

Abstract
Purpose
In March 2007, a US Food and Drug Administration boxed warning was issued for erythropoietin-stimulating agents (ESAs) regarding serious adverse events, such as venous thromboembolism (VTE). We evaluated the US Food and Drug Administration’s boxed warning of ESAs used to treat chemotherapy-induced anemia because evidence on the effectiveness of boxed warnings remains inconclusive.
Patients and Methods
Using 2004 to 2009 SEER-Medicare data, we exploited a natural experiment to examine the effects of ESA boxed warnings on utilization and risk of VTE. The intervention group included Medicare fee-for-services patients diagnosed with colorectal, breast, or lung cancers targeted by this warning and undergoing chemotherapy; the control group included patients with myelodysplastic syndromes not targeted by this warning. The period from January 2004 to September 2006 was used as the prewarning period; the period from April 2007 to September 2009 was used as the postwarning period. The two binary dependent variables included ESA use and hospitalized VTE. Linear probability models with a difference-in-differences specification were used for estimation.
Results
Our sample consisted of 45,319 unique patients between 2004 and 2009. The trends in ESA use remained similar between the intervention and control groups before the warning, but started declining sharply in the intervention group only after the warning. The trends in hospitalized VTE were relatively stable. Regressions showed that the ESA boxed warning was associated with a 20.2-percentage-point reduction (P < .001) in the likelihood of ESAs being used to treat cancers targeted by the warning, but not significantly associated with the likelihood of hospitalized VTE.
Conclusion
Our study showed that the warning was effective in reducing ESA utilization. Future studies should examine other regulatory drug safety actions, such as the Risk Evaluation and Mitigation Strategy initiative, whose effectiveness remains unknown.
INTRODUCTION
Before the passage of the 2007 US Food and Drug Administration (FDA) Amendments Act, boxed warnings were considered the strongest FDA mechanism for communicating drug safety concerns to the public. However, critics question the effectiveness of boxed warnings, contending that providers and patients may not actually receive or agree with safety concerns in the warnings and concerned that the FDA may not have adequate authority and resources for postwarning surveillance.1 Although the number of boxed warnings issued by the FDA has increased rapidly, available evidence on the effectiveness of these warnings remains inconclusive, largely because almost all of them were drawn from observational studies using pre-post designs without control groups.1,2 Furthermore, there have been concerns about the lack of evidence on the impact of boxed warnings on serious risks advised by these warnings.2
We evaluated the effectiveness of the FDA boxed warning of erythropoietin-stimulating agents (ESAs), a class of biologic agents including epoetin and darbepoetin, used to treat chemotherapy-induced anemia. Sales of ESAs in the United States exceeded $6.4 billion in 2002, and Medicare expenditures for ESAs in the oncology setting surpassed $1.5 billion in 2004.3 Beginning in 2003, however, studies found serious risks, such as venous thromboembolism (VTE) and mortality, associated with ESA use.4-7 After these studies, an FDA boxed warning was issued for ESAs in March 2007. Shortly thereafter, changes in Medicare payment policy, developed under the guidelines of the FDA ESA boxed warning, were implemented to ensure more prudent ESA use. In March 2010, the FDA placed ESAs under a Risk Evaluation and Mitigation Strategy (REMS) program, a heightened pharmacovigilance mechanism authorized by the 2007 FDA Amendments Act.8
Using 2004 to 2009 SEER-Medicare data, we exploited a natural experiment that incorporated a control group into a pre-post design to examine the effects of the ESA boxed warning on the use of ESAs and risk of VTE. We hypothesized that the warning would reduce the use of ESAs and therefore lower the risk of VTE. In our study, the intervention group included elderly Medicare fee-for-service (FFS) patients who were diagnosed with colorectal, breast, or lung cancers targeted by this warning and underwent chemotherapy. A control group included patients with myelodysplastic syndromes (MDS; ie, bone marrow disorders associated with higher risks of leukemia), whose ESA use, representing off-label use, was not targeted by the warning. The period from January 2004 to September 2006 was used as the prewarning period; the period from April 2007 to September 2009 was used as the postwarning period. Linear probability models with a difference-in-differences (DD) specification were used in the regression analysis.
