

A metabolomic, biochemical, fluxomic, and metabolic modeling approach developed using 19 genetically distant maize lines links leaf physiology to kernel yield.
Abstract
A combined metabolomic, biochemical, fluxomic, and metabolic modeling approach was developed using 19 genetically distant maize (Zea mays) lines from Europe and America. Considerable differences were detected between the lines when leaf metabolic profiles and activities of the main enzymes involved in primary metabolism were compared. During grain filling, the leaf metabolic composition appeared to be a reliable marker, allowing a classification matching the genetic diversity of the lines. During the same period, there was a significant correlation between the genetic distance of the lines and the activities of enzymes involved in carbon metabolism, notably glycolysis. Although large differences were observed in terms of leaf metabolic fluxes, these variations were not tightly linked to the genome structure of the lines. Both correlation studies and metabolic network analyses allowed the description of a maize ideotype with a high grain yield potential. Such an ideotype is characterized by low accumulation of soluble amino acids and carbohydrates in the leaves and high activity of enzymes involved in the C4 photosynthetic pathway and in the biosynthesis of amino acids derived from glutamate. Chlorogenates appear to be important markers that can be used to select for maize lines that produce larger kernels.
INTRODUCTION
Maize (Zea mays) is now ranked first among cereal crops, accounting for 41% of the total world cereal production. Maize production has doubled over the last 30 years, with almost 1000 million metric tons (38,105 bushels) produced in 2015–2016 (https://corn3blog.wordpress.com/global-comparison/). With yields of over 10 metric tons per ha, maize also ranks first in terms of grain yield in Europe and the US, although the yield is much lower in the rest of the world, accounting for ∼5 to 6 metric tons per ha (http://www.agprofessional.com/news/A-comparison-of-world-maize-yields-227415201.html). Half of the world maize production consists of silage used for animal feed. The other half contributes grain for seed propagation and for a wide variety of commercial products used mostly to feed humans and animals. Among the products not used for food, bioethanol is one of the most important (Ranum et al., 2014).
Maize is an annual monoecious crop, which for most genotypes cultivated at present, requires a life cycle of around 6 months. Maize was domesticated in Mexico from teosinte around 7000 years ago (Matsuoka et al., 2002). Maize was then distributed over the American continent and the rest of the world, reaching Europe around the time of the discovery of the New World at the end of the 15th century (Rebourg et al., 2003). During this expansion, maize acquired properties related to climatic adaptation, such as insensitivity to photoperiod and early flowering, thus allowing optimal growth under lower temperature conditions in most temperate countries. Maize serves not only as a major crop, but also as a model species that is well adapted for fundamental research. This use of maize is particularly important for increasing our understanding of the agronomic and the genetic bases of yield performance, related to the genomic changes that have occurred with domestication and breeding (Solomon et al., 2014; Shi and Lai, 2015). Studies on maize can take advantage of its wide genetic diversity and the availability of mutant collections, recombinant inbred lines, and straightforward transformation protocols. In addition, physiological, biochemical and “omics” data, as well as genome sequences (Hirel et al., 2007) and, more recently, genome-scale metabolic models (Simons et al., 2014), have become accessible.
Due to the complexity of the biological systems involved in the control of yield and biomass production, several complementary approaches have been undertaken to improve our understanding of the genetic and physiological basis of plant productivity as a function of environmental constraints, at the cellular, organ, and whole-plant levels (Wuyts et al., 2015). One of these approaches was based on maize quantitative genetics, including the development of powerful genome-wide association studies (GWAS) and the insights they bring to the molecular and physiological mechanisms underlying complex agronomic traits (reviewed in Wallace et al., 2014). GWAS use the huge genetic variety existing in maize, as well as the identification of genes or loci involved in crop evolution, which may have determined the genetics of domestication and diversification (Meyer and Purugganan, 2013).
Some metabolic pathways, notably those involved in carbon (C) assimilation, have a direct effect on crop yield. As these metabolic pathways also exhibit wide plasticity, according to the environmental conditions and the genetic background, it is essential to obtain a better understanding of the underlying molecular and physiological mechanisms controlling crop productivity (Rossi et al., 2015). Thus, in parallel to quantitative genetic studies, integrated approaches termed “systems biology” take advantage of numerous transcriptome, proteome, metabolome, and fluxome data sets, developed using mathematical, bioinformatic, and computational tools. Such integrated analyses, combined with whole-plant physiology and quantitative genetic studies, may ultimately allow the identification of key elements involved in the control of complex biological processes and agronomic traits such as yield and grain quality (Saito and Matsuda, 2010; Schilmiller et al., 2012).
Among the data sets necessary to develop systems biology, metabolomic, proteomic, and enzyme activity profiles have been increasingly used. These profiles establish whether relationships exist between metabolite and protein accumulation, enzyme activities, and phenotypic traits related to plant growth and development (Fukushima and Kusano, 2013; Stitt, 2013). Until now, the majority of high-throughput biochemical and metabolic approaches have been performed using Arabidopsis thaliana. More recently, these approaches have been extended to a wider range of plants including cereals such as rice (Oryza sativa) and maize (Kusano et al., 2011; Lisec et al., 2011; Amiour et al., 2014; Riedelsheimer et al., 2012a). Thus, interesting perspectives have arisen concerning the use of metabolome-assisted breeding techniques to narrow the genotype/phenotype gap of complex traits, such as yield and biomass production (DellaPenna and Last, 2008; Fernie and Shauer, 2009; Lisec et al., 2011; Kusano and Fukushima, 2013; Wen et al., 2015).
In particular, metabolite-based GWAS have recently been very useful for dissecting complex traits both in model and crop species (Luo, 2015). Such GWAS were successfully conducted using leaves and kernels of maize lines and hybrids. The use of GWAS identified novel biochemical insights and candidate genes involved in controlling the agronomic traits of interest, related to plant productivity or kernel quality (Riedelsheimer et al., 2012a, 2012b; Wen et al., 2014; Zhang et al., 2015).
Although very powerful for examining the genetic and biochemical bases of crop metabolism, these studies did not provide detailed interpretation of the underlying physiology, either in terms of metabolite accumulation or metabolic flux. To fill this gap, we have developed a combined metabolomic, biochemical, fluxomic, and metabolic modeling approach, taking advantage of the genetic diversity of a core panel of 19 American and European maize lines, which have been classified as Maize Belt Dent, European Flint, Northern Flint, Stiff Stalk, and Tropical lines on the basis of their genetic relatedness (Camus-Kulandaivelu et al., 2006; Bouchet et al., 2013). The aim of this work was to determine if analyses of metabolites and enzyme activities at two key stages of plant development can be used as selection markers for breeding maize with a superior agronomic performance.
RESULTS
Leaf Metabolite and Enzyme Activity Profiles of Genetically Distant Maize Lines Show Large Variability at Two Stages of Development
In this study, gas chromatography coupled to mass spectrometry (GC-MS) analyses of the leaf metabolome were performed on 19 maize lines that had been grown in the field. The plants were grown under the optimal nitrogen (N) supply currently used in field experiments to ensure maximal vegetative growth and grain production of maize under temperate climatic conditions. The 19 lines originated from different northern and southern countries of both America and Europe, which had been used as a core collection for genome wide association genetic studies by Camus-Kulandaivelu et al. (2006) and Bouchet et al. (2013) (for details, see Methods and Supplemental Table 1). When the proportion of alleles shared between the 19 lines was analyzed, we found that they were not closely related, since their genetic dissimilarity ranged from ∼20% (between line HP301 and line SA24U) to 42% (between line NYS302 and line CML254). A genetic relatedness study revealed that the 19 lines were spread within the whole S1P9 genetic diversity panel originally characterized by Camus-Kulandaivelu et al. (2006), thus indicating that the genetic diversity of S1P9 was well covered (Supplemental Figure 1).
In the leaf samples taken at the vegetative (V) stage of plant development and during the grain-filling period 15 d after silking (DAS), 155 water-soluble leaf metabolites were detected. However, after ANOVA statistical analysis (P ≤ 0.05) followed by a Bonferroni post-hoc test and correction, 107 metabolites were found to be significantly different between the 19 lines at the V stage and 124 metabolites at 15 DAS (Supplemental Data Set 1). For most of the metabolites, a quantitative estimation of their abundance was performed using standards as described in Methods. The total leaf soluble metabolite content at the V stage was 4.2 nmol mg−1 leaf fresh weight (FW), whereas at 15 DAS it was 3.2 nmol mg−1 leaf FW.
At the V stage, the average proportion of the leaf soluble metabolite content in the 19 lines was 52% for carbohydrates, 32% for organic acids, and 9% for amino acids, with the remainder (7%) being represented by various molecules such as lipids, polyamines, vitamins, and secondary metabolites (Supplemental Figures 2A and 2B). A number of unidentified compounds (listed as unk.) were also detected. Their relative proportions in the 19 lines can be found in Supplemental Data Set 1, taking into account that their concentration was 30% higher at the V stage compared with 15 DAS. For carbohydrates, sucrose was by far the most abundant, representing 68% of the total, followed by glucose (17%). Fructose, myo-inositol, and raffinose were less abundant, representing 4, 3, and 1%, respectively, of the total leaf soluble carbohydrate. For the soluble amino acids, alanine at a relative concentration of 35% of the total, predominated followed by glutamate (15%) and glycine (14%). Glutamine was present as only 6% of the total leaf soluble amino acids. As observed in previous studies, aconitate (65%) was the most abundant organic acid in the leaf (Brauer and Teel, 1981; Sicher and Barnaby, 2012; Yesbergenova-Cuny et al., 2016), followed by malate (12%) and pyruvate (7%).
In the 19 lines, the relative proportions of the most abundant C- and N-containing molecules at 15 DAS were very similar to that of the V stage, except for glycine and raffinose, which were approximately 3 times lower and higher, respectively, at 15 DAS. However, differences between the V and the 15 DAS growth stages were observed in leaf metabolites present in low concentrations, such as quinate and 5-caffeoylquinate-trans, respectively (Supplemental Figures 2A and 2B).
The range of variation observed between the leaves of the 19 lines is illustrated in Figure 1. At the V stage, a large variability was observed for the amino acids asparagine (Asn) and proline (Pro), with coefficients of variation of 200 and 70% in their content, respectively. The range of variation observed for the most abundant amino acids, such as alanine, glutamate, and aspartate, was much lower (20−30%). When the range of soluble carbohydrates in the 19 lines was examined, the most abundant (sucrose) was the least variable (16% at the V stage and 30% 15 DAS). Although glucose and fructose were present in much lower amounts compared with sucrose, their range of variation was much higher (40 and 200% at the V stage and 15 DAS, respectively). Almost no differences in total C and N and plant water content were observed.
Figure 1.

Differences in the Metabolite Content, Enzyme Activities, and Biomass-Related Components in the Leaves of 19 Maize Lines Originating from Europe and America at Two Key Stages of Plant Development.
The top of the figure shows the coefficients of variations (expressed as percentage) of the biomass-related components in red (biomass components are shown as: C, total carbon; N, total nitrogen), including yield. Enzyme activities are in blue (GS, glutamine synthetase; MDH, NADP-dependent malate dehydrogenase; GOGAT: ferredoxin-dependent glutamate synthase; AspAT, aspartate aminotransferase). Metabolites and classes of metabolites are in black (2-OG, 2-oxoglutarate; Cit, citrate; Pyr, pyruvate; AA, total amino acids; Unknown, unidentified metabolites). An overview representation of the average of the coefficients of variations (from 0 to 80%) for the main classes of metabolites, enzyme activities, and biomass components is shown at the bottom of the figure. Anions correspond to nitrate and phosphate and biomass components to C, N, and water contents.
Differences between the 19 lines were detected in the activities of a number of enzymes involved in primary N and C metabolism (listed in Supplemental Table 2), including C4 photosynthesis for the latter (Figure 1). The largest variations in these enzyme activities, ranging from 20 to 30%, were detected for nitrate reductase (NR), glutamate dehydrogenase (GDH), and ferredoxin-dependent glutamate synthase (Fd-GOGAT), both at the V stage and 15 DAS. Such variations are likely due to the fact that enzyme activities were measured in vitro, thus probably reflecting their maximal activities (Biais et al., 2014). However, almost no differences in phosphoenolpyruvate carboxylase (PEPC) activity were observed. During the grain filling period 15 DAS, the range of variation observed for most of the enzyme activities was comparable to that observed at the V stage.
Hierarchical Clustering Analysis Demonstrates That the Leaf Metabolite and Enzyme Activity Profiles Are Specific to the Developmental Stage of the Plant
A hierarchical clustering analysis (HCA) showed that metabolites exhibiting significant differences between the 19 lines and that were common to the two developmental stages displayed a clear difference in the level of accumulation between the V stage and 15 DAS (Supplemental Figure 3).
Three main groups of metabolites exhibiting opposing patterns of accumulation between the V stage and 15 DAS were clearly identified. The first group contained metabolites that accumulated to a higher degree at 15 DAS in all the lines. The second group contained metabolites that were present in higher amounts at the V stage compared with 15 DAS in most of the lines. In the third group, the difference in metabolite content between the two plant developmental stages was less marked (Supplemental Figure 3).
Specific differences in the activity of the portfolio of enzymes representative of plant primary and secondary metabolism (Supplemental Table 2) were observed between the V stage and 15 DAS (Table 1). The same pattern of enzyme activity (higher or lower) was observed between the V stage and 15 DAS across all five Tropical lines, irrespective of their physiological function. In the lines of the four other groups (defined on the basis of their genetic distance; see Methods and Supplemental Table 1), the differences in most of the enzyme activities between the two developmental stages were similar among the lines, when compared within a group.
Table 1. Changes in the Activity of the Main Enzymes Involved in Central and Secondary Metabolism during Plant Development.
| Line Origin |
Tropicala |
Corn Belt |
European |
Northern |
Stiff Stalk |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Developmental Stage | V | 15 | V | 15 | V | 15 | V | 15 | V | 15 | |
| Classes |
Enzymes |
||||||||||
| C4 | PPDKb | + | − | + | − | + | − | + | − | + | − |
| NADP-MDHb | + | − | + | − | + | − | + | − | + | − | |
| NAD-MEb | + | − | + | − | + | − | + | − | + | − | |
| NADP-ME | + | − | − | + | 0 | 0 | + | − | + | − | |
| PEPCK | + | − | − | + | − | + | − | + | − | + | |
| PEPC | + | − | + | − | 0 | 0 | 0 | 0 | 0 | 0 | |
| C | FK | + | − | + | − | + | − | − | + | − | + |
| N-invertase | + | − | + | − | + | − | + | − | + | − | |
| GK | + | − | − | + | − | + | + | − | |||
| SPS | + | − | + | − | + | − | + | − | − | + | |
| PFK-PPi (PFP) | − | + | − | + | − | + | − | + | − | + | |
| GL | F1,6BP | − | + | 0 | 0 | + | − | 0 | 0 | − | + |
| PFK-ATP | + | − | + | − | + | − | − | + | + | − | |
| Enolase | + | − | 0 | 0 | 0 | 0 | − | + | 0 | 0 | |
| N | NRmaxb | + | − | + | − | + | - | + | − | + | − |
| NR-Pb | + | − | + | − | + | - | + | − | + | − | |
| NR%activb | + | − | + | − | + | - | + | − | + | − | |
| AlaATb | − | + | − | + | − | + | − | + | − | + | |
| NAD-GDHb | − | + | − | + | − | + | − | + | − | + | |
| GS | + | − | + | − | − | + | − | + | + | − | |
| Fd-GOGAT | + | − | + | − | 0 | 0 | − | + | 0 | 0 | |
| AspAT | + | − | − | + | 0 | 0 | 0 | 0 | + | − | |
| PP | G6PDH | − | + | + | − | + | − | − | + | + | − |
| SM | ShikDHb | + | − | + | − | + | − | + | − | − | + |
| TCA | Fumaraseb | − | + | − | + | − | + | − | + | − | + |
| Aconitase | − | + | − | + | + | − | − | + | − | + | |
| CS | − | + | + | − | − | + | + | − | − | + | |
| IDH | + | − | 0 | 0 | 0 | 0 | − | + | − | + | |
| NAD-MDH | + | − | + | − | 0 | 0 | 0 | 0 | + | − | |
Enzyme activities were measured at the vegetative stage (V) in young fully developed leaves and at the grain filling stage 15 DAS (15). The 19 maize lines covering European and American maize genetic diversity were classified into five groups of different geographical origin based on the microsatellite genotyping performed by Camus-Kulandaivelu et al. (2006). The enzymes are involved in C4 = C4 photosynthetic metabolism; C = carbon metabolism; GL = glycolysis; N = nitrogen metabolism; PP = pentose phosphate pathway; SM = secondary metabolism; TCA = tricarboxylic acid cycle. The “+” indicates that that the enzyme activity is higher and “−” that it is lower at the vegetative stage (V) or at 15 DAS (15). The “0” indicates that there is no difference in the enzyme activity between the two developmental stages. NAD-ME, NAD-dependent malic enzyme; PEPCK, phosphoenolpyruvate carboxykinase; FK, fructokinase; SPS, sucrose phosphate synthase; F1,6BP, fructose 1,6 bisphosphatase; PFK-ATP, phosphofrutokinase ATP-dependent; AspAT, aspartate amino tranferase; G6PDH, glucose-6-phosphate dehydrogenase; CS, citrate synthase; IDH, isocitrate dehydrogenase; NAD-MDH, NAD-dependent malate dehydrogenase; GK, glucokinase.
