MFC and NGS early after induction therapy had a greatly superior 5-year overall survival (>80%) compared to those cases MRD positive by NGS but not MFC (40%), or MRD positive by NGS and MFC (<10%).
NGS has previously been compared to flow cytometry in pediatric T-ALL and B-ALL, where it appeared to be more sensitive in detecting MRD than MFC 17, 21, 22. Disease burden pre-treatment showed a very high correlation between the two methods, but post-treatment MRD levels showed significant discordance, with approximately an additional 33% of total samples positive by NGS but negative by MFC (there were no cases of positive MFC and negative NGS). In B cell ALL, NGS has been compared to quantitative PCR, again showing similar quantitative results when both measurements were positive. Discordance occurred only in 15% of samples, with some samples showing higher levels of MRD by PCR, while others higher with NGS 24.
Despite more extensive data needs to be gathered to clarify the significance of very low levels of MRD, our study shows patients with no MRD by NGS have an excellent survival of >80%, compared to those with MRD detected only by NGS. This is consistent with a recent report suggesting any amount of MRD detected after stem cell transplantation increases relapse risk 25. If these data are confirmed in larger studies, NGS will be useful to fine tune criteria for transplantation in adult ALL patients, as the excellent outcome of patients with no disease detectable by NGS could favor a less aggressive treatment path.
Clonal evolution in B-ALL is a well-described phenomenon26, 27. Relapse clones can be genetically identical to diagnostic clones, completely distinct, or evolved either from the diagnostic clone or an ancestor27. NGS at diagnosis has identified multiple clonal IgH rearrangements at high frequency or (index clones) in most pediatric B-ALL cases, as well as IgH evolved sub-clones (ranging from 0–4,000 clones) 28. We detected sub-clones related to “index clones” in some of our patients, though none of the 8 patients examined had an expansion of these low level minor clones at relapse. In one case the clonal profile at relapse was “inverted” from diagnosis: two index clones were described at diagnosis, one clone being four-fold more abundant than the second one. At relapse, 145 days after diagnosis, both clones were identified but the proportions were switched and 95% of VDJ sequences were the originally minor clone’s, suggesting therapy-driven clonal selection.
In B-ALL, all modalities used to track MRD detect a higher level of disease burden in BM than in PB29–31, so marrow testing is the standard for MRD monitoring. As expected, we found higher levels of MRD in BM than in PB. However, NGS in PB missed the detection of MRD in only 17% of cases in our series, and those are predominantly cases with low MRD levels in BM: in 8/9 pairs with BM positive but PB negative for MRD, the BM disease levels was detected at a level of 1:104 total cells, or lower (Figure 1B). Since NGS sequencing appears to be more sensitive than MFC or PCR technologies, could frequent peripheral blood monitoring with NGS be used rather than (or, strategically supplementing) marrow measurements? This provocative question could be integrated in future clinical trials, for example as a more frequent schedule of MRD testing in PB by NGS, complemented by BM testing when PB was found to be negative.
The accuracy and sensitivity of the NGS assay holds a promise for MRD investigation in ALL and potentially can be used in studies of disease kinetics, MRD, and clonal selection. Our study suggests a future role of NGS detection of MRD in ALL. Early identification of patients at risk for relapse using MRD is already leading to modifications of therapy in pediatric ALL trials. In the future, such MRD-driven decisions may occur in adult ALL, and in addition, reliable MRD measurements may provide surrogate endpoints for clinical trials allowing more rapid assessment of novel therapies.
HIGHLIGHTS.
NGS of IgH VDJ in adult ALL is an accurate and sensitive measure of MRD.
Patients MRD negative by NGS and MFC have superior outcome vs. cases MRD positive.
Peripheral blood testing for MRD in ALL may complement BM testing.