PATIENTS AND METHODS
Data Sources
This study used SEER-Medicare data from 2004 to 2009, starting in the year immediately after the evidence on serious risks associated with ESA use started emerging and ending in the year immediately before the new FDA ESA REMS took effect. The SEER program is a cancer registry that covered approximately one fourth of the U.S. population from 12 states during our study period.9 The SEER data, which included detailed clinical information on incident cancers, were linked to Medicare claims from 2004 to 2010. The linked data provided Medicare enrollment information, such as Part A and B eligibility status, and monthly FFS status. Linked claims data are available for Medicare beneficiaries > 65 years of age enrolled in FFS plans. The claims information pertinent to our study included International Classification of Diseases (9th revision, clinical modification) diagnosis and procedural codes and Current Procedural Terminology (4th edition) codes from inpatient, physician, and outpatient claims. (The specific codes used for selecting the study sample and constructing variables were based on the previously published work.10-12) This study was approved by our institutional review board.
Study Sample
Using the SEER-Medicare data, we constructed a sample consisting of patients ≥ 66 years of age at diagnosis with first-ever incident colorectal, female breast, or non–small-cell lung cancer or MDS between January 2004 and September 2009, excluding the period from October 2006 to March 2007. The two additional inclusion criteria required that patients (1) were enrolled in Medicare for eligibility on the basis of age only and in FFS plans with both Part A and B benefits within 6 months postdiagnosis, and (2) were diagnosed with colorectal, breast, or lung cancer of known staging presentation and received chemotherapy treatment in the physician or outpatient claims within 6 months postdiagnosis (note that MDS has no staging presentation).
Dependent Variables
Our analysis had two key binary dependent variables. The first measured ESA use, equal to 1 if any ESA use was recorded in the physician or outpatient claims within 6 months postdiagnosis or 0 otherwise. The second measured hospitalized VTE, equal to 1 if any VTE events (deep vein thrombosis or pulmonary embolism) indicated by the primary or secondary diagnosis codes were recorded in the inpatient claims within 6 months postdiagnosis or 0 otherwise. A third dependent variable measured the use of granulocyte/granulocyte-macrophage colony-stimulating factors (G/GM-CSFs), equal to 1 if any G/GM-CSF use was recorded in the physician or outpatient claims within 6 months postdiagnosis or 0 otherwise. We included G/GM-CSFs as a dependent variable to conduct a falsification test as part of statistical analysis.
Independent Variables
The key independent variable used to capture the effects of the ESA boxed warning on the use of ESAs and risk of VTE was the interaction of the intervention and the postwarning period variables, which equaled 1 if a patient was diagnosed with colorectal, breast, or lung cancer from April 2007 to September 2009 or 0 otherwise. Other independent variables were all at the patient level. Demographic variables included age and race/ethnicity; gender was not included because only female patients with breast cancer were retained for our analysis. Age was grouped into five levels, representing patients ages 66 to 70, 71 to 75, 76 to 80, 81 to 85, and ≥ 86 years. Race or ethnicity was categorized into three groups: white, black, or other. The Charlson index, calculated from the primary and secondary diagnosis codes available in the inpatient claims within 6 months postdiagnosis, was categorized into three groups: equal to 0, equal to 1, or > 1.13 We also created a patient-level variable representing Medicare state buy-in program entitlement status, a proxy for socioeconomic status, coded as 1 if a patient was eligible for buy-in subsidies for at least 1 month in the year of diagnosis or 0 otherwise.14
Statistical Analysis