The same pattern of enzyme activity (higher or lower) was observed between the vegetative stage and 15 DAS across all five Tropical lines, irrespective of their physiological function.
Common differences between the vegetative stage and 15 DAS within a single group of maize lines or across the five groups of maize lines were observed for the enzymes PPDK, NADP-MDH, NAD-ME, NRmax, NR-P, NR%activ, AlaAT, NAD-GDH, ShikDH, and fumarase.
The changeover from the vegetative to the 15 DAS stage is representative of the transition from sink to source leaf, since irrespective of the genetic background, the content of most of the physiological metabolites in the leaves was strongly modified.
Relationship between Plant Physiology and Plant Genetic Distance
To investigate the possible relationships that may exist between the genetic distance and plant physiology (metabolite accumulation, enzyme activities, and physiological traits) in the leaves of the maize lines, a HCA was first performed using the results presented in Supplemental Data Set 1. The main results from the clustering analysis are summarized in Figure 2, using the detailed HCA presented in Supplemental Figure 4.
Figure 2.

Graphic Representation of the Relationships Existing between the Relative Amounts of Leaf Metabolites in 19 Lines of Maize Originating from Europe and America.
The metabolite content was measured by GC-MS analysis of young developing leaves at the vegetative stage (V) and of the leaves below the ear, 15 DAS, during the kernel-filling period. HCA was performed using the metabolome data presented in Supplemental Data Set 1. For each metabolite exhibiting a significant difference between the 19 lines (P ≤ 0.05), the ratio (content for each line/mean value of the 19 lines) was calculated and transformed into a log2 ratio before clustering analysis. The vertical green, blue, orange, red, and yellow boxes represent the five groups of maize lines in different countries of Europe and America (Tropical, orange; European Flint, blue; Northern Flint, red; Maize Belt Dent, green; Stiff Stalk, yellow). At the right of the panel, the silking dates are indicated (J = July, A = August, S = September), along with the grain yield (g−1 plant). Grain yield was determined for the 19 maize lines grown in the field (Supplemental Data Set 1). Compared with the others lines, the Tropical line Argl256 had a very low yield. It was also observed that the kernels of the other Tropical line, CML254, did not reach full maturity at the time of harvest (nm = not measured). Details of the HCA are presented in Supplemental Figure 3.
At the V stage of plant development, it can be seen that the different lines belonging to the Tropical and Maize Belt Dent groups did not exhibit a common pattern of metabolite accumulation. By contrast, the European and Northern Flint lines were grouped in a common cluster, indicating that their metabolite composition was very similar, while the Stiff Stalk line B73 was close to the Northern Flint lines in terms of metabolite content (Figure 2). The metabolite distribution pattern was different between the five groups of lines 15 DAS, compared with that at the V stage. However, all the lines belonging to each of the five groups were clustered, which indicates that their leaf metabolite contents were very similar and, thus, tightly linked to the classification based on their genetic distance (Figure 2).
Although there was a major genetic variability in the activity of a number of enzymes involved in C4 photosynthesis, and primary C and N metabolism in the five groups of lines, a clear distribution pattern for these enzymes either at the V stage or 15 DAS could be seen following HCA (Supplemental Figure 5). At the V stage, in lines CML245 and C105, most of the enzymes involved in N metabolism and several enzymes involved in C assimilation were more active. During the grain filling period (15 DAS), most of the enzymes were more active in the Tropical line ND36.
A sparse partial least squares-discriminant analysis (sPLS-DA) was then performed on the 124 metabolites detected at 15 DAS (Supplemental Data Set 1) to discriminate in a supervised way which metabolites permitted differentiation of the five groups of lines. The sPLS-DA (Supplemental Figure 7) showed the similarities and dissimilarities between the lines clustered in the five groups in score plots corresponding to the four first components of sPLS. The loading vectors for each dimension reflected metabolites that can be used as biomarkers for differentiating the five groups of lines. The exhaustive list of metabolites belonging to each of the four loading vectors is presented in Supplemental Data Set 2. Among them, various chlorogenates, β-alanine, allantoin, and β-sitosterol, were representative of component 1. Amino acids, such as glutamine, histidine, and lysine; carbohydrates, such as mannose, arabinose, and fructose; and chlorogenates different from those belonging to component 1 were representative of component 2. Various disaccharides, stigmasterol, and galactonic acid were representative of component 3. Dopamine, phytol, and organic acids, such as aconitate, ribonate, and ferulate, were representative of component 4. In addition, using the normalized raw data (Supplemental Data Set 1), the sPLS-DA also allowed a characterization of the differences in the content of the four main classes of metabolites that included carbohydrates, amino acids, organic acids, and chlorogenates, among the five groups of lines (Figure 3).
Figure 3.

Example of Leaf Metabolic Signatures Representative of the Five Groups of Maize Lines during the Grain Filling (15 DAS) Period.
sPLS-DA was used to quantify the relationship between the leaf metabolite content and the five groups of maize lines to detect putative leaf metabolite biomarkers at 15 DAS.
(A) Relative content of four main classes of metabolites, including carbohydrates, organic acids, chlorogenates, and amino acids, in the five groups of maize lines.
(B) Amount of soluble carbohydrates detected in the five groups of maize lines. From left to right: fructose (dark gray), glucose (pale gray), and sucrose (gray).
Finally, an attempt was made to determine which differences in metabolites, enzyme activities, and physiological traits resulted from the genetic relatedness of the 19 lines. First, it was checked to see if there was any correlation between the genetic distance of the 19 lines (based on molecular markers) and the Euclidean phenotypic distance (based on leaf metabolite contents and enzyme activities). At both the V stage and 15 DAS, the amounts of metabolites and of enzyme activities of the 19 lines were grouped using the genetic and phenotypic distances as the two parameters of the HCA. There was only a slight relationship between the genetic distance of the 19 lines and their enzyme activities 15 DAS (Figure 4). Such a low correlation could be explained by the fact that maximal enzyme activities were measured in vitro, thus not necessarily reflecting their activity in vivo. The low genetic relatedness of the 19 lines could also explain why the relationship between their genetic distance and their enzyme activities was also very low, suggesting that genetically related lines have similar enzymatic capacities.
Figure 4.

Relationship between the Genetic Distance of the 19 Lines Originating from Europe and America and the Phenotypic Distance of Enzyme Activities.
Heat map showing the standardized level of enzyme activities of the 19 maize lines at 15 DAS. Two HCAs were performed to group the lines and the enzymatic pathways according to their genetic distances based on molecular markers (A_IBD) and according to their Euclidean phenotypic distance based on enzyme activities, respectively. At the left of the HCA, the vertical bar represents the five groups of maize lines (Tropical, orange; European Flint, blue; Northern Flint, red; Corn Belt Dent, green; Stiff Stalk, yellow). At the top of the HCA, colored bars represent the main classes of enzymes (C4 cycle, red; TCA, yellow; glycolysis, pale green; N-assimilation, turquoise; pentose-P, pale blue; secondary metabolism, dark blue; carbohydrate metabolism, purple). The top left scale represents the relative higher (yellow) or lower values (red) for enzyme activities compared with the median.
A Mantel Test was then performed to determine if in the 19 lines there was any correlation between the two genetic distance matrices based on single nucleotide polymorphism (SNP) markers (see Methods) and the phenotypic distance matrices (including metabolite contents, enzyme activities, and the metabolic pathway to which they belong; Supplemental Data Set 3). Variations in enzyme activities were significantly correlated between the 19 lines for three different pathways, which included the tricarboxylic acid (TCA) cycle, carbohydrate biosynthesis, and glycolysis, the highest correlation being observed for the latter at 15 DAS (r = 0.28, P value <0.01). For metabolites, a high correlation with the genetic distance was observed at 15 DAS only for α-tocopherol (vitamin E; r = 0.17, P value = 0.013). At this stage of plant development, such a significant correlation was also observed for physiological traits such as the leaf N content (%N; r = 0.19, P value = 0.02) and kernel number (KN; r = 0.18, P value = 0.03; see Supplemental Data Set 4 for details).
To further refine which part of the variation of the enzyme activities and the metabolite contents could be explained by genetic variation between the 19 lines, an ANOVA test was used to estimate the repeatability of the different biochemical and physiological traits at the two stages of plant development (Supplemental Data Set 4). Large variations were observed between the metabolite contents and enzyme activities involved, irrespective of the plant developmental stage, but they were more consistent 15 DAS.
Genetic Variability of Ammonia Assimilation: 15N-Labeling Experiments
A 15NH4+ labeling experiment was conducted using detached leaves collected at the V stage of plant development. Primary ammonia assimilation was examined in a pulse experiment following 8 and 16 h labeling that quantified the amount of preexisting amino acids (14N-amino acids) and the amount of newly synthesized amino acids (15N-amino acids) formed during the pulse period. A calculation of the 15N enrichment of each amino acid was performed on a relative basis to identify any differences in the dynamics of amino acid accumulation.
The 15N-labeling study was only performed at the V stage, during which time active N assimilation takes place, allowing the measurement of fluxes using 15N-labeled molecules. Such a study was not performed during the remobilization phase 15 DAS, since most of the metabolites originate from degraded proteins and from C translocation from source organs (Uhart and Andrade, 1995; Hirel and Gallais, 2006). Measuring the 15N-enrichment into individual amino acids originating from 15N-labeled degraded proteins is much more difficult. Additionally, the genome-scale metabolic model was analyzed at the V stage to ensure that the necessary pseudo-steady state assumptions were valid.
Since similar results were obtained after the two labeling periods, the results are presented only for the 8 h time point (Supplemental Data Set 5). Graphs showing examples of the amount of 15N incorporation into glutamine, glutamate, asparagine, and alanine, four amino acids of major importance during N assimilation and export, are presented in Figure 5. In all the lines, 15NH4Cl was also incorporated into most of the protein amino acids and γ-aminobutyrate, thus indicating the presence of a functional ammonia assimilatory pathway. The highest 15N-labeling was detected in alanine, the amino acid that predominated in the soluble pool of all the lines, although it was significantly higher in line ND283 and lower in line ND36 and FV2. Additionally, it was observed that the leaf content of 15N-alanine was higher than that of its labeled precursors 15N-glutamine and 15N-glutamate. Despite this, the highest 15N-glutamine and 15N-glutamate labeling was observed in line Mo17 and line ND283, respectively. The lowest 15N-glutamine and 15N-glutamate labeling was observed in line ND36, thus exhibiting a pattern of accumulation similar to that found for alanine (Figure 5). The three 15N-labeled branched chain amino acids leucine, isoleucine, and valine, which use pyruvate as a C skeleton, had much lower 15N accumulation compared with alanine. Although maize is a C4 plant that carries out limited photorespiration (Dai et al., 1995), serine and glycine were among the amino acids that were considerably labeled in all genotypes. In addition, labeling in asparagine was at least 2-fold higher in lines HP301 and SA24 (Figure 5) and the amino acids of the aspartate biosynthetic pathway were all labeled. However, the amounts of 15N-methionine and 15N-lysine were very low compared with that of 15N-threonine and of the branched chain amino acid 15N-isoleucine, indicating that N was channeled preferentially through the threonine branch of the aspartate pathway (Azevedo et al., 2006; Joshi et al., 2010). Cysteine was labeled with 15N in only eight genotypes (FV252, MBS847, ND283, Lo3, Lo32, C105, Argl256, and CML254) belonging to different geographical origins. By contrast, no 15N-labeling was found in arginine in all genotypes, probably due to difficulties in defining the fragmentation pattern arising from the loss of guanido-N atoms (Patterson et al., 1993; Allen and Ratcliffe, 2009). Moreover, low or zero amounts of arginine have generally been detected in maize leaves (Martin et al., 2005; Amiour et al., 2012; Obata et al., 2015).
Figure 5.

Example of the Differences in 15N Amino Acid Content in the 19 Maize Lines Originating from Europe and America.
The 15N-labeled glutamine, glutamate, asparagine, and alanine contents were measured at the end of the labeling period when the isotope 15N was replaced by 14N. The data for the 15N-labeling experiment are presented in Supplemental Data Set 5. Young developing leaves at the vegetative stage of plant development were labeled for 8 h with 15NH4Cl. Values expressed as μmol g−1 FW are the mean of three individual leaves of (±sd bars) each harvested from three different plants grown in the field. Letters a, b, and c represent the result of an ANOVA statistical analysis performed with a Student-Newman-Keuls test and used to identify groups of lines exhibiting a similar pattern of 15N-labeling (P ≤ 0.05).
Flux Range Comparison Using a Compartmentalized Leaf Genome-Scale Metabolic Model
The 19 maize lines were modeled by incorporating enzyme activity data into the published leaf genome-scale metabolic (GSM) model (Simons et al., 2014). Flux balance analysis (FBA) was used to predict the flow of metabolites through each reaction (i.e., the flux value) at the maximum rate of sucrose export for each line. FBA assumes that the production and consumption rates of each metabolite are equivalent at pseudo-steady state, which is justified during vegetative growth. Biomass production was modeled as the ratio of experimentally measured biomass components (see Methods). The leaf biomass was constrained to 35% of the rate of biomass production for each maize line estimated from the growth rate at the 7- to 8-leaf stage, compared with the maximum growth rate among all stages (Bender et al., 2013). At the maximum rate of sucrose production, reactions with overlapping flux ranges are not required to vary among the maize lines; however, reactions without overlapping flux ranges warrant further analysis, as these reactions differ between lines. All flux range analyses were completed in a pairwise manner by comparing each individual maize line to the remaining maize lines, resulting in 171 total pairwise combinations. Based on the change in maximal enzyme activity levels, 34 reactions (not including duplicates due to compartmentalization) were constrained. However, the in silico metabolic differences observed can be recapitulated by imposing only the constraints on alanine aminotransferase (AlaAT) and pyrophosphate-dependent phosphofructokinase (PFK-PPi; also known as pyrophosphate-fructose 6-phosphate 1-phosphotransferase [PFP]). Of the 2561 reactions that were active in at least one line at the maximum rate of biomass production, 249 reactions comprised the differing reaction set (i.e., those reactions with nonoverlapping flux ranges between at least two maize lines; Supplemental Data Set 6). The pathways associated with these 249 differing reactions encompassed 37 Kyoto Encyclopedia of Gene and Genome (KEGG) pathways, including amino acid biosynthesis, purine metabolism, photosynthetic CO2 fixation, the TCA cycle, and fatty acid biosynthesis.