Acknowledgments
Support: This investigation was supported in part by the following PHS Cooperative Agreement grants awarded by the National Cancer Institute, DHHS: CA32102, CA38926, CA20319, and CA 18029. ClinicalTrials.gov Identifier: NCT00109837
Footnotes
Conflicts of Interest Disclosure: David W. Williamson and Ilan Kirsch own stock and are employees of Adaptive Biotechnologies. Harlan Robins owns stock in and receives consulting fees from Adaptive Biotechnologies.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bassan R, Spinelli O, Oldani E, et al. Improved risk classification for risk-specific therapy based on the molecular study of minimal residual disease (MRD) in adult acute lymphoblastic leukemia (ALL) Blood. 2009;113:4153–4162. doi: 10.1182/blood-2008-11-185132. [DOI] [PubMed] [Google Scholar]
- 2.Bruggemann M, Schrauder A, Raff T, et al. Standardized MRD quantification in European ALL trials: proceedings of the Second International Symposium on MRD assessment in Kiel, Germany, 18–20 September 2008. Leukemia. 2010;24:521–535. doi: 10.1038/leu.2009.268. [DOI] [PubMed] [Google Scholar]
- 3.Conter V, Bartram CR, Valsecchi MG, et al. Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood. 2010;115:3206–3214. doi: 10.1182/blood-2009-10-248146. [DOI] [PubMed] [Google Scholar]
- 4.Gokbuget N, Kneba M, Raff T, et al. Adult patients with acute lymphoblastic leukemia and molecular failure display a poor prognosis and are candidates for stem cell transplantation and targeted therapies. Blood. 2012;120:1868–1876. doi: 10.1182/blood-2011-09-377713. [DOI] [PubMed] [Google Scholar]
- 5.Holowiecki J, Krawczyk-Kulis M, Giebel S, et al. Status of minimal residual disease after induction predicts outcome in both standard and high-risk Ph-negative adult acute lymphoblastic leukaemia. The Polish Adult Leukemia Group ALL 4-2002 MRD Study. Br J Haematol. 2008;142:227–237. doi: 10.1111/j.1365-2141.2008.07185.x. [DOI] [PubMed] [Google Scholar]
- 6.Vidriales MB, Perez JJ, Lopez-Berges MC, et al. Minimal residual disease in adolescent (older than 14 years) and adult acute lymphoblastic leukemias: early immunophenotypic evaluation has high clinical value. Blood. 2003;101:4695–4700. doi: 10.1182/blood-2002-08-2613. [DOI] [PubMed] [Google Scholar]
- 7.Bruggemann M, Raff T, Flohr T, et al. Clinical significance of minimal residual disease quantification in adult patients with standard-risk acute lymphoblastic leukemia. Blood. 2006;107:1116–1123. doi: 10.1182/blood-2005-07-2708. [DOI] [PubMed] [Google Scholar]
- 8.Ribera JM, Oriol A, Morgades M, et al. Treatment of high-risk Philadelphia chromosome-negative acute lymphoblastic leukemia in adolescents and adults according to early cytologic response and minimal residual disease after consolidation assessed by flow cytometry: final results of the PETHEMA ALL-AR-03 trial. J Clin Oncol. 2014;32:1595–1604. doi: 10.1200/JCO.2013.52.2425. [DOI] [PubMed] [Google Scholar]
- 9.Toft N, Birgens H, Abrahamsson J, et al. Risk group assignment differs for children and adults 1–45 yr with acute lymphoblastic leukemia treated by the NOPHO ALL-2008 protocol. Eur J Haematol. 2013;90:404–412. doi: 10.1111/ejh.12097. [DOI] [PubMed] [Google Scholar]
- 10.Gaipa G, Cazzaniga G, Valsecchi MG, et al. Time point-dependent concordance of flow cytometry and real-time quantitative polymerase chain reaction for minimal residual disease detection in childhood acute lymphoblastic leukemia. Haematologica. 2012;97:1582–1593. doi: 10.3324/haematol.2011.060426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Irving J, Jesson J, Virgo P, et al. Establishment and validation of a standard protocol for the detection of minimal residual disease in B lineage childhood acute lymphoblastic leukemia by flow cytometry in a multi-center setting. Haematologica. 2009;94:870–874. doi: 10.3324/haematol.2008.000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malec M, van der Velden VH, Bjorklund E, et al. Analysis of minimal residual disease in childhood acute lymphoblastic leukemia: comparison between RQ-PCR analysis of Ig/TcR gene rearrangements and multicolor flow cytometric immunophenotyping. Leukemia. 2004;18:1630–1636. doi: 10.1038/sj.leu.2403444. [DOI] [PubMed] [Google Scholar]
- 13.Ryan J, Quinn F, Meunier A, et al. Minimal residual disease detection in childhood acute lymphoblastic leukaemia patients at multiple time-points reveals high levels of concordance between molecular and immunophenotypic approaches. Br J Haematol. 2009;144:107–115. doi: 10.1111/j.1365-2141.2008.07429.x. [DOI] [PubMed] [Google Scholar]
- 14.Denys B, van der Sluijs-Gelling AJ, Homburg C, et al. Improved flow cytometric detection of minimal residual disease in childhood acute lymphoblastic leukemia. Leukemia. 2013;27:635–641. doi: 10.1038/leu.2012.231. [DOI] [PubMed] [Google Scholar]