To minimize threats to internal validity from unobserved confounders in evaluating the impact of a policy change, a natural experimental design may be preferred to a pre-post design.15-17 In our study, we compared an intervention targeted by the warning with a nonequivalent control not targeted by the warning. We assumed that the pre-post differences in our control group were appropriate estimates of what the pre-post differences in our intervention would have been if the warning had not been issued. We chose colorectal, breast, and lung cancers as the intervention and MDS as the control. ESAs used by patients with MDS are not under the FDA ESA boxed warning.3 Although the association of ESAs with the risk of VTE is unclear among patients with MDS, one phase II study of patients with MDS given ESAs was terminated earlier because an unexpected high number of VTE events was found.18
Linear probability models (LPMs) with a DD specification were used as the main regression analysis, in which the unit of analysis was a unique patient. (We used LPMs instead of logit models because the boxed warning effects were captured from the interaction term that may be complicated in estimating average effects in logit models.19) We regressed ESA use or hospitalized VTE on the interaction variable of the intervention and the postwarning period and on a set of patient-level covariables. (We controlled for the use of ESAs in estimating the specification with hospitalized VTE as the dependent variable.) Additional specifications included diagnosis (MDS and three cancers) and year fixed effects. The diagnosis fixed effects captured all diagnosis-specific, time-invariant differences (eg, disease characteristics, natural history of disease, and treatment patterns, such as duration of ESA use) as potential confounders between the intervention and the control, and the year fixed effects captured secular trends (eg, across-the-board policy changes) that similarly affected the use of ESAs or risk of VTE in the two groups. A state fixed effects specification was also used to control for all time-invariant state-level characteristics.20
We performed two additional regression analyses. The first one was a sensitivity analysis of whether the effects of the boxed warning might vary by type of cancer in the intervention. The second one was a falsification test of whether the ESA boxed warning only affected the use of ESAs intended by the warning. To do so, we used the same main regression analysis specification to assess the effect of the FDA ESA boxed warning on the use of G/GM-CSFs. Because the use of G/GM-CSFs is not under any FDA boxed warning, we would expect no effect of ESA boxed warning on the use of G/GM-CSFs. (If this is the case, we would be more confident that our main regression analysis did not capture a spurious relationship between ESA boxed warning and the use of ESAs.)
RESULTS
Descriptive Statistics
There were 45,319 unique patients from 12 states between 2004 and 2009, which included 3,375 patients with MDS, 10,243 with colorectal cancer, 9,375 with breast cancer, and 22,344 with lung cancer. (Note that the sample size in 2006, 2007, and 2009 only accounted for the patients diagnosed in three quarters of a year.) Table 1 lists the mean statistics of the sample by diagnosis over time.
Table 1.
Mean Descriptive Statistics of the Study Sample by Diagnoses (myelodysplastic syndromes and three cancers) From 2004 to 2009

The trends in ESA use within 6 months postdiagnosis among the four diagnoses, highlighted in Figure 1, were similar before the warning, but started to decline sharply after the warning, except for patients with MDS. For example, the proportions for patients with breast cancer receiving ESAs ranged from 49.1% to 54.9% during the 3-year period before the warning and declined to 30.5% in 2007, 16.4% in 2008, and 8.8% in 2009. In contrast, the proportions for patients with MDS were 39.3% to 41.7%, 34.6%, 32.3%, and 32.0%, respectively. The trends in hospitalized VTE (Fig 2) also remained stable before the warning. However, the proportion of patients with lung cancer with hospitalized VTE increased after the warning, whereas the proportion decreased slightly among patients with colorectal cancer. Furthermore, the proportions of patients with breast cancer and MDS with hospitalized VTE were much lower than those with colorectal or lung cancer. The trends of other patient-level variables, such as demographic characteristics, Charlson index, and Medicare state buy-in program, differed across the four diagnoses, but remained stable within each diagnosis.
Fig 1.

Trends in the use of erythropoietin-stimulating agents by diagnoses.
Fig 2.

Trends of hospitalized venous thromboembolism by diagnoses.