The leaf GSM model was used to corroborate the hypotheses based on the results of the 15N-labeling experiment. In agreement with the 15N-labeling experiments, the in silico results indicated that the photorespiratory glycolate pathway and the ammonia assimilatory pathway may be active in all maize lines with flux ranges spanning from zero to positive flux values at the maximum rate of biomass production. Additionally, by analyzing the two major branch points in the aspartate pathway, the model supported the hypothesis that the flux from aspartate is predominantly diverted toward threonine compared with lysine and methionine. At the first branch, the model predicted over a 200-fold higher maximum possible flux across the bundle sheath and mesophyll cells in all lines through homoserine dehydrogenase (which yields homoserine, the precursor to threonine and methionine) compared with the flux through dihydrodipicolinate synthase (which produces the lysine precursor). At the second branch point, O-phosphohomoserine is converted to threonine by threonine synthase with over a 500-fold higher maximum flux compared with the conversion to methionine via cystathionine γ-synthase (Azevedo et al., 2006; Joshi et al., 2010). However, in all lines, the largest flux from aspartate is probably through arginine synthesis and the urea cycle (Winter et al., 2015).
Further analysis of the reactions that varied revealed that reaction fluxes could fluctuate in a large proportion of the maize line combinations or could be specific for a small subset of maize lines. The reaction catalyzed by PEPC varied in 93 of 171 total pairwise combinations, mainly due to the flux ranges associated with the Argl256, FV2, MBS847, ND283, ND36, and NYS302 lines. While the reaction catalyzed by PEPC had a flux range that was distinct in many maize lines (i.e., the flux range in the maize line did not overlap with any other maize line), the variation in fluxes between all lines was small, with only a 0.50-fold variation in the minimum achievable flux compared with the maximal achievable flux across all maize lines. A tightly controlled distinct range of PEPC is expected, as all C in C4 plants must be shuttled through PEPC (Chollet et al., 1996; Cousins et al., 2007; Paulus et al., 2013).
The enolase reaction had a distinct flux range in only 34 pairwise combinations; however, the flux range varied 5.33-fold among the maize lines. Along with the enolase reaction, PEP can be produced by the pyruvate phosphate dikinase (PPDK) reaction. The reaction catalyzed by PPDK had a distinctive flux in 60 pairwise combinations; however, only four pairwise combinations were shared with the enolase reaction. While all of the distinct pairwise combinations for the enolase reaction involved the FV2 and NYS302 lines, many of the distinct pairwise combinations for the PPDK reaction involved Argl256, MBS847, ND283, and ND36.
The similarity matrix (Supplemental Figure 6) displays the ratios of active reactions at the maximum rate of biomass production with flux ranges that overlapped between two maize lines. Based on the additional constraints applied by the enzyme activities and updated biomass equations, all pairwise combinations displayed over 89% of reactions with overlapping flux ranges. While other factors (e.g., regulation and metabolite concentration) can influence metabolism, this study specifically targeted the influence of stoichiometric constraints based on varying biomass compositions and enzymatic differences between the lines and their effect on metabolism. The F64 and FV2 lines were the most disparate from the other maize lines. The overlapping flux ranges were compared in a pairwise manner to determine the similarities in their metabolism. These metabolic similarities were compared with their plant genetic diversity determined using simple sequence repeats and SNPs (Supplemental Data Set 3). The genetic diversity study revealed that the HP301 and SA24 lines were the most similar. Although these lines were not the most similar from the GSM model analysis, a relatively high percentage (97.3%) of their reaction flux ranges were overlapping, ranking 24th most similar among the 171 pairwise comparisons.
Additionally, the genetic diversity study identified NYS302 and CML254 as the most disparate lines, while the GSM model analysis revealed a relatively low metabolic similarity of 90.1% compared with all pairwise metabolic similarity comparisons, ranking 17th most dissimilar among the 171 pairwise comparisons. From the genetic diversity analysis (Supplemental Table 1), several lines were identified with more than one origin or structural group. The C105 line was identified with the Northern Flint and Maize Belt Dent structural groups, a result that mirrored the closely related metabolism of the C105 line with lines in the Maize Belt Dent structural groups. Additionally, the P465P line, identified with the Tropical and Maize Belt Dent structural groups based on genetic diversity, had a high metabolic similarity to the Maize Belt Dent lines. Overall, no clear patterns existed between the genetic diversity and the metabolic similarities determined by GSM modeling at the V stage.
Correlation Studies between Physiological and Agronomic Traits
Grain yield (GY) and its components KN and thousand kernel weight (TKW) were determined for the 19 maize lines grown in the field (Supplemental Data Set 1). Compared with the other lines, the Tropical line Argl256 had a very low yield under the Versailles field experimental conditions. In addition, the kernels of the other Tropical line, CML254, had not reached full maturity at the time of harvest. Among the 19 lines examined, except for lines Argl256 and CML254, a significant genetic variability was observed for the traits related to yield, with up to a 2-fold variation in GY between the lowest- and the highest-producing lines.
Pearson correlation coefficients between agronomic traits related to grain yield, the leaf physiological status, and the geographical origin of the 19 lines were calculated in order to identify their possible functional relationships. These included the agronomic traits GY, KN, and TKW, and key parameters representative directly or indirectly of the leaf physiological status (kernel and leaf C, N, C/N ratio, leaf protein, PEPC protein, and leaf nitrate contents). Following a Shapiro-Wilk test (Shapiro et al., 1968), the variables used for the Pearson coefficient calculation followed a normal distribution. The results are presented by the means of a heat map in Figure 6A and a Cytoscape visual representation in Figure 6B. Data related to the complete correlation study are presented in Supplemental Data Set 7. The two highest positive correlations were found between GY and the total C and N contents in the kernels (close to 1 and 0.94, respectively). In addition, yield components (GY, KN, and TKW) and the physiological traits related to the C and N contents of the kernels showed significant positive correlations. This finding is illustrated in Figure 6B, in which it can be seen that nearly all the traits related to the kernels were positively interconnected. Interestingly, the C% in the kernels was the trait exhibiting the highest number of significant correlations (nine correlations) with the other leaf physiological and yield-related traits. Moreover, there were eight correlations between GY, TKW, and the kernel N content as well as the other kernel and leaf traits. Among the yield components, GY and KN showed a positive correlation (0.81). At 15 DAS, two leaf traits (total N and N%) were significantly correlated with GY (0.52) and TKW (0.76), whereas the leaf soluble protein content was correlated only with TKW (0.60). The leaf protein content 15 DAS also exhibited positive correlations with the leaf N content (N%) 15 DAS (0.83) and with the kernel N% (0.50). By contrast, the leaf C/N ratio at 15 DAS exhibited eight negative correlations, with GY (−0.63), TKW (−0.79), the kernel C and N contents (−0.55 with C%, −0.60 with total C, −0.55 with N%, and −0.62 with total N), the leaf N% 15 DAS (−0.97), and the leaf protein content 15 DAS (−0.80). Notably, the leaf C content at the V stage and the leaf N content 15 DAS exhibited a high positive correlation of 0.51. A positive correlation was found with the leaf protein content at the V stage and the leaf C% (0.53) and N% (0.56) at the V stage. At the V stage, only negative correlations were obtained with the leaf nitrate content and a number of traits such as the kernel C/N ratio (−0.50), the kernel C% (−0.53), and the leaf C % (−0.52). Only the leaf C content exhibited a high negative correlation of −0.6 with the geographical origin of the lines.
Figure 6.

Pearson Correlations between Agronomic and Physiological Traits Representative of the Leaf Physiological and Kernel Physiological Status of the 19 Maize Lines.
(A) Heat map showing the significant correlations (adjusted P values <0.05) found between kernel yield traits (GY, KN, and TKW) and key parameters representative of the kernel and leaf physiological status (C = carbon, N = nitrogen, C/N ratio, leaf soluble protein, PEPC protein, and nitrate contents). The negative and positive correlation coefficient values are indicated in each colored box of the heat map using the scale on the left side of the panel.
(B) Network diagram illustrating the most significant correlations found between agronomic and physiological traits. Traits with a higher number of correlations are represented by larger and darker red dots. Thicker and red lines represent the highest positive correlations. Thicker and blue lines represent the highest negative correlations.
Network Analysis of Physiological Traits
To uncover the putative mechanisms underlying the leaf physiology of the different maize lines, network analyses were employed. At the V stage, the first step of the network analysis allowed the generation of eight modules (encompassing coregulated metabolites or enzymes as module components). Each of these eight modules was represented by a different color (pink, red, brown, green, turquoise, yellow, black, and blue). An additional module component that did not meet the threshold criteria (gray module) was also identified (Supplemental Data Set 8). At 15 DAS, 11 modules and a gray module were obtained from the metabolite and enzyme activity data set (Supplemental Data Set 9). Modules obtained for the V stage contained 10 to 27 components, while for 15 DAS there was a range from 5 to 44 components. The module component annotations are presented in the Supplemental Data Sets 8 and 9 for the V stage and 15 DAS, respectively. In these two data sets, the module colors are specific to each of the two developmental stages.
Pearson correlations were then calculated to establish possible relationships between the different modules identified at the V stage and 15 DAS, yield-related traits (GY, KN, and TKW), and physiological parameters (kernel and leaf C, N, C/N ratio and leaf protein, PEPC protein, and nitrate contents). Correlations found at the V stage and 15 DAS are shown in Supplemental Data Set 10. For clarity, only those with a Bonferroni adjusted P value lower than 0.05 have been considered (Supplemental Figure 8 for the V stage and Figure 7 for 15 DAS). At the V stage, there was no significant correlation between GY and any of the identified modules.
Figure 7.

Heat Map and Pearson Correlations with the Modules and Agronomic Traits (GY, KN, and TKW) and Physiological Parameters Representative of the Kernel and Leaf Physiological Status 15 DAS.
Correlations with the modules containing the generic physiological traits 15 DAS. The color names correspond to the 12 modules that were obtained from the 15 DAS metabolite and enzyme activity data set network analyses (Supplemental Data Set 10). Heat map and Pearson correlations between modules and kernel yield traits (GY, KN, and TKW) and key parameters representative of the kernel and leaf physiological status (C = carbon, N = nitrogen, C/N ratio, leaf soluble protein, PEPC protein, and nitrate contents). Correlations were considered significant with Bonferroni adjusted P values <0.05. Adjusted P values are shown in parentheses. Only significant negative and positive correlation coefficient values are indicated in each colored box. For clarity, only those with a correlation higher than 0.4 have been considered. The box of the heat map using the scale on the right side of the panel. e = exponent base 10. The same analysis was performed at the V stage (Supplemental Figure 8).
One of the main characteristics of the brown vegetative (V) module was the presence of metabolites (such as succinate, 2-oxoglutarate, pyruvate, and alanine) and enzymes (such as NADP- and NAD-malic enzyme) directly or indirectly involved in C4 metabolism. The rest of the components in this module were metabolites (e.g., inositol) and enzymes, such as neutral-invertase (N-invertase), involved in C metabolism. Only a few components of the brown module were related to N metabolism (e.g., methionine or NR activity).
The turquoise V-module contained precursors in the biosynthesis of aromatic amino acids and secondary metabolites (e.g., shikimate and quinate) and carbohydrates and their derivatives (e.g., arabitol, raffinose, and galactonate). Additionally, there were three metabolites related to N metabolism (γ-aminobutyrate, proline, and urea), two main intermediates of the C assimilatory pathway (phosphoenolpyruvate and glycerate-3-phosphate) and three enzymes involved in central C metabolism (PEPCK, PFP, and CS).
The red V-module was mainly composed by carbohydrates (e.g., glucose, fructose, or arabinose) and by two enzyme activities related to C metabolism (e.g., sucrose-phosphate synthase and isocitrate dehydrogenase).
For the physiological traits, the highest positive correlation was found between the brown V-module and the leaf protein content (0.66). For this module, there was also a good correlation with the leaf PEPC protein (0.5) and C contents (0.46). A negative correlation was also found between the brown V-module and the origin of the lines corresponding to the five structural groups defined on the basis of genetic relatedness (−0.51). The turquoise and red V-modules had good positive correlations with the C/N ratio (0.52 and 0.5, respectively) and negative correlations with the leaf N content (−0.51 and −0.47, respectively).
At 15 DAS, the highest positive correlations were found between the blue 15DAS-module and the leaf protein content (0.9) and the leaf N content (0.84). Conversely, there was a high negative correlation between the blue 15DAS-module and the C/N ratio (−0.83). Although less marked, similar correlation profiles were found between the purple 15DAS-module and these three physiological traits. The blue 15DAS-module showed a positive correlation between GY (0.49) and its component TKW (0.6) and with the leaf PEPC protein content (0.48). Negative correlations within the turquoise 15DAS-module included GY (−0.51) and KN (−0.52). In this module, only the correlation with the C/N ratio was positive (0.55). Among the components of the blue 15DAS-module, there were 15 enzymes, including seven enzymes directly involved in C4 metabolism, five enzymes related to central C metabolism, and the two enzymes involved in N assimilation (GS and Fd-GOGAT). With the exception of glutamate, the rest of the components were metabolites related to secondary C metabolism. In the red 15DAS-module, there was an abundance of metabolites involved in lipid metabolism (e.g., campesterol and linoleic acid) and metabolites related to the TCA cycle (e.g., citrate and alanine). Glucokinase activity was found only in the red 15DAS-module. The purple 15DAS-module contained different metabolites, including unidentified carbohydrates, chlorogenates, and lipids such as α-tocopherol and stigmasterol. Finally, the turquoise 15DAS-module contained most of the amino acids (e.g., glutamine and phenylalanine), different carbohydrates and derivative metabolites (e.g., sucrose, glucose, and glycerate-3-phosphate), and several secondary metabolites (e.g., caffeate, chlorogenate, and coumarate). In this module, four enzyme activities related to C and N metabolism (PEPC, AlaAT, GDH, and N-invertase) were also identified.
Weighted Gene Co-Expression Network Analysis (WGCNA) software was used to perform correlation studies between agronomic traits and physiological traits (Supplemental Data Set 1). The relationships between agronomic traits and physiological traits were analyzed using weighted correlations that take into account the module membership and amplify correlations of the members of outcome-related modules (Langfelder and Horvath, 2008). The main results of these correlation studies are presented in Table 2, and their detailed analysis is shown in Supplemental Data Sets 11 (V stage) and 12 (15 DAS). At the V stage, 12 significant correlations were found between GY and physiological traits. One of the most important results was the finding of strong positive correlations between GY and the amounts of 2-oxoglutarate (0.73) and succinate (0.66). Although lower than that found for GY, 2-oxoglutarate also exhibited a positive correlation with TKW (0.38). Interestingly, both the 2-oxoglutarate and succinate contents were positively correlated with the total C and N content of the kernels and the leaf protein content at the V stage. Erythritol exhibited a positive correlation with both GY (0.51) and TKW (0.42). The leaf α-tocopherol content was strongly and positively correlated with TKW (0.85) and to a lesser extent with the leaf C content both at the V stage and 15 DAS, and to the leaf N and protein contents but only at 15 DAS. When the enzyme activities were considered, the highest correlation at the V stage was found between KN and AlaAT in the direction of glutamate synthesis (0.69). Such a positive correlation between AlaAT activity, GY (0.59), and KN (0.48) was also found 15 DAS. Interestingly, the total activities of three enzymes involved in central C metabolism aconitase, enolase, and fructokinase were positively correlated (correlation coefficient around 0.50) with KN at the V stage. During the grain filling period 15 DAS, enolase and fructokinase activities were also positively correlated with GY, whereas for aconitase it was with KN.