- 15.Deininger MW, Kopecky KJ, Radich JP, et al. Imatinib 800 mg daily induces deeper molecular responses than imatinib 400 mg daily: results of SWOG S0325, an intergroup randomized PHASE II trial in newly diagnosed chronic phase chronic myeloid leukaemia. Br J Haematol. 2014;164:223–232. doi: 10.1111/bjh.12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kantarjian HM, Hochhaus A, Saglio G, et al. Nilotinib versus imatinib for the treatment of patients with newly diagnosed chronic phase, Philadelphia chromosome-positive, chronic myeloid leukaemia: 24-month minimum follow-up of the phase 3 randomised ENESTnd trial. Lancet Oncol. 2011;12:841–851. doi: 10.1016/S1470-2045(11)70201-7. [DOI] [PubMed] [Google Scholar]
- 17.Wu D, Sherwood A, Fromm JR, et al. High-throughput sequencing detects minimal residual disease in acute T lymphoblastic leukemia. Sci Transl Med. 2012;4:134ra163. doi: 10.1126/scitranslmed.3003656. [DOI] [PubMed] [Google Scholar]
- 18.Faham M, Zheng J, Moorhead M, et al. Deep-sequencing approach for minimal residual disease detection in acute lymphoblastic leukemia. Blood. 2012;120:5173–5180. doi: 10.1182/blood-2012-07-444042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Larimore K, McCormick MW, Robins HS, Greenberg PD. Shaping of human germline IgH repertoires revealed by deep sequencing. J Immunol. 2012;189:3221–3230. doi: 10.4049/jimmunol.1201303. [DOI] [PubMed] [Google Scholar]
- 20.Mannis GN, Martin TG, 3rd, Damon LE, et al. Quantification of Acute Lymphoblastic Leukemia Clonotypes in Leukapheresed Peripheral Blood Progenitor Cells Predicts Relapse Risk after Autologous Hematopoietic Stem Cell Transplantation. Biol Blood Marrow Transplant. 2016;22:1030–1036. doi: 10.1016/j.bbmt.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 21.Carlson CS, Emerson RO, Sherwood AM, et al. Using synthetic templates to design an unbiased multiplex PCR assay. Nat Commun. 2013;4:2680. doi: 10.1038/ncomms3680. [DOI] [PubMed] [Google Scholar]
- 22.Wu D, Emerson RO, Sherwood A, et al. Detection of Minimal Residual Disease in B Lymphoblastic Leukemia by High-Throughput Sequencing of IGH. Clin Cancer Res. 2014;20:4540–4548. doi: 10.1158/1078-0432.CCR-13-3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wood B. 9-color and 10-color flow cytometry in the clinical laboratory. Arch Pathol Lab Med. 2006;130:680–690. doi: 10.5858/2006-130-680-CACFCI. [DOI] [PubMed] [Google Scholar]
- 24.Ladetto M, Bruggemann M, Monitillo L, et al. Next-generation sequencing and real-time quantitative PCR for minimal residual disease detection in B-cell disorders. Leukemia. 2013 doi: 10.1038/leu.2013.375. [DOI] [PubMed] [Google Scholar]
- 25.Pulsipher MA, Carlson C, Langholz B, et al. IgH-V(D)J NGS-MRD measurement pre- and early post-allotransplant defines very low- and very high-risk ALL patients. Blood. 2015;125:3501–3508. doi: 10.1182/blood-2014-12-615757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosenquist R, Thunberg U, Li AH, et al. Clonal evolution as judged by immunoglobulin heavy chain gene rearrangements in relapsing precursor-B acute lymphoblastic leukemia. Eur J Haematol. 1999;63:171–179. doi: 10.1111/j.1600-0609.1999.tb01765.x. [DOI] [PubMed] [Google Scholar]
- 27.Mullighan CG, Phillips LA, Su X, et al. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008;322:1377–1380. doi: 10.1126/science.1164266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gawad C, Pepin F, Carlton VE, et al. Massive evolution of the immunoglobulin heavy chain locus in children with B precursor acute lymphoblastic leukemia. Blood. 2012;120:4407–4417. doi: 10.1182/blood-2012-05-429811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coustan-Smith E, Sancho J, Hancock ML, et al. Use of peripheral blood instead of bone marrow to monitor residual disease in children with acute lymphoblastic leukemia. Blood. 2002;100:2399–2402. doi: 10.1182/blood-2002-04-1130. [DOI] [PubMed] [Google Scholar]
- 30.van der Velden VH, Jacobs DC, Wijkhuijs AJ, et al. Minimal residual disease levels in bone marrow and peripheral blood are comparable in children with T cell acute lymphoblastic leukemia (ALL), but not in precursor-B-ALL. Leukemia. 2002;16:1432–1436. doi: 10.1038/sj.leu.2402636. [DOI] [PubMed] [Google Scholar]
- 31.Logan AC, Vashi N, Faham M, et al. Immunoglobulin and T cell receptor gene high-throughput sequencing quantifies minimal residual disease in acute lymphoblastic leukemia and predicts post-transplantation relapse and survival. Biol Blood Marrow Transplant. 2014;20:1307–1313. doi: 10.1016/j.bbmt.2014.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]