Regression Results
Table 2 lists the estimates from the two LPMs with the DD specification. The first model showed that the FDA ESA boxed warning was associated with an overall 20.2-percentage-point reduction (P < .001) in the likelihood of ESAs used among patients diagnosed with one of the three types of cancer targeted by the warning. This reduction represented a fall of more than 42.2% from the average ESA use among the patients in the intervention group in the 3 years before the warning. The sensitivity analysis of differential effects of the warning (not shown) indicated that the warning was associated with 17.5-, 26.7-, and 18.8-percentage-point reductions in the likelihood of ESAs among patients with colorectal, breast, or lung cancer, respectively (all P < .001). The falsification test (not shown) demonstrated no association between the ESA boxed warning and the likelihood of G/GM-CSFs used among the patients in the study sample (P > .50). Figure 3 shows the parallel trends in the use of G/GM-CSFs by each of the four diagnoses over time. After examining the associations of other patient-level covariables with the boxed warning, we found that compared with patients 66 to 70 years of age, those at older age categories had a higher likelihood of ESA use (all P < .001) and that patients having a Charlson index equal to zero were more likely to use ESAs than were those having a higher Charlson index. Furthermore, patients with lower socioeconomic status, indicated by their enrollment in a Medicare state buy-in program, had a lower likelihood of receiving ESAs (3.1 percentage points) than did those with a higher socioeconomic status (P < .001).
Table 2.
Estimates From Linear Probability Models With Difference-in-Differences Specification
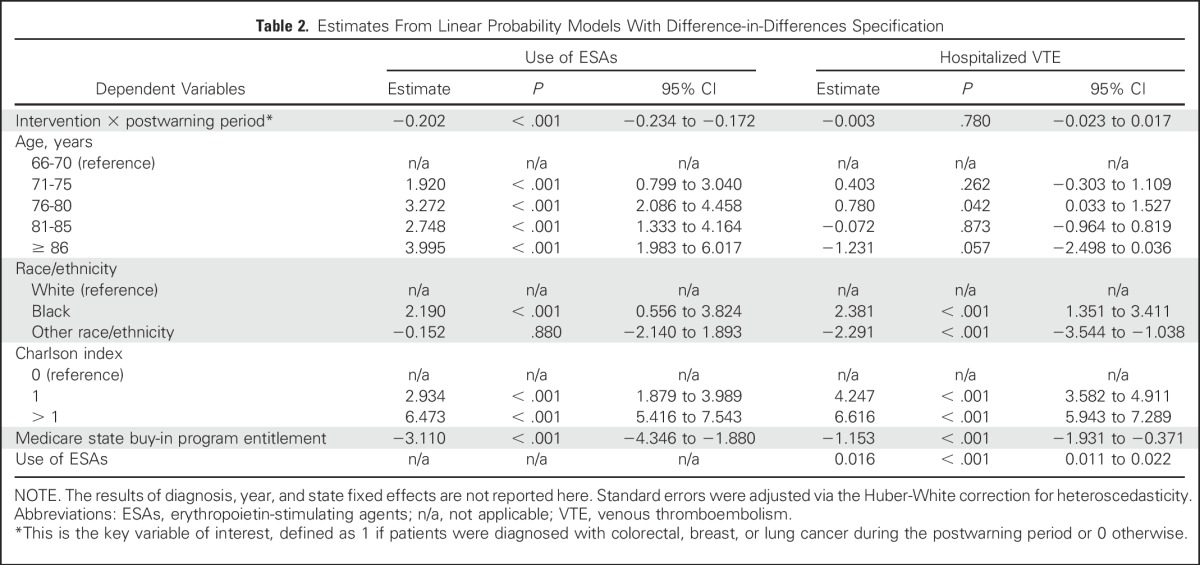
Fig 3.

Trends in the use of granulocyte/granulocyte-macrophage colony-stimulating factors by diagnoses.
The second model that evaluated the effect of the warning on risk of VTE showed no significant association between ESA boxed warning and the likelihood of hospitalized VTE experienced by patients diagnosed with one of the three types of cancer targeted by the warning (P > .50). The corresponding sensitivity analysis did not find any significant associations by the three types of cancer in the intervention (all P > .10). The estimates of other patient-level variables showed that blacks had a higher risk of VTE than whites (P < .001) and that patients having a Charlson index > 0 also had a higher risk of VTE than patients with a Charlson index equal to zero (both P < .001). In addition, ESA use was associated with a 1.6-percentage-point increase in the likelihood of hospitalization for VTE (P < .001).