Table 2. The Most Representative Correlations between Leaf Metabolite Content, Enzyme Activities, and Yield-Related Traits.
| Correlations | V Stage | 15 DAS |
|---|---|---|
| Negative | 3-Coumaroylquinate 2974.6/345 (GY) | 3-Coumaroylquinate 2974.6/345 (GY, KN, TKW) |
| Ethanolamine | 3-Caffeoylquinate-trans (GY, KN) | |
| 3-Hydroxybutyrate | Feruloylquinate 3088.4/249 (GY, KN) | |
| Ethanolamine (GY, KN) | ||
| Amino acids (GY, KN) | ||
| Carbohydrates (GY, KN) | ||
| Positive | 2-Oxoglutarate (GY, TKW) | N-invertase (GY, KN, TKW) |
| α-Tocopherol (TKW) | NAD-GDH (GY, KN, TKW) | |
| AlaAT (KN) | AspAT (GY, TKW) | |
| AlaAT (GY, KN) | ||
| GS/GOGAT (GY, TKW) | ||
| C4 enzymes (GY, TKW) |
Yield and its components were measured on plants grown in the field under optimal N feeding conditions. Leaf metabolites were quantified and enzymes activities were measured at the vegetative stage (V) on a young fully developed leaf and at the grain filling stage (15 DAS) on the leaf below the ear. Weighted correlation studies were performed using the physiological trait data set and yield data set with a q-value ≤0.05. Details of the correlation study are presented in Supplemental Data Sets 8 and 9. Yield components exhibiting a positive or a negative correlation with the various physiological traits are indicated in parentheses. The number 2974.6/345 corresponds to the metabolic signature of the identified 3-coumaroylquinate.
At 15 DAS, there was a clear match between most of the blue and turquoise module physiological components and their correlations found with GY (Figure 8, Table 2; Supplemental Data Set 12). For the blue module components (notably, enzymes of C4 metabolism and ammonia assimilation), correlations with GY were positive, whereas for the turquoise module components (mostly enzymes involved in amino acid and carbohydrate metabolism), most of the correlations were negative. In the turquoise 15DAS-module, only the activities of four enzymes, AlaAT, NAD-GDH (deaminating activity), PEPC, and N-invertase, were positively correlated with GY (Figure 8, Table 2; Supplemental Data Set 12). In addition, it is worth stressing that most of the members of the blue module showed positive correlations with the kernel C and N contents, the leaf N and protein contents, both at the V stage and 15 DAS. Furthermore, the components of the blue module exhibited negative correlations with the leaf nitrate content at the V stage. For a number of traits belonging to the blue module (kernel total C and N contents) and for leaf N content at 15 DAS, it was found that instead of being positive, they were negative in the turquoise module. Irrespective of the composition of the modules, other enzyme activities such as NAD-GDH and N-invertase were also positively correlated with KN and TKW. The finding that a number of secondary metabolites involved in the lignin biosynthetic pathway were positively or negatively correlated with GY and its components is another noteworthy result that arose from this correlation study (Table 2).
Figure 8.

Network Diagram for Relationships between Leaf Metabolites and Enzyme Activities 15 DAS.
Diamonds represent enzymes, and circles represent metabolites. Colors of the circles and diamonds correspond to the different components within a module. The names of metabolites and enzymes positively or negatively correlated with GY are highlighted in bold black and in red characters, respectively. Only the network connections that have a topological overlap above the threshold of 0.1 are shown. The red circle in the center corresponds to a metabolite of unknown function (unk. Fer-quin 3088.4/249) with a structure similar to a feruloylquinate that exhibits a significant negative correlation with GY. Lines represent a significant correlation between two traits. Thicker lines represent the highest positive or negative correlations. The same analysis was performed at 15 DAS for correlations with TKW (Supplemental Figure 9).
DISCUSSION
Is There a Link between Maize Genetic Diversity and Leaf Physiology?
The relative amounts of metabolites present in the leaves of the 19 maize lines representative of American and European maize genetic diversity were quantified in order to determine if they were different according to their genetic background. In addition, physiological markers representative of the leaf physiological status including total C, total N, leaf nitrate, and leaf water contents were also measured. Irrespective of the plant developmental stage and of the genetic background, carbohydrates were the most abundant soluble organic molecules in the leaf (57%), with sucrose notably representing almost 70% of the total carbohydrates. Organic acids, mainly represented by aconitate, were the second most abundant class of metabolites, representing ∼27% of the total. Soluble amino acids were present in lower concentrations in the leaves, representing around 9% of the total metabolites. Alanine was by far the most abundant soluble N containing molecule (Supplemental Figures 1A and 1B), representing 35% of the total soluble leaf amino acid content. However, as previously observed (Amiour et al., 2012), the leaf metabolic profiles of the 19 maize lines were different at the V stage and 15 DAS. In all lines, three main groups of metabolites exhibiting opposing patterns of accumulation between the V stage and 15 DAS were clearly identified, reflecting the transition from sink leaves (accumulating C and N assimilates) to source leaves (exporting C and N assimilates to the grain) (Supplemental Figure 3).
When examining the portfolio of enzymes selected for their key role in primary N and C metabolism and in the C4 photosynthetic pathway, it can be seen that most of the enzymes were more active at the V stage compared with 15 DAS, except PEPCK, PFK-PPi (PFP), aconitase, AlaAT, AspAT, NAD-GDH, and fumarase (mitochondrial and cytoplasmic), which were more active during the 15 DAS grain filling period (Table 1). It has been generally observed that NAD-GDH activity increases during leaf senescence and N remobilization and that such an increase can be variable depending on the genetic background (Dubois et al., 2003; Girondé et al., 2015). Such changes between the two stages of plant development also reflect the transition from sink leaves to source leaves in which C and N assimilatory enzymes are downregulated and replaced by another set of enzymes involved in the remobilization process (Hirel et al., 2005).
Further analyses revealed that there was a wide range of variation for both leaf metabolite composition and leaf enzyme activities. For example, the asparagine contents varied by up to 200% (Figure 1), while differences in PEPC activity were low (10% at the V stage and 25% 15 DAS). It is well established that due to its central role in plant N assimilation and management, the asparagine content can vary considerably from one plant species to another and can be greatly affected by environmental conditions (Lea et al., 2007). PEPC is an enzyme that plays a major role in C4 photosynthetic CO2 assimilation (Maroco et al., 1998; Cousins et al., 2007; Paulus et al., 2013). While the model-predicted flux range of the reaction catalyzed by PEPC was comparable for many maize lines, the variation in the nonoverlapping flux ranges among all maize lines was small. This indicated that the metabolic differences between maize lines were reflected in small changes in the flux through the PEPC-catalyzed reaction.
When examining further the leaf metabolite composition at the V stage and 15 DAS, the most interesting results were obtained following hierarchical clustering analysis. The leaf metabolite profile 15 DAS was similar within each of the five groups of maize lines defined on the basis of their genetic distance. In addition, neither the silking date nor GY production had any direct relationship with the metabolite-clustering pattern (Figure 2). Such observations indicate that leaf metabolite accumulation in source leaves (15 DAS) mostly depends on the genetic background, whatever the sink capacity of the kernels. It is therefore attractive to propose that the metabolite accumulation pattern during the grain filling period could be used as a marker for assessing the genetic dissimilarity of maize lines both in Europe and America.
To identify which metabolites or clusters of metabolites were the most representative of the leaf metabolic signature 15 DAS, sPLS-DA was performed (Supplemental Figure 7; Figure 3). Northern Flint and Tropical lines were characterized by high contents of amino acids and chlorogenates. However, the amount of carbohydrates in the Tropical lines was much higher compared with the other lines. As there was also a good correlation with the leaf C content 15 DAS and the origin of five genetic groups (Figure 6), it would appear that the Tropical lines are more efficient in terms of carbohydrate biosynthesis, thus providing more C skeletons for amino acid production. In addition, the content of three unknown chlorogenates (Unk.Chlorogenate 3105.5/2, Unk.Chlorogenate 2981.0/2, and Unk.Chlorogenate 3118.9) allowed a clear separation of the lines classified as Northern Flint and Tropical, the three secondary metabolites being high in the former and much lower in the latter. The Stiff Stalk line B73 was characterized by lower amounts of chlorogenates and of most of the amino acids. Maize Belt lines and European Flint lines were both characterized by a low accumulation of carbohydrates. In the European Flint lines, the amount of caffeic acid was similar compared with that of the Maize Belt lines, whereas the amounts of ferulic acid were extremely low in the European Flint lines; thus, chlorogenates may be key molecules for differentiating the five groups of lines. Despite the identification of a metabolic signature specific for each of the five groups of lines, its use could be limited by the fact that individual variations have occurred during breeding. Thus, it will be interesting to verify in a larger panel of genotypes which of the components of the leaf metabolic signature are representative of the grain filling period. Such a metabolic signature could also be useful in a study on maize domestication (Riedelsheimer et al., 2012a), with respect to the accumulation and transport of assimilates to the kernels (Sosso et al., 2015).
By contrast, activities of the main enzymes involved in central C and N metabolism did not allow the differentiation of any of the five groups of maize lines except the Maize Belt Dent lines 15 DAS, for which they were grouped in a single cluster (Supplemental Figure 5). Additionally, using genome-scale modeling, the fluxes through metabolic reactions during the V stage could not be clearly differentiated based on the five groups of maize lines. However, the differences in the measured enzyme activities occurring between the V stage and 15 DAS were the same in all Tropical lines (Table 1). The activity of three enzymes related to the C4 photosynthetic pathway (PPDK, NADP-MDH, and NAD-ME), of four enzymes involved in N assimilation (NR, GS, Fd-GOGAT, and AlaAT), and of ShiDH (shikimate dehydrogenase, an enzyme involved in aromatic amino acid biosynthesis and secondary metabolism) were lower 15 DAS compared with that measured at the V stage in the Tropical lines (Table 1). These enzymes probably correspond to a core set of enzymes whose activity needs to be higher during the vegetative stage before silking in genetically distant maize lines. It will be interesting to test if the activities of the enzymes involved in C4 photosynthesis and N assimilatory pathways are, like those involved in starch biosynthesis (Doebley et al., 2006), representative of maize domestication and potentially useful for breeding.
A preliminary study was conducted to determine if there were any relationships between the genetic distances of the 19 lines and the various leaf biochemical and physiological traits. Although low (r = 0.28), the genetic distances of the 19 lines were significantly correlated with the glycolytic pathway at 15 DAS represented by the enzymes F1,6BP, PFK-PPi (PFP), PFK-ATP, and enolase (Figure 4). For metabolites, a significant correlation was observed only with the α-tocopherol content during grain filling, which also exhibited a positive correlation with TKW at the V stage (Table 2). At the V stage of plant development, a low but significant correlation was also observed for physiological traits such as the leaf N content (%N) and KN (r = 0.19; Supplemental Data Set 3). It is therefore likely that the three types of phenotypic traits, TKW, %N, and KN, could be putative markers representative of European and American genetic diversity of maize.
Is There a Link between the Genetic Variability of Metabolic Fluxes and Maize Genetic Diversity?
It is generally agreed that compared with fluxomic studies, metabolomic and enzyme activity profiling only provide a narrow and static picture of the physiological status of a given organ, at a particular stage of plant development (Fernie and Stitt, 2012). Therefore, a 15NH4Cl-labeling experiment was performed and the metabolic fluxes were analyzed using a compartmentalized leaf genome-scale metabolic model. The aim of the experiment was to determine if there was any relationship between the genetic variability of the metabolic fluxes (in particular those involved in amino acid biosynthetic pathways) and the five groups of lines.
Irrespective of the classification of the lines into five groups, the highest labeling with the 15N tracer (μmol 15N-amino acid g−1 FW) was detected in the most abundant leaf amino acid, alanine (Figure 5D). Such a result is consistent with the fact that the rate of turnover of both glutamine and glutamate is higher than that of alanine, as they act as amino donors for the synthesis of most other amino acids. On a relative basis, the highest 15N-labeling was in glutamine (44.9% as a mean value of %15N-glutamine for the 19 lines) due to the small size of the soluble pool coupled to a high turnover, while alanine had a lower relative 15N-labeling (18.3% as a mean value of 15N-alanine for the 19 lines). Interestingly, asparagine had the lowest 15N-labeling (4.9% as a mean value of %15N-asparagine for the 19 lines) due to a large size of the pool and a low turnover. Alanine was the most abundant newly synthesized amino acid, probably due to the dual role of this amino acid as a N storage compound (McAllister et al., 2012; Limami et al., 2014) and as an intermediate in the CO2 assimilatory pathway in C4 plants (Wang et al., 2014). Overall, the Maize Belt Dent lines exhibited the highest 15N-labeling in all the amino acids, suggesting that the flux of ammonium going through the synthesis of glutamine was much higher in these lines.
Glycine and serine were among the amino acids that were highly labeled in all lines, despite the fact that their accurate measurement is often difficult (Novitskaya et al., 2002). Glycine and serine have recently been shown to accumulate in maize leaves under drought stress conditions (Obata et al., 2015). Low rates of photorespiration have been detected in maize by gas exchange measurements (Dai et al., 1995), and the enzymes required for the photorespiratory glycolate pathway have recently been identified in Sorghum bicolor leaves predominantly in the bundle sheath cells (Döring et al., 2016). In addition, studies with mutants of Amaranthus edulis (Lacuesta et al., 1997; Wingler et al., 1999) and more recently with maize (Zelitch et al., 2009) have also indicated that there can be an active photorespiratory glycolate pathway operating in C4 plants. Therefore, it is likely that glycine and serine can be generated from glycolate through the process of photorespiration. However, incorporation of 15N was higher in serine than in glycine, with a 4-fold higher labeling in lines SA24 and C105. Furthermore, the mean values for the 19 lines showed higher values for serine (14.6%) compared with glycine (9.6%). This may be due to the rapid metabolism of glycine to serine in photorespiration (Novitskaya et al., 2002) or the synthesis of serine via the nonphotorespiratory 3-phosphoglycerate pathway (Ros et al., 2013). The elevated labeling in serine may reflect the fact that there are multiple pools of glycine, only one of which is highly labeled and involved in photorespiration. The other glycine pools, containing low or zero 15N-label, would be able to dilute out the photorespiratory glycine, giving the appearance of glycine that is less labeled than serine.
On some occasions, connections can be drawn between the metabolic similarities of two maize lines and the five genetic groups to which they belong. However, there was no observable link between their genetic relatedness and the metabolism predicted through genome-scale models that account for varying biomass compositions and the change in maximal enzyme activities among maize lines. Nevertheless, the metabolism of the FV2 and NYS302 lines were the most disparate, which may be related to the low enzymatic activity of AlaAT observed in these lines.
Predictive Value of Physiological Markers for Yield and Its Components
To assess the predictive value of leaf physiological traits for grain yield and its components, we performed correlation studies and network analyses. Such an approach has already been developed to identify single or multiple metabolites as potential markers for the breeding of abiotic stress-tolerant maize grown in the field (Obata et al., 2015). Pearson correlations were first analyzed between the yield-related traits and markers representative of the leaf physiological status, including the C, N, C/N ratio, the soluble protein, the PEPC protein, and the nitrate contents. Across the 19 lines, there was as expected a high positive correlation between GY and both KN and TKW. Such a finding indicates that despite the large genetic variability of KN and TKW (Bertin and Gallais, 2000), both traits can be further used for identifying predictive physiological traits for yield in maize (Figure 6).
The positive relationship observed between the leaf N content, the leaf protein, the kernel N content, and both GY and TKW during the grain filling period was one of the most notable results of the correlation study. Such a finding indicates that when the protein N accumulated in source leaves is high, it will have a good predictive value for selecting high yielding maize lines containing more N both from Europe and America. This conclusion is in agreement with the strong correlation generally observed between plant N uptake at silking and kernel yield in ancient and recent maize hybrids (Ciampitti and Vyn, 2012). Conversely, an accumulation of C in leaves during the grain filling period will be detrimental to yield, as revealed by the negative relationship found between KN and total C content, but also between GY, TKW, and the C/N ratio. This could be related to evidence that carbohydrate accumulation occurs when sink capacity is limited, for example, when there are less or smaller kernels (Uhart and Andrade, 1995). As for C, nitrate accumulation in young leaves can also be considered as a negative marker for maize GY, probably resulting from an inefficient metabolic activity in terms of N utilization.
We performed additional correlation studies to identify relationships between GY and its components, the leaf metabolite content, and the activities of the main enzymes involved in C and N assimilation (Table 2).
At both the V and 15 DAS stages, there were negative correlations of the three yield components with 3-coumaroylquinate (metabolic signature 274.6/345) and a positive correlation with the enzyme activity of AlaAT. We therefore propose that these two physiological markers can be measured either before or after silking for the selection of high yielding maize lines. Of the enzymes constrained in the metabolic model with experimental data, AlaAT was observed to be one of two main enzymes responsible for the in silico differences in metabolism. Additionally, it has been shown that increasing the activity of the enzyme AlaAT enhanced the productivity of several crops (Good and Beatty, 2011; Han et al., 2016). The direct effect of the leaf chlorogenate content on crop yield is not well documented. However, it has been reported that chlorogenates are key molecules involved in pest resistance (Dhillon et al., 2013; Ferruz et al., 2016). They can also be used as marker molecules for maize improvement (Butrón et al., 2001; Sood et al., 2014). In line with this proposal, the accumulation of chlorogenates was also negatively correlated with GY and KN 15 DAS. Taken together, these results indicate that the most productive lines in terms of yield components contain lower amounts of secondary metabolites in the leaves.
Other molecules, such as α-tocopherol (vitamin E) and 2-oxoglutarate, could also represent good markers for TKW, when they accumulate at the V stage. Vitamin E has been found to be of importance for the quality of maize kernels (Wong et al., 2003). It is therefore not surprising to find that the 2-oxoglutarate content was positively correlated with both GY and TKW at the V stage, since it is a key molecule involved in ammonia assimilation, transamination, and the TCA cycle. The activities of several enzymes, notably AlaAT and GDH, two enzymes that metabolize or synthesize 2-oxoglutarate (Good and Beatty, 2011; Fontaine et al., 2012), were correlated with TKW.
Another of the key results obtained through these correlation studies was the number of positive correlations found at 15 DAS between yield-related traits such as GY and TKW and enzyme activities involved in C4 metabolism and ammonia assimilation, notably PEPC for the former and GS and Fd-GOGAT for the latter. Moreover, our correlation-based network analyses enabled the identification of a hub of coregulations containing the activities of most of the enzymes involved in the C4 pathway and the two enzymes GS and Fd-GOGAT involved in the assimilation of ammonia (blue 15DAS-module in Figure 8). In maize, the importance of the genes encoding GS in controlling GY and TKW has been previously demonstrated using both forward and reverse genetic approaches (Martin et al., 2006). It has also been shown that in C3 cereals the enzyme NADH-GOGAT is important for yield performance (Tamura et al., 2011). The C4 photosynthetic pathway is considered a key biotechnological target for crop improvement (Covshoff and Hibberd, 2012; Kandoi et al. 2016).
Another network module (turquoise-15DAS module) comprising most of the major amino acids and essential carbohydrates was identified during the grain filling period 15 DAS. All components of this module were negatively correlated with both GY and TKW, except the activity of four enzymes, AlaAT, NAD-GDH, PEPC, and N-invertase. PEPC plays a major role in C4 CO2 assimilation (Maroco et al., 1998; Cousins et al., 2007; Paulus et al., 2013) and invertase in sucrose metabolism in sink organs (Sturm and Tang, 1999). However, the negative correlations observed between the yield-related traits with most soluble carbohydrates and amino acids suggests that even when there is efficient C and N assimilation, an accumulation of the resulting products in the leaves will be detrimental for both kernel set and kernel filling. In line with this hypothesis, it has been shown that whatever the sink capacity of the developing ear, an accumulation of C and N metabolites in source organs such as leaves, negatively affects GY (Cliquet et al., 1990; Yang et al., 2004). In turn, it is likely that most of the metabolites and enzyme activities that exhibit positive or negative relationships with kernel yield and its components could be used as representative markers for the metabolic or physiological status of the plant. Such an idea is further supported by the fact that for most of the components found in the blue and turquoise 15DAS-modules, there were positive or negative correlations between yield-related traits and both leaf and kernel physiological traits (Supplemental Data Set 12).
Although NAD-GDH activity 15 DAS is not shown in the graphical representations of the network (due to the selected threshold of significance over 0.1), it was found to belong to the turquoise 15DAS-module, thus being positively correlated with the three yield components GY, KN, and TKW (notably TKW with a correlation higher than 0.8). It has been proposed that NAD-GDH, by virtue of its role in replenishing a shortage of carbohydrates through the synthesis of 2-oxoglutarate is a key enzyme involved in the control of plant productivity (Dubois et al., 2003, Tercé-Laforgue et al., 2015).
As well as providing putative physiological markers for maize grain production, our network analyses allowed the identification of regulatory modules in which metabolite accumulation and enzyme activities were coregulated during grain filling (15 DAS), irrespective of the genetic background (Figure 8). In addition to the two main network modules (blue 15DAS and turquoise 15DAS) exhibiting densely connected regions and correlations with GY and its components, a number of other minor modules were also identified, although these will not be described in detail. In the green 15DAS-module, there were strong relationships between NR activity and a number of organic acids involved in the TCA cycle, in line with the finding that the accumulation of nitrate and C skeletons used for amino acid transamination are coregulated (Scheible et al., 1997). The occurrence of a strong relationship between different chlorogenates of known or unknown structure both in the magenta 15DAS-module and in the brown 15DAS-module was in agreement with the previous finding that there are modules of coexpressed genes that determine their accumulation (Joët et al., 2010). The network analysis approach is also consistent with the occurrence of interactions existing between C and N metabolism (Nunes-Nesi et al., 2010), highlighted in the blue 15DAS- and turquoise 15DAS-modules.
Conclusion: Toward a Maize Ideotype for Optimal Grain Production
A combined metabolomic and enzyme activity profiling approach has stimulated correlation studies and network analyses to identify regulatory modules depicting the interaction occurring between a large set of leaf physiological traits. Members of these regulatory modules exhibit positive or negative correlations with yield-related traits over all lines, irrespective of which of the five structural groups they belong. Such a finding allows the construction of a maize ideotype exhibiting optimal high yielding characteristics in maize lines of European and American origin, having a wide genetic diversity. The beginning of the grain filling period (15 DAS) appears to be the best time to define such an ideotype. This period must be characterized by at least two features. First, there must be little accumulation of the major soluble amino acids and carbohydrates in the leaves, indicating that these two main classes of organic molecules are being efficiently assimilated and exported to the developing kernels. Second, there must be high activity of the leaf enzymes involved in the C4 photosynthetic pathway and in glutamate, glutamine, and alanine biosynthesis. These three amino acids are the most important for transport and management of N in maize (Weiner et al., 1991; Martin et al., 2006). Both a low chlorogenate content and an accumulation of sterols in the leaves appear to be important marker traits that can be used to select maize lines producing larger kernels. The use of enzyme activities and the genes controlling their biosynthesis could constitute interesting new tools for future breeding strategies.
We propose that a number of metabolites and enzyme activities could be used as physiological markers for breeding purposes. However, modulation of the expression levels of these markers individually or simultaneously, using, for example, genetic manipulation techniques and genome-wide association genetics studies, will be required to fully validate their predictive value. Another alternative will be to develop user-friendly and affordable high-throughput tools aimed at monitoring the nutritional status of the plant in relation to its genetic origin. However, considering the decreasing costs of analyses, genetic screens based on the use of genotyping arrays can be used in parallel as they increase the ability to perform genetic mapping and marker-assisted breeding. In any case, integrated systems biology approaches like those described in this study will help in deciphering the physiological and regulatory mechanisms underlying genetic variability and exploiting them to improve maize productivity.
METHODS
Plant Material for Agronomic and Physiological Studies
Nineteen selected maize (Zea mays) inbred lines including races that are representative of American and European plant genetic diversity (Supplemental Table 1) have been used previously as a core collection for association genetic studies (Camus-Kulandaivelu et al., 2006; Bouchet et al., 2013). Seeds of the 19 maize lines were obtained from INRA (Saint-Martin-de-Hinx, France). These lines were classified into five main maize groups named Maize Belt Dent (six lines), European Flint (four lines), Northern Flint (three lines), Stiff Stalk (one line), and Tropical (five lines), by Camus-Kulandaivelu et al. (2006) and Bouchet et al. (2013). This original classification was organized on the basis of the genetic diversity of the lines using simple sequence repeat microsatellite markers and later using SNP markers as presented in Supplemental Table 1. The group named Tropical contained lines from Argentina, Mexico, and Spain, whereas the Maize Belt Dent lines, which mostly originated from North America, contained two Popcorn lines. In the European Flint lines, there was also one line from Argentina, whereas all the Northern Flint lines came from North America.
The plants were grown in the field at INRA, Versailles, France (N 48°48.133’, E 2°04.942’) in deep silt loam without any stone. The level of N fertilization was 175 kg/ha and N provided by the soil was estimated at 60 kg/ha. Both phosphorus (P205) and potassium (K20) were also applied at 100 kg/ha.
The 19 lines were grown side by side in two separate rows of 25 plants in three separate blocks of 25 × 25 m with an outside boarder area of 3 m (line MBS857) included in each block. The plants were sown on May 15, 2011. To measure yield traits in each block at plant maturity, the ears from 10 individual plants from each line were harvested, making 30 replicates per sample point and year. In this study, agronomic traits used for correlation studies were GY and its components: KN/plant and TKW. For more details about the procedure used to measure the agronomic traits, see Bertin and Gallais (2000, 2001).
For all biochemical analyses at the V stage and 15 DAS, three halves of leaves cut vertically were harvested from three individual plants in each of the three blocks, making nine replicates in total. For the metabolite analyses, the sampling procedure was the same as for the biochemical analyses, except that the three leaf samples from each block were pooled making three replicates in total. At the vegetative (V) stage, half of the 6th fully emerged leaf without the main central midrib was harvested at the 7- to 8-leaf stage between 9 am and noon on July 2, 2011. For the grain filling stage, half of the leaf below the ear of each individual plant was harvested 15 DAS, each plant being harvested at the same developmental stage and the harvesting date for each plant is indicated in Figure 2. The plant developmental stage at 15 DAS has been shown to provide a good indication of the transition occurring when both C and N metabolites start to be actively translocated to the developing kernels (Martin et al., 2005; Amiour et al., 2012). Moreover, the leaf below the ear was selected since it provides a good indication of the sink-to-source transition during grain filling (Prioul and Schwebel-Dugué, 1992). The delay in the silking date between the 19 lines was ∼4 weeks, starting from the 5th of August, 2011. However, a metabolite profiling study revealed that from 15 DAS and onwards, there were no major differences in the metabolites of the leaf below the ear within this period (L. Brulé and B. Hirel, unpublished data). Leaf samples were harvested from three different plants, exhibiting a similar pattern of development in each of the three blocks and pooled. The samples of the three replicates for each metabolite and enzyme activity were immediately placed in liquid N2 and stored at −80°C until further analysis.
Protein Extraction, Enzyme Assays, Metabolite Extraction, and Analyses
Frozen leaf tissues were reduced to a homogenous powder and stored at −80°C until required for the metabolite and enzyme activity measurements. Each sample contained the equivalent of 500 mg FW of the 6th emerged leaf (V) or the 7- to 8-leaf stage of the leaf below the ear (15 DAS), as described above for biochemical analyses. Proteins for enzyme activity measurements were extracted from 25 mg aliquots of frozen leaf material stored at −80°C using the protocol described by Gibon et al. (2004). The extraction buffer consisted of 500 mM HEPES, pH 7.5, containing 100 mM MgCl2, 10 mM EDTA, 10 mM EGTA, 2 mM leupeptin, 0.5 mM DTT, 0.1% (v/v) Triton, and 1% (w/v) polyvinylpolypyrrolidone. All extractions were performed at 4°C. The soluble protein concentration was determined using a commercially available kit (Coomassie protein assay reagent; Bio-Rad), with BSA as a standard (Bradford, 1976). A robot-based platform was used to measure the activity of 29 enzymes that are involved in C4 photosynthetic, central C, and N metabolism, using the protocols described by Gibon et al. (2004). In addition the following enzymes measurement methods were employed: PEP carboxykinase, NADP-MDH, NAD-ME, NADP-ME, and enolase (Biais et al., 2014), N-Invertase (Desnoues et al., 2014), SPS (Lunn and Hatch, 1997), PFK-PPi (PFP) and PFK-ATP (Keurentjes et al., 2008), aconitase (Piques et al., 2009), CS (Nunes-Nesi et al., 2007), NAD-IDH (Zhang et al., 2010), and NAD-MDH (Jenner et al., 2001). NRmax corresponds to maximal NR activity, NR-P to phosphorylated NR, and NR% to the percentage of active NR. The list of enzymes analyzed for their activity in soluble protein leaf extracts are presented in Supplemental Table 2.
Soluble leaf proteins were separated by SDS-PAGE (Laemmli, 1970). The percentage of polyacrylamide in the running gels was 8%. At the end of the electrophoresis period, proteins were stained with SYPRO Ruby protein gel stain (Bio-Rad). Gels were then destained with a solution containing 10% ethanol (v/v) and 7% acetic acid (v/v). Stained proteins were visualized using a Typhoon FLA 9500 laser scanner (GE Healthcare Life Sciences) with the following settings (excitation, 450 nm; emission, 640 nm; photo multiplication = 600, and pixel size = 100 µm). Total leaf and PEPC proteins were quantified using ImageQuant TL software (GE Healthcare Life Sciences). The total C and N content of 25 mg of frozen leaf material and dry kernels was determined in an elemental analyzer using the combustion method of Dumas (Flash 2000; Thermo Scientific). For nitrate measurements, 500 mg of frozen leaf powder was extracted in 1 mL of 80% ethanol at room temperature for an hour. The samples were continuously agitated during extraction and centrifuged at 12,000g for 5 min. The supernatant was removed and the pellet subjected to further extractions in 60% ethanol and finally in water. All supernatants were combined to form the water/ethanol extract. Nitrate was determined by the method of Cataldo et al. (1975).
For the leaf metabolome analyses, all steps were adapted from the original protocol described by Fiehn (2006), following the procedure described by Amiour et al. (2012). The ground frozen leaf samples (25 mg fresh weight) were resuspended in 1 mL of frozen (−20°C) water:chloroform:methanol (1:1:2.5) and extracted for 10 min at 4°C with shaking at 1400 rpm in an Eppendorf Thermomixer. Insoluble material was removed by centrifugation, and 900 μL of the supernatant was mixed with 20 μL of 200 µg/mL ribitol in methanol. Water (360 µL) was added, and after mixing and centrifugation, 50 μL of the upper polar phase was collected and dried for 3 h in a Speed-Vac and stored at −80°C. For derivatization, samples were removed from −80°C storage, warmed for 15 min before opening, and SpeedVac dried for 1 h before the addition of 10 μL of 20 mg/mL methoxyamine in pyridine. The reactions with the individual samples, blanks, and amino acid standards were performed for 90 min at 28°C with continuous shaking. N-methyl-N-trimethylsilyl-trifluoroacetamide (90 µL) was then added and the reaction continued for 30 min at 37°C. After cooling, 50 μL of the reaction mixture was transferred to an Agilent vial for injection. For the analyses, 3 h and 20 min after derivatization, 1 μL each of the derivatized samples was injected in the splitless mode onto an Agilent 7890A gas chromatograph coupled to an Agilent 5975C mass spectrometer. The column used was an Rxi-5Sil MS from Restek (30 m with 10 m Integra-Guard column). The oven temperature ramp was 70°C for 7 min, then 10°C/min up to 325°C, which was maintained for 4 min. For data processing, Raw Agilent data files were converted into the NetCDF format and analyzed with AMDIS (http://chemdata.nist.gov/dokuwiki/doku.php?id=chemdata:amdis). Peak areas were then determined using quanlynx software (Waters) after conversion of the NetCDF file into the masslynx format. Statistical analyses were performed with TMEV (http://mev.tm4.org/#/welcome). Univariate analyses by permutation (one- and two-way ANOVA) were first used to select the metabolites exhibiting significant changes in their concentration (P ≤ 0.05). Amino acid standards were injected at the beginning and end of the analyses, for the monitoring of derivatization stability. An alkane mixture (C10, C12, C15, C19, C22, C28, C32, and C36) was injected in the middle of the run for external retention index calibration. For the analysis of the leaf samples, metabolite standards were injected at the beginning and end of each analysis. The metabolite concentration is expressed as nmol mg−1 leaf FW.