DISCUSSION
We have found that the FDA ESA boxed warning issued in March 2007 was associated with a reduction in ESAs used among elderly Medicare FFS patients who were diagnosed with colorectal, breast, or lung cancers and received chemotherapy within 6 months postdiagnosis. This estimated reduction represented a > 40% decrease in the use of ESAs among the studied patients after the warning took effect. However, our analysis did not show significant associations of the warning with the risk of hospitalized VTE.
Although our results were broadly consistent with the evidence of the impact of boxed warnings on utilization in general, they differed from the evidence on ESAs used by patients with cancer.2,21 One study of veterans diagnosed with lung or colorectal cancer only found a slight decline in utilization after the ESA boxed warning.22 Furthermore, a second study suggested that this boxed warning had little impact on the use of ESAs.12 Comparative evidence of the impact of boxed warnings on adverse health outcomes has not been reported. To the best of our knowledge, ours is the first study of the impact of boxed warnings on outcomes showing results similar to the limited evidence on the impact of regulatory drug safety actions on clinical outcomes.2,23,24
Although natural experiments may have a stronger causal implication than pre-post designs, our study has several limitations. First, there is a concern about whether MDS was an appropriate control. A DD specification assumes parallel time trends in dependent variables between the intervention and the control before a change in policy.16 In our descriptive analyses, the trends in ESA use were similar between the two groups before the warning (Fig 1). The time trends in patients hospitalized with VTE before the warning remained stable and similar, although not as apparent as with ESA use (Fig 2). In the regression analysis, recognizing inherent differences between the intervention and control groups, we further specified diagnosis fixed effects to control for all time-invariant, diagnosis-specific differences as potential confounders. However, the remaining unobserved time-varying, diagnosis-specific confounders may still be a potential source of bias.
Second, although the DD specification with year fixed effects controlled for secular trends that similarly affected the intervention and the control groups, additional biases may arise if some of the changes might have affected the two groups differently. (Note that it is important to differentiate mediators from confounders, in which the latter may cause biases if not controlled. One study showed that the change in Medicare payment policy for ESAs may have influenced ESA use.12 However, this change may be viewed as a mediator because it was developed under the guidelines of the FDA ESA boxed warning and implemented after the warning was issued. Although not explicitly controlling for this change would not lead to biases in our regression analysis, our estimates of the boxed warning might have also captured any influence of this change on the use of ESAs.)
Third, we are nonetheless concerned about suspected spillover effects of the ESA boxed warning. For example, health care providers who treated patients with a diagnosis not targeted by the warning may be aware of serious risks associated with ESAs used by patients with cancer and renal disease, given that a great amount of scrutiny surrounded the safety of these biologic agents. As a result, it was possible that ESAs used by patients with MDS might have been slightly affected. In this case, our DD specification may underestimate the impact of the boxed warning on ESA use. Fourth, accuracy of variable measurement may be another concern. Because metastatic solid-tumor cancers may not receive ESAs within the 6 months postdiagnosis during the postwarning period, we performed a sensitivity analysis by excluding patients with stage IV disease, but found similar results. Another example is that the claims data did not allow us to measure ESA dosage that could have implications for the finding of no association of the warning with risk of VTE. Furthermore, Medicare started to require hemoglobin value reporting for reimbursement of ESAs used by patients with cancer in 2008. However, hemoglobin value reporting was not available before 2008 and has not been required for ESAs used by patients with MDS. As a result, our findings do not shed light on the appropriateness of ESA use. Finally, this study may have limited generalizability. Our results may not be applicable to the boxed warnings of other drugs or to different populations.
This is the first study using a natural experiment to examine the effects of FDA boxed warnings on utilization and adverse outcomes. Although our analysis indicated that the 2007 FDA ESA boxed warning was followed by a significant reduction in ESAs used by patients diagnosed with cancers targeted by the warning, our results found little impact of the warning on the risk of VTE, a serious risk associated with ESA use. In an era of ever-faster FDA drug approvals, likely fueled by the 21st Century Cures Act and manifested by a growing number of boxed warnings, our study has filled a critical gap in evidence on effectiveness of one commonly used drug safety warning mechanism. Future studies should continue to investigate the impact of boxed warnings on serious adverse events and to examine other regulatory actions (such as the FDA REMS initiative) to promote the safe use of drugs whose effectiveness remains largely unknown.