15N-Labeling Experiment, Gas Chromatography, and Mass Spectrometry
Three entire 6th emerged leaves were harvested from individual plants grown in the field in 2011 and then incubated for 8 and 16 h in glass tubes containing 50 mL of a nutrient solution containing 4 mM NH4Cl, enriched with 50% 15NH4Cl (Euriso-top; Les Algorithmes). The nutrient solution also contained 1.25 mM K+, 0.25 mM Ca2+, 0.25 mM Mg2+, 1.25 mM H2PO4−, 0.75 mM SO42−, 21.5 µM Fe2+ (Sequestrene; Ciba-Geigy), 23 µM B3+, 9 µM Mn2+, 0.3 µM Mo2+, 0.95 µM Cu2+, and 3.5 µM Zn2+. Leaves in the nutrient tubes were placed in a controlled environment growth chamber (16 h light, 350–400 μmol photons m−2 s−1 at 26°C). During the labeling period, the pH of the nutrient solution remained stable from 5.6 to 5.8. After the labeling period, leaf samples were stored at −80°C for further analysis.
Both the changes in the amino acid content and their respective 15N-enrichment were determined at the different time points of the labeling period by GC-MS. Amino acids were extracted using the following procedure: frozen leaf samples (100 mg fresh weight) were ground in liquid nitrogen and extracted at 4°C in 2.7 mL of methanol-chloroform-distilled water (1/2.5/1 [v/v]) for 30 min. Prior to the extraction, 1.25 μL of 2.5 mM 2-aminobutyric acid (internal standard) was added to the extraction medium. The homogenate was centrifuged at 27,000g for 20 min and 0.6 mL of distilled water was added to the recovered supernatant. The upper methanol-water fraction was collected, freeze-dried, and redissolved in 1 mL of distilled water. The resulting samples were then filtered with a 0.20-mm filter (Pall Life Science). Distilled water (550 μL) was added to 450 μL of the filtered solution and acidified with 27 μL of 1 M HCL. The acidified solution was then applied to a 5.0 × 0.5-cm syringe containing 2 mL of Dowex-50WX8-200 ion-exchange resin (Sigma-Aldrich). Prior to the loading of the acidified solution onto the column, the resin was rinsed three times with 4 mL of distilled water, 2 mL of 10 mM HCl, and then 8 mL of distilled water. Amino acids were eluted with 7 mL of 6 M NH4OH. The amino acid fraction was lyophylized and redissolved in a suitable buffer for either quantification of total amino acids or GC-MS analysis. Amino acid quantities were determined as described earlier. For GC-MS analysis, lyophylized amino acid samples were resuspended in 0.1 n HCl, dried under N2, and derivatized with N-methyl-N-(tert-butyldimethylsilyl)-trifluoroacetamid (Pierce) as described by Rhodes et al. (1989). The atom %15N of each amino acid was then determined by GC-MS analysis (MD800; Fisons). ANOVA statistical analysis performed with a Student-Newman-Keuls test and used to identify groups of lines exhibiting a similar pattern of 15N-labeling (P ≤ 0.05) (Supplemental Data Set 5).
Determining the Metabolic Changes through Modeling
The previously published genome-scale maize leaf model of Simons et al. (2014) was combined with the vegetative enzyme activity and metabolite data to evaluate the metabolic differences between the maize lines. The model is composed of the stoichiometric constraints of all known metabolic reactions within the maize leaf, as well as thermodynamic constraints represented by the directionality of the reactions. FBA was used to determine the conversion of reactants to products by assuming that every metabolite is equally produced and consumed at a pseudo-steady state (Orth et al., 2010). To represent maximum cell growth, biomass components were maximized in a defined proportion based on metabolite amounts specific to each line or experimental evidence based on line B73 (Simons et al., 2014). Flux boundaries were modified for reactions with corresponding enzyme activity data to represent the regulation for each maize line. The fold change for each enzyme in each maize line was calculated by dividing the rate of enzyme activity in the maize line by the maximum enzyme activity observed over all maize lines. These fold changes were incorporated into the model by constraining the allowed maximum and minimum flux ranges calculated with only stoichiometric and thermodynamic constraints (Colijn et al., 2009; Dash et al., 2014). For each maize line, the maximum rate of sucrose export was calculated by incorporating the new reaction boundaries based on the enzyme activity, as described in the following equation:
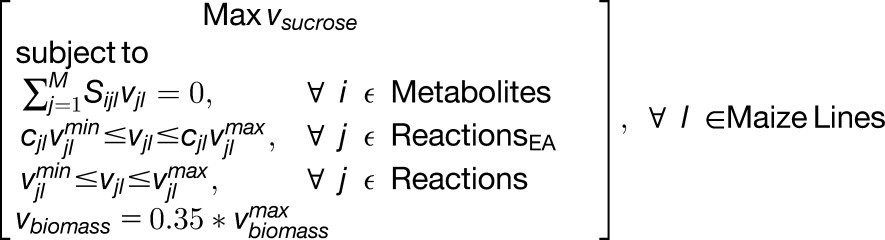 |
where Sijl represents the stoichiometric coefficient of metabolite i in reaction j in maize line l, and vjl represents the flux through reaction j in maize line l. Here, vjlmax and vjlmin are the largest and smallest flux values for the maize line l resulting from the flux variability analysis performed on the model with only stoichiometric and thermodynamic constraints. The enzyme level fold change of reaction j in maize line l is represented by cjl for reactions corresponding to measured enzymes (ReactionsEA). The upper and lower boundaries were calculated when only stoichiometric and thermodynamic constraints are enforced by vjmax and vjmin, respectively. The model was constrained to require production of at least 35% of the in silico maximum biomass. This biomass production was determined using the growth rates approximated from the change in dry weight over a growing degree day (GDD) at the V sample collection stage (V7/V8) and the maximum leaf blade growth rate, which occurs during the V12 stage (Bender et al., 2013). The change in GDD over time was calculated from the reported daily temperatures (Bender et al., 2013) and was found to be relatively stable between the V7/V8 stage and the V12 stage, so the ratio of the V7/V8 stage to the V12 stage is approximated as the change in dry weight over GDD. The similarity between two maize lines was determined based on the number of overlapping flux ranges compared with the total number of reactions that are active in either maize line. The flux range for each reaction in each maize line was calculated with stoichiometric, thermodynamic, maximum biomass, and the adjusted flux range constraints.
Statistical and Hierarchical Clustering Analysis
The results presented in the Supplemental Data Set 1 were analyzed using the ANOVA function of the Multi Experiment Viewer (MeV) software version 4.9 (http://mev.tm4.org/#/welcome). The ANOVA statistical analyses (P ≤ 0.05) were performed using metabolite (three replicates, each containing three pooled leaf samples) and enzyme activity measurements (three replicates, each containing three pooled leaf samples), along with yield and its components (GY, GN, and TKW: 30 replicates per line) in the 19 maize lines. When the ANOVA tests returned an overall level of significance at a P value ≤ 0.05, the data were further submitted to the Bonferroni post-hoc test. To establish if the classification of the maize lines into five groups was correlated with the different agronomic and biochemical traits, HCA was performed using the MeV software, version 4.9. Correlation studies were performed using the XLStat-Pro 7.5 software (Addinsoft). Pearson correlations between agronomic and physiological traits were calculated and the P values were submitted to the Bonferroni correction. For studying the correlation between agronomic traits and physiological traits related to the leaf physiological status at the V stage and 15 DAS, a heat map of the Pearson correlation matrix was obtained using Excel software with the results presented in Supplemental Data Set 1. Network diagrams of the Pearson correlation matrix were obtained using Cytoscape 3.2.1 software (Smoot et al., 2011) with the network analyzer plug-in (Assenov et al., 2008).
Analysis of the Relationship between Plant Genetic Distance and Plant Physiological Characteristics
Pairwise relationship coefficients between the 19 lines were calculated using the 27,681 Panzea SNP markers from a 50K Illumina Array (Ganal et al., 2011). The calculations were performed using two genomic relationship matrices obtained (1) by averaging the proportion of shared alleles over all the SNP markers (Identity-By-State, A_IBS) and (2) by using the method described by Astle and Balding (2009), in which the weight of each locus is inversely proportional to the genetic diversity of the locus in the population (A_IBD). Such methods give more weight to the rarest SNP for calculating relationship coefficients in a relationship matrix called A_Freq (Supplemental Data Set 3). For studying the relationship between the genetic distance of the 19 lines and the different physiological traits, the 19 lines were ordered on the basis of their genetic distance (distance = 1-A_Freq) using the unweighted pair group method with arithmetic mean hierarchical clustering method. Enzyme activities and metabolites were ordered by hierarchical clustering using Ward’s method (Ward, 1963). This method is based on the calculation of the phenotypic Euclidean distance of the two physiological traits using centered and scaled values for leaf enzyme activities and leaf metabolite contents both at the V stage and 15 DAS.
The repeatability of the different physiological traits was calculated at the two stages of plant development using the R-Package heritability test (Kruijer et al., 2015). In this R-package, the repeatability ĥ2 was estimated using a classical ANOVA model in which the phenotype P was fitted to estimate the genotype effect (G) and environmental effect (Env) using the following model: Pij = Gi + eij, where eij is the residual error for the genotype i and the replicate j. Repeatability ĥ2 was calculated using the following formula: ĥ2 = Vg/(Vg +Ve), where the genetic variance Vg were estimated using Vg = (MS(G)-MS(Env))/r and Ve = MS(Env) = mean sum (MS) of square for residual errors and MS(G) = mean sum of square for the genotype obtained from the ANOVA.
Metabolite Biomarker Analysis for Genetic Relatedness
sPLS-DA was used to quantify the relationship between the leaf metabolite content and the genetic relatedness of the lines in order to detect putative metabolite biomarkers. The mixOmics library (version 5.1.2) in R was used to carry out sPLS-DA (Lê Cao et al., 2011) for the metabolite data set at the grain filling stage 15 DAS (Supplemental Data Set 1). For sPLS-DA, two parameters were adjusted: the total number of components onto which the data were projected, and the number of variables selected for each component. In the analysis, G-1 was considered, G being the number of classes necessary for determining which optimal number of components needed to be used (Lê Cao et al., 2011). Since the five groups of lines (Maize Belt Dent, European Flint, Northern Flint, Stiff Stalk, and Tropical) corresponded to five G classes, the analysis was focused on four main components identified following sPLS-DA analysis. For the parameter tuning, the original data set was divided into training and testing sets from the initial individuals. Ten plants were randomly selected to constitute the training set. A sPLS-DA classifier was then built using the metabolite training sets and tested on the remaining individuals. The number of variables for a parsimonious model was selected for the first (smallest) model. The average classification error rate fell to within one se of the average of the different computed minima. The classification error rate based on 100 times 5-fold cross validation was run on the training data. To identify the four main components, 6, 12, 44, and 58 metabolites, respectively, were selected.
Network Analysis Using the WGCNA Software
Relationships between metabolite content, enzyme activities, and traits related to yield and the leaf physiological status were analyzed using the R package WGCNA. Such a package originally used for studying gene coexpression networks (Langfelder and Horvath, 2008) can also be used for analyzing metabolite data sets (DiLeo et al., 2011) and integrating morphological traits (Toubiana et al., 2013; Liseron-Monfils and Ware, 2015). The WGCNA scripts used in this study are described in the Supplemental File 1. Two different coexpression module networks corresponding to the V stage and to 15 DAS were constructed for studying biologically meaningful relationships between the different metabolite and enzyme data. Before the networks were constructed, the appropriate soft-threshold power was first determined by performing an analysis of the network topology. For the V stage, the one-step network construction option was used with a soft-threshold power value of 5 and a minModuleSize of 5. For 15 DAS, the same one-step network construction option was used with the same parameters, but with a soft-threshold power value of 4. Physiological traits with a k Module (kME) >0.3 were assigned to a colored coexpression module or to a gray module, if they did not meet the threshold criteria described above (Supplemental Data Sets 8 and 9). The connectivity and module membership for each trait are presented in Supplemental Data Sets 13 and 14 for the V stage and for 15 DAS, respectively. Network screening was performed using weighted and Pearson correlations of the WGCNA networkScreening function. The q-values (false discovery rates) were then calculated (Storey et al., 2004). Correlations between the mean values of the modules (metabolite content and enzyme activity), their individual values and agronomical traits (GY, KN, and TKW), and physiological parameters representative of the plant physiological status (C, N, C/N ratio, and nitrate contents) at the V stage and 15 DAS were considered significant when the q-value was <0.05.
Supplemental Data
Supplemental Figure 1. Genetic relatedness of the 19 maize lines within the origin of the five groups of lines.
Supplemental Figure 2. Average proportions of different metabolite classes in the 19 maize lines representative of American and European plant diversity (Camus-Kulandaivelu et al., 2006).
Supplemental Figure 3. Hierarchical clustering analysis of metabolites and at the V stage and 15 DAS in the 19 maize lines.
Supplemental Figure 4. Hierarchical clustering analysis to investigate the relationship between the five groups of maize lines and metabolites.
Supplemental Figure 5. Hierarchical clustering analysis used to investigate the relationship between the five groups of maize lines of the maize lines and enzyme activities.
Supplemental Figure 6. Metabolism similarity matrix indicating the ratio of active reactions with shared flux ranges between the 19 maize lines in a pairwise manner.
Supplemental Figure 7. The five groups of maize lines identified by means of specific leaf metabolic signature during the grain filling period.
Supplemental Figure 8. Heat map and Pearson correlations with the modules and agronomic traits (GY, KN, and TKW) and physiological parameters representative of the kernel and leaf physiological status at the V stage.
Supplemental Figure 9. Network diagram of relationships between leaf metabolites and enzyme activities 15 DAS.
Supplemental Table 1. Classification of the 19 maize lines originating from Europe and America on the basis of their genetic relatedness.
Supplemental Table 2. The enzymes involved in central C and N metabolism used for studying genotypic variation in their activity and for determining the metabolic changes through modeling.
Supplemental File 1. WGCNA script.
Supplemental Data Set 1. Database containing the relative values of the three replicates for the different agronomic, biochemical, and metabolic traits measured in the panel of 19 maize lines representative of American and European plant genetic diversity.
Supplemental Data Set 2. List of metabolites selected from the four components of the sPLS-DA.
Supplemental Data Set 3. Genetic distance matrices based on SNP markers and phenotypic distances matrices used for the Mantel test.
Supplemental Data Set 4. Analysis of variance to estimate the repeatability for metabolites, enzyme activities, and physiological traits at the V stage and 15 DAS.
Supplemental Data Set 5. Incorporation of 15NH4+ in a young fully developed leaf at the vegetative stage of plant development.
Supplemental Data Set 6. Flux range associated with each reaction in the genome scale model for each maize line.
Supplemental Data Set 7. Correlations between agronomical and physiological traits at the V stage and 15 DAS.
Supplemental Data Set 8. Metabolite and enzyme annotations including module membership and weighted correlation with agronomical and physiological traits at the V stage.
Supplemental Data Set 9. Metabolite and enzyme annotations including module membership and weighted correlation with agronomical and physiological traits 15 DAS.
Supplemental Data Set 10. Correlations between modules and agronomic and physiological traits at the V stage and 15 DAS.
Supplemental Data Set 11. Weighted correlations between leaf metabolite content, enzyme activities, and GY-related traits.
Supplemental Data Set 12. Weighted correlations between leaf metabolite content, enzyme activities, and GY-related traits.
Supplemental Data Set 13. Connectivity and module component values at the V stage.
Supplemental Data Set 14. Connectivity and module component values 15 DAS.
Supplementary Material
Acknowledgments
We thank Gilles Clément for performing the metabolome analyses, which were carried out at the Obervatoire du Végétal Chimie Metabolisme of the Institute Jean-Pierre Bourgin, INRA, Versailles-Grignon. We also thank Guillaume Ménard and Patricia Ballias for technical support at the Bordeaux Metabolome Platform and Christophe Montagnier from the INRA Versailles field experiment unit for setting up the plant culture.
AUTHOR CONTRIBUTIONS
R.A.C. and Z.Y.-C. performed the metabolomic, enzymatic, physiological, and agronomic traits studies and analysis of the data. M.S. performed the metabolic modeling using the data generated by R.A.C., Z.Y.-C., and S.N. F.C. performed the study on the biomarkers. P.A. participated to the enzyme activity measurements and the analysis of the data. I.Q. conducted the field trials and yield measurements. C.C. and A.M.L. performed and analyzed the 15N-labeling studies, respectively. Y.G. supervised the enzyme activity measurements on the robot-based platform. S.N. performed the genetic distance studies. L.B. performed the leaf protein and seed nitrogen content analysis. P.J.L., C.D.M., and B.H. designed the experiment and participated to the analysis of the data. P.J.L. and B.H. wrote the article.
Glossary
- GWAS
genome-wide association studies
- GC-MS
gas chromatography coupled to mass spectrometry
- DAS
days after silking
- FW
fresh weight
- NR
nitrate reductase
- GDH
glutamate dehydrogenase
- Fd-GOGAT
ferredoxin-dependent glutamate synthase
- PEPC
phosphoenolpyruvate carboxylase
- HCA
hierarchical clustering analysis
- sPLS-DA
sparse partial least squares-discriminant analysis
- SNP
single nucleotide polymorphism
- TCA
tricarboxylic acid
- KN
kernel number
- GSM
genome-scale metabolic
- FBA
flux balance analysis
- AlaAT
alanine aminotransferase
- PFK-PPi
pyrophosphate-dependent phosphofructokinase
- PFP
pyrophosphate-fructose 6-phosphate 1-phosphotransferase
- PPDK
pyruvate phosphate dikinase
- GY
grain yield
- TKW
thousand kernel weight
- WGCNA
Weighted Gene Co-Expression Network Analysis
- GDD
growing degree day
References
- Allen D.K., Ratcliffe R.G. (2009). Quantification of isotope label. In Plant Metabolic Networks, Schwender J., ed (New York: Springer; ), pp. 105–149. [Google Scholar]
- Amiour N., Imbaud S., Clément G., Agier N., Zivy M., Valot B., Balliau T., Armengaud P., Quilleré I., Cañas R., Tercet-Laforgue T., Hirel B. (2012). The use of metabolomics integrated with transcriptomic and proteomic studies for identifying key steps involved in the control of nitrogen metabolism in crops such as maize. J. Exp. Bot. 63: 5017–5033. [DOI] [PubMed] [Google Scholar]
- Amiour N., Imbaud S., Clément G., Agier N., Zivy M., Valot B., Balliau T., Quilleré I., Tercé-Laforgue T., Dargel-Graffin C., Hirel B. (2014). An integrated “omics” approach to the characterization of maize (Zea mays L.) mutants deficient in the expression of two genes encoding cytosolic glutamine synthetase. BMC Genomics 15: 1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assenov Y., Ramírez F., Schelhorn S.E., Lengauer T., Albrecht M. (2008). Computing topological parameters of biological networks. Bioinformatics 24: 282–284. [DOI] [PubMed] [Google Scholar]
- Astle W., Balding D.J. (2009). Population structure and cryptic relatedness in association genetics studies. Stat. Sci. 24: 451–471. [Google Scholar]
- Azevedo R.A., Lancien M., Lea P.J. (2006). The aspartic acid metabolic pathway, an exciting and essential pathway in plants. Amino Acids 30: 143–162. [DOI] [PubMed] [Google Scholar]
- Bender R.R., Haegele J.W., Ruffo M.L., Below F.E. (2013). Nutrient uptake, partitioning, and remobilization in modern, transgenic insect-protected maize hybrids. Agron. J. 105: 161–170. [Google Scholar]
- Bertin P., Gallais A. (2000). Physiological and genetic basis of nitrogen use efficiency in maize. I. Agrophysiological results. Maydica 45: 53–66. [Google Scholar]
- Bertin P., Gallais A. (2001). Physiological and genetic basis of nitrogen use efficiency in maize. II. QTL detection and coincidences. Maydica 46: 53–68. [Google Scholar]
- Biais B., et al. (2014). Remarkable reproducibility of enzyme activity profiles in tomato fruits grown under contrasting environments provides a roadmap for studies of fruit metabolism. Plant Physiol. 164: 1204–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchet S., Servin B., Bertin P., Madur D., Combes V., Dumas F., Brunel D., Laborde J., Charcosset A., Nicolas S. (2013). Adaptation of maize to temperate climates: mid-density genome-wide association genetics and diversity patterns reveal key genomic regions, with a major contribution of the Vgt2 (ZCN8) locus. PLoS One 8: e71377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford M.M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72: 248–254. [DOI] [PubMed] [Google Scholar]
- Brauer D., Teel M.R. (1981). Metabolism of trans-aconitic acid in maize 1: purification of two molecular forms of citrate dehydrase. Plant Physiol. 68: 1406–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butrón A., Li R.G., Guo B.Z., Widstrom N.W., Snook M.E., Cleveland T.E., Lynch R.E. (2001). Molecular markers to increase corn earworm resistance in a maize population. Maydica 46: 117–124. [Google Scholar]
- Camus-Kulandaivelu L., Veyrieras J.B., Madur D., Combes V., Fourmann M., Barraud S., Dubreuil P., Gouesnard B., Manicacci D., Charcosset A. (2006). Maize adaptation to temperate climate: relationship between population structure and polymorphism in the Dwarf8 gene. Genetics 172: 2449–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo D.A., Haroon M., Schrader L.E., Youngs V.L. (1975). Rapid colorimetric determination of nitrate in plant tissue by nitration of salicylic acid. Commun. Soil Sci. Plant Anal. 6: 71–80. [Google Scholar]
- Chollet R., Vidal J., O’Leary M.H. (1996). Phophoenolpyruvate carboxylase: a ubiquitous, highly regulated enzyme in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 47: 273–298. [DOI] [PubMed] [Google Scholar]
- Ciampitti I.A., Vyn T.J. (2012). Physiological perspectives of changes over time in maize yield dependency on nitrogen uptake and associated nitrogen efficiency: a review. Field Crops Res. 133: 48–67. [Google Scholar]
- Cliquet J.B., Deléens E., Mariotti A. (1990). C and N mobilization from stalk and leaves during kernel filling by 13C and 15N tracing in Zea mays L. Plant Physiol. 94: 1547–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colijn C., Brandes A., Zucker J., Lun D.S., Weiner B., Farhat M.R., Cheng T.Y., Moody D.B., Murray M., Galagan J.E. (2009). Interpreting expression data with metabolic flux models: predicting Mycobacterium tuberculosis mycolic acid production. PLOS Comput. Biol. 5: e1000489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousins A.B., Baroli I., Badger M.R., Ivakov A., Lea P.J., Leegood R.C., von Caemmerer S. (2007). The role of phosphoenolpyruvate carboxylase during C4 photosynthetic isotope exchange and stomatal conductance. Plant Physiol. 145: 1006–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covshoff S., Hibberd J.M. (2012). Integrating C4 photosynthesis into C3 crops to increase yield potential. Curr. Opin. Biotechnol. 23: 209–214. [DOI] [PubMed] [Google Scholar]
- Dai Z., Ku M., Edwards G.E. (1995). C4 photosynthesis - the effects of leaf development on the CO2-concentrating mechanism and photorespiration in maize. Plant Physiol. 107: 815–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash S., Mueller T.J., Venkataramanan K.P., Papoutsakis E.T., Maranas C.D. (2014). Capturing the response of Clostridium acetobutylicum to chemical stressors using a regulated genome-scale metabolic model. Biotechnol. Biofuels 7: 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DellaPenna D., Last R.L. (2008). Genome-enabled approaches shed new light on plant metabolism. Science 320: 479–481. [DOI] [PubMed] [Google Scholar]
- Desnoues E., Gibon Y., Baldazzi V., Signoret V., Génard M., Quilot-Turion B. (2014). Profiling sugar metabolism during fruit development in a peach progeny with different fructose-to-glucose ratios. BMC Plant Biol. 14: 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiLeo M.V., Strahan G.D., den Bakker M., Hoekenga O.A. (2011). Weighted correlation network analysis (WGCNA) applied to the tomato fruit metabolome. PLoS One 6: e26683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon M.K., Kalia V.K., Gujar G.T. (2013). Insect-pest and their management: current status and future need of research in quality maize. In Maize: Nutrition Dynamics and Novel Uses, P. Chaudhary, S. Kumar, and S. Langyan, eds (New Delhi, India: Springer; ), pp. 95–103. [Google Scholar]
- Doebley J.F., Gaut B.S., Smith B.D. (2006). The molecular genetics of crop domestication. Cell 127: 1309–1321. [DOI] [PubMed] [Google Scholar]
- Döring F., Streubel M., Bräutigam A., Gowik U. (2016). Most photorespiratory genes are preferentially expressed in the bundle sheath cells of the C4 grass Sorghum bicolor. J. Exp. Bot. 67: 3053–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois F., Tercé-Laforgue T., Gonzalez-Moro M.B., Estavillo M.B., Sangwan R., Gallais A., Hirel B. (2003). Glutamate dehydrogenase in plants: is there a new story for an old enzyme? Plant Physiol. Biochem. 41: 565–576. [Google Scholar]
- Fernie A.R., Schauer N. (2009). Metabolomics-assisted breeding: a viable option for crop improvement? Trends Genet. 25: 39–48. [DOI] [PubMed] [Google Scholar]
- Fernie A.R., Stitt M. (2012). On the discordance of metabolomics with proteomics and transcriptomics: coping with increasing complexity in logic, chemistry, and network interactions scientific correspondence. Plant Physiol. 158: 1139–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferruz E., Loran S., Herrera M., Gimenez I., Bervis N., Barcena C., Carramiñana J.J., Juan T., Herrera A., Ariño A. (2016). Inhibition of Fusarium growth and mycotoxin production in culture medium and maize kernels by natural phenolic acids. J. Food Prot. 79: 1753–1758. [DOI] [PubMed] [Google Scholar]
- Fiehn O. (2006). Metabolite profiling in Arabidopsis. In Methods in Molecular Biology: Arabidopsis Protocols, 2nd ed, J. Salinas and J.J. Sanchez-Serrano, eds (Totowa NJ: Humana Press; ), pp. 439–447. [DOI] [PubMed] [Google Scholar]
- Fontaine J.X., Tercé-Laforgue T., Armengaud P., Clément G., Renou J.P., Pelletier S., Catterou M., Azzopardi M., Gibon Y., Lea P.J., Hirel B., Dubois F. (2012). Characterization of a NADH-dependent glutamate dehydrogenase mutant of Arabidopsis demonstrates the key role of this enzyme in root carbon and nitrogen metabolism. Plant Cell 24: 4044–4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushima A., Kusano M. (2013). Recent progress in the development of metabolome databases for plant systems biology. Front. Plant Sci. 4: 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganal M.W., et al. (2011). A large maize (Zea mays L.) SNP genotyping array: development and germplasm genotyping, and genetic mapping to compare with the B73 reference genome. PLoS One 6: e28334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibon Y., Blaesing O.E., Hannemann J., Carillo P., Höhne M., Hendriks J.H., Palacios N., Cross J., Selbig J., Stitt M. (2004). A Robot-based platform to measure multiple enzyme activities in Arabidopsis using a set of cycling assays: comparison of changes of enzyme activities and transcript levels during diurnal cycles and in prolonged darkness. Plant Cell 16: 3304–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girondé A., et al. (2015). The contrasting N management of two oilseed rape genotypes reveals the mechanisms of proteolysis associated with leaf N remobilization and the respective contributions of leaves and stems to N storage and remobilization during seed filling. BMC Plant Biol. 15: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good A.G., Beatty P.H. (2011). Biotechnological approaches to improving nitrogen use efficiency in plants: alanine aminotransferase as a case of study. In Molecular and Physiological Basis of Nutrient Use Efficiency in Crops, M.J. Hawkesford and P. Barraclough, eds (Chichester, UK: Science Publishers, Wiley-Blackwell; ), pp. 165–192. [Google Scholar]
- Han M., Wong J., Beatty P.H., Good A.G. (2016). Identification of nitrogen use efficiency genes in barley: searching for QTLs controlling complex physiological traits. Front. Plant Sci. 7: 1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirel B., Martin A., Tercé-Laforgue T., Gonzalez-Moro M.B., Estavillo J.M. (2005). Physiology of maize I: A comprehensive and integrated view of nitrogen metabolism in a C4 plant. Physiol. Plant. 124: 167–177. [Google Scholar]
- Hirel B., Gallais A. (2006). Rubisco synthesis, turnover and degradation: some new thoughts on an old problem. New Phytol. 169: 445–448. [DOI] [PubMed] [Google Scholar]
- Hirel B., Le Gouis J., Bernard M., Perez P., Falque M., Quétier F., Joets J., Montalent P., Rogwoski P., Murigneux A., Charcosset A. (2007). Genomics and plant breeding: maize and wheat. In Functional Plant Genomics, J.F. Morot-Gaudry, P.J. Lea, and J.F. Briat, eds (Enfield, NH:Science Publishers; ), pp. 614–635. [Google Scholar]
- Jenner H.L., Winning B.M., Millar A.H., Tomlinson K.L., Leaver C.J., Hill S.A. (2001). NAD malic enzyme and the control of carbohydrate metabolism in potato tubers. Plant Physiol. 126: 1139–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joët T., Salmona J., Laffargue A., Descroix F., Dussert S. (2010). Use of the growing environment as a source of variation to identify the quantitative trait transcripts and modules of co-expressed genes that determine chlorogenic acid accumulation. Plant Cell Environ. 33: 1220–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi V., Joung J.G., Fei Z., Jander G. (2010). Interdependence of threonine, methionine and isoleucine metabolism in plants: accumulation and transcriptional regulation under abiotic stress. Amino Acids 39: 933–947. [DOI] [PubMed] [Google Scholar]
- Kandoi D., Mohanty S., Govindjee, Tripathy B.C. (2016). Towards efficient photosynthesis: overexpression of Zea mays phosphoenolpyruvate carboxylase in Arabidopsis thaliana. Photosynth. Res. 130: 47–72. [DOI] [PubMed] [Google Scholar]