Footnotes
Supported in part by grants from the American Cancer Society (IRG13-043-01), the National Institutes of Health (R01CA165609), the University of South Carolina and the Centers for Economic Excellence program of the state of South Carolina (11150-13-32600), and philanthropic gifts from Doris Levkoff Meddin and Frank P. and Josie M. Fletcher to the Smart State Center for Medication Safety.
AUTHOR CONTRIBUTIONS
Conception and design: John Bian, Brian Chen, Dawn L. Hershman, LeAnn Norris, Richard Schulz, Charles L. Bennett
Collection and assembly of data: John Bian
Data analysis and interpretation: John Bian, Brian Chen, Norman Marks, Richard Schulz, Charles L. Bennett
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Effects of the US Food and Drug Administration Boxed Warning of Erythropoietin-Stimulating Agents on Utilization and Adverse Outcome
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
John Bian
No relationship to disclose
Brian Chen
No relationship to disclose
Dawn L. Hershman
No relationship to disclose
Norman Marks
Employment: Medical Product Safety
Leadership: Medical Product Safety
Stock or Other Ownership: Medical Product Safety
Consulting or Advisory Role: Quincy Bioscience
LeAnn Norris
Consulting or Advisory Role: Bristol-Myers Squibb, Taiho Pharmaceutical
Richard Schulz
Stock or Other Ownership: Mylan, Eli Lilly, Merck, Amgen
Consulting or Advisory Role: BDI
Charles L. Bennett
No relationship to disclose
REFERENCES
- 1.Frank C, Himmelstein DU, Woolhandler S, et al. Era of faster FDA drug approval has also seen increased black-box warnings and market withdrawals. Health Aff (Millwood) 2014;33:1453–1459. doi: 10.1377/hlthaff.2014.0122. [DOI] [PubMed] [Google Scholar]
- 2.Piening S, Haaijer-Ruskamp FM, de Vries JTN, et al. Impact of safety-related regulatory action on clinical practice: A systematic review. Drug Saf. 2012;35:373–385. doi: 10.2165/11599100-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 3.Steinbrook R. Erythropoietin, the FDA, and oncology. N Engl J Med. 2007;356:2448–2451. doi: 10.1056/NEJMp078100. [DOI] [PubMed] [Google Scholar]
- 4.Henke M, Laszig R, Rübe C, et al. Erythropoietin to treat head and neck cancer patients with anaemia undergoing radiotherapy: Randomised, double-blind, placebo-controlled trial. Lancet. 2003;362:1255–1260. doi: 10.1016/S0140-6736(03)14567-9. [DOI] [PubMed] [Google Scholar]
- 5.Leyland-Jones B. Breast cancer trial with erythropoietin terminated unexpectedly. Lancet Oncol. 2003;4:459–460. doi: 10.1016/s1470-2045(03)01163-x. [DOI] [PubMed] [Google Scholar]
- 6.Bennett CL, Silver SM, Djulbegovic B, et al. Venous thromboembolism and mortality associated with recombinant erythropoietin and darbepoetin administration for the treatment of cancer-associated anemia. JAMA. 2008;299:914–924. doi: 10.1001/jama.299.8.914. [DOI] [PubMed] [Google Scholar]
- 7.Bohlius J, Schmidlin K, Brillant C, et al. Recombinant human erythropoiesis-stimulating agents and mortality in patients with cancer: A meta-analysis of randomised trials. Lancet. 2009;373:1532–1542. doi: 10.1016/S0140-6736(09)60502-X. [DOI] [PubMed] [Google Scholar]
- 8.Mitka M. New oversight put in place for physicians giving anemia drugs to patients with cancer. JAMA. 2010;303:1355–1356. doi: 10.1001/jama.2010.359. [DOI] [PubMed] [Google Scholar]
- 9.Bian J, Lipscomb J, Mello MM. Spillover effects of state mandated benefit laws: The case of outpatient breast cancer surgery. Inquiry. 2009-2010/2010;46:433–447. doi: 10.5034/inquiryjrnl_46.4.433. [DOI] [PubMed] [Google Scholar]