- Keurentjes J.J.B., Sulpice R., Gibon Y., Steinhauser M.C., Fu J., Koornneef M., Stitt M., Vreugdenhil D. (2008). Integrative analyses of genetic variation in enzyme activities of primary carbohydrate metabolism reveal distinct modes of regulation in Arabidopsis thaliana. Genome Biol. 9: R129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruijer W., Boer M.P., Malosetti M., Flood P.J., Engel B., Kooke R., Keurentjes J.J.B., van Eeuwijk F.A. (2015). Marker-based estimation of heritability in immortal populations. Genetics 199: 379–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusano M., Fukushima A., Redestig H., Saito K. (2011). Metabolomic approaches toward understanding nitrogen metabolism in plants. J. Exp. Bot. 62: 1439–1453. [DOI] [PubMed] [Google Scholar]
- Kusano M., Fukushima A. (2013). Current challenges and future potential of tomato breeding using omics approaches. Breed. Sci. 63: 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacuesta M., Dever L.V., Muñoz-Rueda A., Lea P.J. (1997). A study of photorespiratory ammonia production in the C4 plant Amaranthus edulis, using mutants with altered photosynthetic capacities. Physiol. Plant. 99: 447–455. [Google Scholar]
- Laemmli U.K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685. [DOI] [PubMed] [Google Scholar]
- Langfelder P., Horvath S. (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9: 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lea P.J., Sodek L., Parry M.A.J., Shewry P., Halford N. (2007). Asparagine in plants. Ann. Appl. Biol. 150: 1–26. [Google Scholar]
- Lê Cao K.A., Boitard S., Besse P. (2011). Sparse PLS discriminant analysis: biologically relevant feature selection and graphical displays for multiclass problems. BMC Bioinformatics 12: 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limami A.M., Diab H., Lothier J. (2014). Nitrogen metabolism in plants under low oxygen stress. Planta 239: 531–541. [DOI] [PubMed] [Google Scholar]
- Lisec J., Römisch-Margl L., Nikoloski Z., Piepho H.P., Giavalisco P., Selbig J., Gierl A., Willmitzer L. (2011). Corn hybrids display lower metabolite variability and complex metabolite inheritance patterns. Plant J. 68: 326–336. [DOI] [PubMed] [Google Scholar]
- Liseron-Monfils C., Ware D. (2015). Revealing gene regulation and associations through biological networks. Curr. Plant Biol. 3–4: 30–39. [Google Scholar]
- Lunn J.E., Hatch M.D. (1997). The role of sucrose-phosphate synthase in the control of photosynthate partitioning in Zea mays leaves. Aus. J. Plant Physiol. 24: 1–8. [Google Scholar]
- Luo J. (2015). Metabolite-based genome-wide association studies in plants. Curr. Opin. Plant Biol. 24: 31–38. [DOI] [PubMed] [Google Scholar]
- Maroco J.P., Ku M.S.B., Lea P.J., Dever L.V., Leegood R.C., Furbank R.T., Edwards G.E. (1998). Oxygen requirement and inhibition of C4 photosynthesis - An analysis of C4 plants deficient in the C3 and C4 cycle. Plant Physiol. 116: 823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A., Belastegui-Macadam X., Quilleré I., Floriot M., Valadier M.H., Pommel B., Andrieu B., Donnison I., Hirel B. (2005). Nitrogen management and senescence in two maize hybrids differing in the persistence of leaf greenness: agronomic, physiological and molecular aspects. New Phytol. 167: 483–492. [DOI] [PubMed] [Google Scholar]
- Martin A., et al. (2006). Two cytosolic glutamine synthetase isoforms of maize are specifically involved in the control of grain production. Plant Cell 18: 3252–3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka Y., Vigouroux Y., Goodman M.M., Sanchez G J., Buckler E., Doebley J. (2002). A single domestication for maize shown by multilocus microsatellite genotyping. Proc. Natl. Acad. Sci. USA 99: 6080–6084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister C.H., Beatty P.H., Good A.G. (2012). Engineering nitrogen use efficient crop plants: the current status. Plant Biotechnol. J. 10: 1011–1025. [DOI] [PubMed] [Google Scholar]
- Meyer R.S., Purugganan M.D. (2013). Evolution of crop species: genetics of domestication and diversification. Nat. Rev. Genet. 14: 840–852. [DOI] [PubMed] [Google Scholar]
- Novitskaya L., Trevanion S.J., Driscoll S., Foyer C.H., Noctor G. (2002). How does photorespiration modulate leaf amino acid contents? A dual approach through modelling and metabolite analysis. Plant Cell Environ. 25: 821–831. [Google Scholar]
- Nunes-Nesi A., Sweetlove L.J., Fernie A.R. (2007). Operation and function of the tricarboxylic acid cycle in the illuminated leaf. Physiol. Plant. 129: 45–56. [Google Scholar]
- Nunes-Nesi A., Fernie A.R., Stitt M. (2010). Metabolic and signaling aspects underpinning the regulation of plant carbon nitrogen interactions. Mol. Plant 3: 973–996. [DOI] [PubMed] [Google Scholar]
- Obata T., Witt S., Lisec J., Palacios-Rojas N., Florez-Sarasa I., Yousfi S., Araus J.L., Cairns J.E., Fernie A.R. (2015). Metabolite profiles of maize leaves in drought, heat and combined stress field trials reveal the relationship between metabolism and grain yield. Plant Physiol. 169: 2665–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth J.D., Thiele I., Palsson B.O. (2010). What is flux balance analysis? Nat. Biotechnol. 28: 245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson B.W., Carraro F., Wolfe R.R. (1993). Measurement of 15N enrichment in multiple amino acids and urea in a single analysis by gas chromatography/mass spectrometry. Biol. Mass Spectrom. 22: 518–523. [DOI] [PubMed] [Google Scholar]
- Paulus J.K., Schlieper D., Groth G. (2013). Greater efficiency of photosynthetic carbon fixation due to single amino-acid substitution. Nat. Commun. 4: 1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piques M., Schulze W.X., Höhne M., Usadel B., Gibon Y., Rohwer J., Stitt M. (2009). Ribosome and transcript copy numbers, polysome occupancy and enzyme dynamics in Arabidopsis. Mol. Syst. Biol. 5: 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prioul J.L., Schwebel-Dugué N. (1992). Source-sink manipulations and carbohydrate metabolism in maize. Crop Sci. 32: 751–756. [Google Scholar]
- Ranum P., Peña-Rosas J.P., Garcia-Casal M.N. (2014). Global maize production, utilization, and consumption. Ann. N. Y. Acad. Sci. 1312: 105–112. [DOI] [PubMed] [Google Scholar]
- Rebourg C., Chastanet M., Gouesnard B., Welcker C., Dubreuil P., Charcosset A. (2003). Maize introduction into Europe: the history reviewed in the light of molecular data. Theor. Appl. Genet. 106: 895–903. [DOI] [PubMed] [Google Scholar]
- Rhodes D., Rich P.J., Brunk D.G. (1989). Amino acid metabolism of Lemna minor L. IV. 15N labellling kinetics of the amide and amino groups of glutamine and asparagine. Plant Physiol. 89: 1161–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedelsheimer C., Czedik-Eysenberg A., Grieder C., Lisec J., Technow F., Sulpice R., Altmann T., Stitt M., Willmitzer L., Melchinger A.E. (2012a). Genomic and metabolic prediction of complex heterotic traits in hybrid maize. Nat. Genet. 44: 217–220. [DOI] [PubMed] [Google Scholar]
- Riedelsheimer C., Lisec J., Czedik-Eysenberg A., Sulpice R., Flis A., Grieder C., Altmann T., Stitt M., Willmitzer L., Melchinger A.E. (2012b). Genome-wide association mapping of leaf metabolic profiles for dissecting complex traits in maize. Proc. Natl. Acad. Sci. USA 109: 8872–8877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ros R., Cascales-Miñana B., Segura J., Anoman A.D., Toujani W., Flores-Tornero M., Rosa-Tellez S., Muñoz-Bertomeu J. (2013). Serine biosynthesis by photorespiratory and non-photorespiratory pathways: an interesting interplay with unknown regulatory networks. Plant Biol. (Stuttg.) 15: 707–712. [DOI] [PubMed] [Google Scholar]
- Rossi M., Bermudez L., Carrari F. (2015). Crop yield: challenges from a metabolic perspective. Curr. Opin. Plant Biol. 25: 79–89. [DOI] [PubMed] [Google Scholar]
- Saito K., Matsuda F. (2010). Metabolomics for functional genomics, systems biology, and biotechnology. Annu. Rev. Plant Biol. 61: 463–489. [DOI] [PubMed] [Google Scholar]
- Shapiro S.S., Wilk M.B., Chen M.H.J. (1968). A comparative study of various tests for normality. J. Am. Stat. Assoc. 63: 1343–1372. [Google Scholar]
- Scheible W.R., Gonzalez-Fontes A., Lauerer M., Müller-Röber B., Caboche M., Stitt M. (1997). Nitrate acts as a signal to induce organic acid metabolism and repress starch metabolism in tobacco. Plant Cell 9: 783–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilmiller A.L., Pichersky E., Last R.L. (2012). Taming the hydra of specialized metabolism: how systems biology and comparative approaches are revolutionizing plant biochemistry. Curr. Opin. Plant Biol. 15: 338–344. [DOI] [PubMed] [Google Scholar]
- Shi J., Lai J. (2015). Patterns of genomic changes with crop domestication and breeding. Curr. Opin. Plant Biol. 24: 47–53. [DOI] [PubMed] [Google Scholar]
- Sicher R.C., Barnaby J.Y. (2012). Impact of carbon dioxide enrichment on the responses of maize leaf transcripts and metabolites to water stress. Physiol. Plant. 144: 238–253. [DOI] [PubMed] [Google Scholar]
- Simons M., Saha R., Amiour N., Kumar A., Guillard L., Clément G., Miquel M., Li Z., Mouille G., Lea P.J., Hirel B., Maranas C.D. (2014). Assessing the metabolic impact of nitrogen availability using a compartmentalized maize leaf genome-scale model. Plant Physiol. 166: 1659–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoot M.E., Ono K., Ruscheinski J., Wang P.L., Ideker T. (2011). Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27: 431–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood S., Flint-Garcia S., Willcox M.C., Holland J.B. (2014). Mining natural variation for maize improvement: selection on phenotypes and genes. In Genomics of Plant Resources, R. Tuberosa, A. Graner, and E. Frison eds (Dordrecht, The Netherlands: Springer Science + Business Media; ), pp. 615–649. [Google Scholar]
- Sosso D., et al. (2015). Seed filling in domesticated maize and rice depends on SWEET-mediated hexose transport. Nat. Genet. 47: 1489–1493. [DOI] [PubMed] [Google Scholar]
- Solomon B.D., Birchler J., Goldman S.L., Zhang Q. (2014). Basic information on maize. In Compendium of Bioenergy Plants: Corn, S.L. Goldman and C. Kole, eds (Boca Raton, FL: CRC Press; ), pp. 1–32. [Google Scholar]
- Stitt M. (2013). Systems-integration of plant metabolism: means, motive and opportunity. Curr. Opin. Plant Biol. 16: 381–388. [DOI] [PubMed] [Google Scholar]
- Storey J.D., Taylor J.E., Siegmund D. (2004). Strong control, conservative point estimation and simultaneous conservative consistency of false discovery rates: a unified approach. J. R. Stat. Soc. B 66: 187–205. [Google Scholar]
- Sturm A., Tang G.Q. (1999). The sucrose-cleaving enzymes of plants are crucial for development, growth and carbon partitioning. Trends Plant Sci. 4: 401–407. [DOI] [PubMed] [Google Scholar]
- Tamura W., Kojima S., Toyokawa A., Watanabe H., Tabuchi-Kobayashi M., Hayakawa T., Yamaya T. (2011). Disruption of a novel NADH-glutamate synthase2 gene caused marked reduction in spikelet number of rice. Front. Plant Sci. 2: 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tercé-Laforgue T., Clément G., Marchi L., Restivo F.M., Lea P.J., Hirel B. (2015). Resolving the role of plant NAD-glutamate dehydrogenase: III. Overexpressing individually or simultaneously the two enzyme subunits under salt stress induces changes in the leaf metabolic profile and Increases plant biomass production. Plant Cell Physiol. 56: 1918–1929. [DOI] [PubMed] [Google Scholar]
- Toubiana D., Fernie A.R., Nikoloski Z., Fait A. (2013). Network analysis: tackling complex data to study plant metabolism. Trends Biotechnol. 31: 29–36. [DOI] [PubMed] [Google Scholar]
- Uhart S.A., Andrade F.H. (1995). Nitrogen and carbon accumulation and remobilization during grain filling in maize under different source/sink ratio. Crop Sci. 35: 183–190. [Google Scholar]
- Wallace J.G., Larsson S.J., Buckler E.S. (2014). Entering the second century of maize quantitative genetics. Heredity (Edinb) 112: 30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Bräutigam A., Weber A.P.M., Zhu X.G. (2014). Three distinct biochemical subtypes of C4 photosynthesis? A modelling analysis. J. Exp. Bot. 65: 3567–3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward J.H., Jr. (1963). Hierarchical grouping to optimize an objective function. J. Am. Stat. Assoc. 58: 236–244. [Google Scholar]
- Weiner H., Blechschmidt-Schneider S., Mohme H., Eschrich W., Heldt H.W. (1991). Phloem transport of amino acids. Comparison of amino acid contents of maize leaves and of sieve tube exudate. Plant Physiol. Biochem. 29: 19–23. [Google Scholar]
- Wen W., Li D., Li X., Gao Y., Li W., Li H., Liu J., Liu H., Chen W., Luo J., Yan J. (2014). Metabolome-based genome-wide association study of maize kernel leads to novel biochemical insights. Nat. Commun. 5: 3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen W., et al. (2015). Genetic determinants of the network of primary metabolism and their relationships to plant performance in a maize recombinant inbred line population. Plant Cell 27: 1839–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingler A., Lea P.J., Leegood R.C. (1999). Photorespiratory metabolism of glyoxylate and formate in glycine accumulating mutants of barley and Amaranthus edulis. Planta 207: 518–526. [Google Scholar]
- Winter G., Todd C.D., Trovato M., Forlani G., Funck D. (2015). Physiological implications of arginine metabolism in plants. Front. Plant Sci. 6: 534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong J.C., Lambert R.J., Tadmor Y., Rocheford T.R. (2003). QTL associated with accumulation of tocopherols in maize. Crop Sci. 43: 2257–2266. [Google Scholar]
- Wuyts N., Dhondt S., Inzé D. (2015). Measurement of plant growth in view of an integrative analysis of regulatory networks. Curr. Opin. Plant Biol. 25: 90–97. [DOI] [PubMed] [Google Scholar]
- Yang H.S., Dobermann A., Lindquist J.L., Walters D.T., Arkebauer T.J., Cassman K.G. (2004). Hybrid-maize—a maize simulation model that combines two crop modeling approaches. Field Crops Res. 87: 131–154. [Google Scholar]
- Yesbergenova-Cuny Z., Dinant S., Martin-Magniette M.L., Quilleré I., Armengaud P., Monfalet P., Lea P.J., Hirel B. (2016). Genetic variability of the phloem sap metabolite content of maize (Zea mays L.) during the kernel-filling period. Plant Sci. 252: 347–357. [DOI] [PubMed] [Google Scholar]
- Zelitch I., Schultes N.P., Peterson R.B., Brown P., Brutnell T.P. (2009). High glycolate oxidase activity is required for survival of maize in normal air. Plant Physiol. 149: 195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N., Gur A., Gibon Y., Sulpice R., Flint-Garcia S., McMullen M.D., Stitt M., Buckler E.S. (2010). Genetic analysis of central carbon metabolism unveils an amino acid substitution that alters maize NAD-dependent isocitrate dehydrogenase activity. PLoS One 5: e9991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N., et al. (2015). Genome-wide association of carbon and nitrogen metabolism in the maize nested association mapping population. Plant Physiol. 168: 575–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.