- 10.Hershman D, Neugut AI, Jacobson JS, et al. Acute myeloid leukemia or myelodysplastic syndrome following use of granulocyte colony-stimulating factors during breast cancer adjuvant chemotherapy. J Natl Cancer Inst. 2007;99:196–205. doi: 10.1093/jnci/djk028. [DOI] [PubMed] [Google Scholar]
- 11.Hershman DL, Buono DL, Malin J, et al. Patterns of use and risks associated with erythropoiesis-stimulating agents among Medicare patients with cancer. J Natl Cancer Inst. 2009;101:1633–1641. doi: 10.1093/jnci/djp387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hershman DL, Neugut AI, Shim JJ, et al. Erythropoiesis-stimulating agent use after changes in medicare reimbursement policies. J Oncol Pract. 2014;10:264–269. doi: 10.1200/JOP.2013.001255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Charlson ME, Pompei P, Ales KL, et al. A new method of classifying prognostic comorbidity in longitudinal studies: Development and validation. J Chronic Dis. 1987;40:373–383. doi: 10.1016/0021-9681(87)90171-8. [DOI] [PubMed] [Google Scholar]
- 14.Bian J, Bennett C, Cooper G, et al. Assessing colorectal cancer screening adherence of Medicare fee-for-service beneficiaries age 76 to 95 years. J Oncol Pract. 2016;12:e670–e680. doi: 10.1200/JOP.2015.009118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bian J, Bennett CL, Fisher DA, et al. Unintended consequences of health information technology: Evidence from veterans affairs colorectal cancer oncology watch intervention. J Clin Oncol. 2012;30:3947–3952. doi: 10.1200/JCO.2011.39.7448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dimick JB, Ryan AM. Methods for evaluating changes in health care policy: The difference-in-differences approach. JAMA. 2014;312:2401–2402. doi: 10.1001/jama.2014.16153. [DOI] [PubMed] [Google Scholar]
- 17.Loehrer AP, Song Z, Haynes AB, et al. Impact of health insurance expansion on the treatment of colorectal cancer. J Clin Oncol. 2016;34:4110–4115. doi: 10.1200/JCO.2016.68.5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steurer M, Sudmeier I, Stauder R, et al. Thromboembolic events in patients with myelodysplastic syndrome receiving thalidomide in combination with darbepoietin-alpha. Br J Haematol. 2003;121:101–103. doi: 10.1046/j.1365-2141.2003.04252.x. [DOI] [PubMed] [Google Scholar]
- 19.Ai C, Norton EC. Interaction terms in logit and probit models. Econ Lett. 2003;80:123–129. [Google Scholar]
- 20.Wright JD, Neugut AI, Wilde ET, et al. Physician characteristics and variability of erythropoiesis-stimulating agent use among Medicare patients with cancer. J Clin Oncol. 2011;29:3408–3418. doi: 10.1200/JCO.2010.34.5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bradford WD, Kleit AN. Impact of FDA actions, DTCA, and public information on the market for pain medication. Health Econ. 2015;24:859–875. doi: 10.1002/hec.3067. [DOI] [PubMed] [Google Scholar]
- 22.Tarlov E, Stroupe KT, Lee TA, et al. Trends in anemia management in lung and colon cancer patients in the US Department of Veterans Affairs, 2002-2008. Support Care Cancer. 2012;20:1649–1657. doi: 10.1007/s00520-011-1255-0. [DOI] [PubMed] [Google Scholar]
- 23.Wheeler BW, Metcalfe C, Gunnell D, et al. Population impact of regulatory activity restricting prescribing of COX-2 inhibitors: Ecological study. Br J Clin Pharmacol. 2009;68:752–764. doi: 10.1111/j.1365-2125.2009.03500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang C, Kane R, Levenson M, et al. Association between changes in CMS reimbursement policy and drug labels for erythrocyte-stimulating agents with outcomes for older patients undergoing hemodialysis covered by fee-for-service Medicare. JAMA Intern Med. 2016;176:1818–1825. doi: 10.1001/jamainternmed.2016.6520. [DOI] [PubMed] [Google Scholar]