

Abstract
Proliferative glomerulonephritis with monoclonal immunoglobulin G deposit (PGNMID), a recently described pathologic entity in native kidneys, has been recognized in kidney transplant patients, where it can present as either recurrent or de novo disease. There is no definitive treatment to date, in either population. Here, we present two cases of PGNMID in kidney allografts that illustrate the challenges of diagnostic approach and highlight the allograft outcome after treatment with rituximab as a potential treatment of this condition.
Keywords: glomerulonephritis, graft function, graft survival, kidney biopsy, kidney transplantation, rituximab
Introduction
Renal disease related to the glomerular deposition of monoclonal immunoglobulins containing both heavy and light chains can occur in light and heavy chain deposition disease, type 1 cryoglobulinemia, immunotactoid glomerulopathy and fibrillary glomerulonephritis (GN) [1]. In 2004, Nasr et al. first described a novel GN related to monoclonal immunoglobulin G (IgG) deposition termed ‘proliferative GN with monoclonal IgG deposits (PGNMID)’ in native kidneys [2]. The same authors then described biopsy-proven recurrent PGNMID in patients at a mean of 2 years post-kidney transplant [3]. Similar to disease in the native kidneys, the allograft biopsies showed endocapillary or membranoproliferative GN, with or without membranous features on light microscopy. Ultrasturctural findings included amorphous, non-organized electron-dense deposits, typically in a subendothelial and mesangial distribution on electron microscopy, and monoclonal staining by direct immunofluorescence (IF) for a single light-chain isotype and a single heavy-chain subtype, most commonly IgG3κ [3]. In this article, we report two cases of PGNMID occurring in male patients: a case of PGNMID with IgG1κ restriction that was likely recurrent, and a case of a de novo PGNMID with IgG3κ restriction occurring early after transplantation. We discuss the outcome of the allografts after 2 years of follow-up with rituximab being the treatment regimen used in both patients.
Cases
Patient 1
This is a 53-year-old male patient with a past medical history of hypertension, coronary artery disease, insulin-dependent diabetes mellitus and cerebrovascular disease. He developed end-stage renal disease (ESRD) secondary to an ill-defined crescentic GN with immune complex deposits, which stained for IgG, C3 and kappa light chain, and also had a positive ANCA titer. He underwent a deceased donor kidney transplant, and presented for evaluation of hematuria and proteinuria 1.5 years later. His baseline immunosupressive regimen included prednisone, tacrolimus and mycophenolic acid. His urinalysis was positive for blood [>180 red blood cell/high power field (RBC/HPF)] with sub-nephrotic-range proteinuria [urine protein/creatinine ratio (UPCR) 1.3–1.9 g/g], but stable allograft function with serum creatinine (sCr) 1.6 mg/dL. The first kidney allograft biopsy (7 months posttransplant) demonstrated patchy acute tubular injury without signs of acute rejection or glomerular disease. Serologies including antinuclear antibody (ANA), anti- ds-DNA, anti-GBM, ANCA as well as SPEP/UPEP were negative, with normal complement levels. Because proteinuria became nephrotic-range (UPCR of 4.5 g/g) with persistent hematuria (>180 RBC/HPF) and stable allograft function, a repeat kidney allograft biopsy was performed (12 months posttransplant), showing an ill-defined mesangial proliferative GN with immune complex deposition that could not be further characterized due to limited tissue. The patient’s prednisone dose was increased to 10 mg and lisinopril 10 mg daily was added to his regimen. A third allograft biopsy, performed 18 months posttransplant) confirmed the diagnosis of PGNMID with IgG1κ restriction. Specifically, light microscopy showed predominantly mesangial and focal endocapillary proliferative GN (Figure 1), and direct IF showed granular mesangial and capillary wall staining for IgG (2–3+), C1q (trace) and kappa light chain (2–3+). Immunoglobulin subclass staining confirmed monoclonality for IgG1 (Figure 2). Ultrastructural evaluation showed numerous amorphous mesangial immune-type electron-dense deposits, as well as occasional subendothelial and subepithelial deposits without substructure (Figure 3). There was no histologic evidence of rejection, and there was no C4d deposition in the peritubular capillaries by indirect IF microscopy. The patient was treated with 3 days of solumedrol and received two courses of rituximab 375 mg/m2 2 weeks apart. He was kept on his same immunosuppressive medications with higher prednisone dose (10 mg daily). He underwent follow-up allograft biopsy after 1 year of treatment with rituximab (24 months posttransplant), which showed very similar histologic findings but an apparent decrease in the intensity and extent of glomerular immune complex deposition by IF and electron microscopy. Similar to the previous biopsy, there was no evidence of rejection with no C4d deposition in the peritubular capillaries by indirect IF with negative DSA. On routine follow-up, his proteinuria had decreased to 0.5–0.8 g/g with improving allograft function and stable sCr at 1.1 mg/dL 2 years after treatment with rituximab, with urinalysis showing improving hematuria with only 1+ blood (7 RBC/HPF) and 100 protein (Table 1).
Fig. 1.
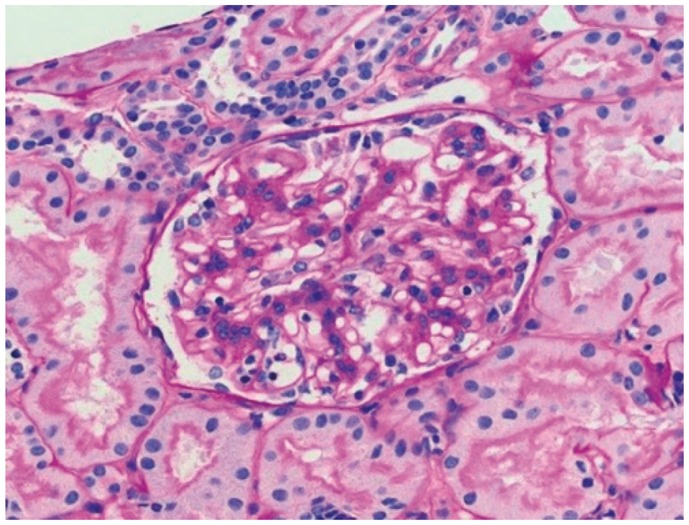
Proliferative GN with monoclonal IgG deposits in the kidney allograft biopsy specimen from patient 1, 2 years posttransplantation. Light microscopy showed a mesangial and focal endocapillary GN (periodic acid–Schiff stain; original magnification, 400×).
Fig. 2.
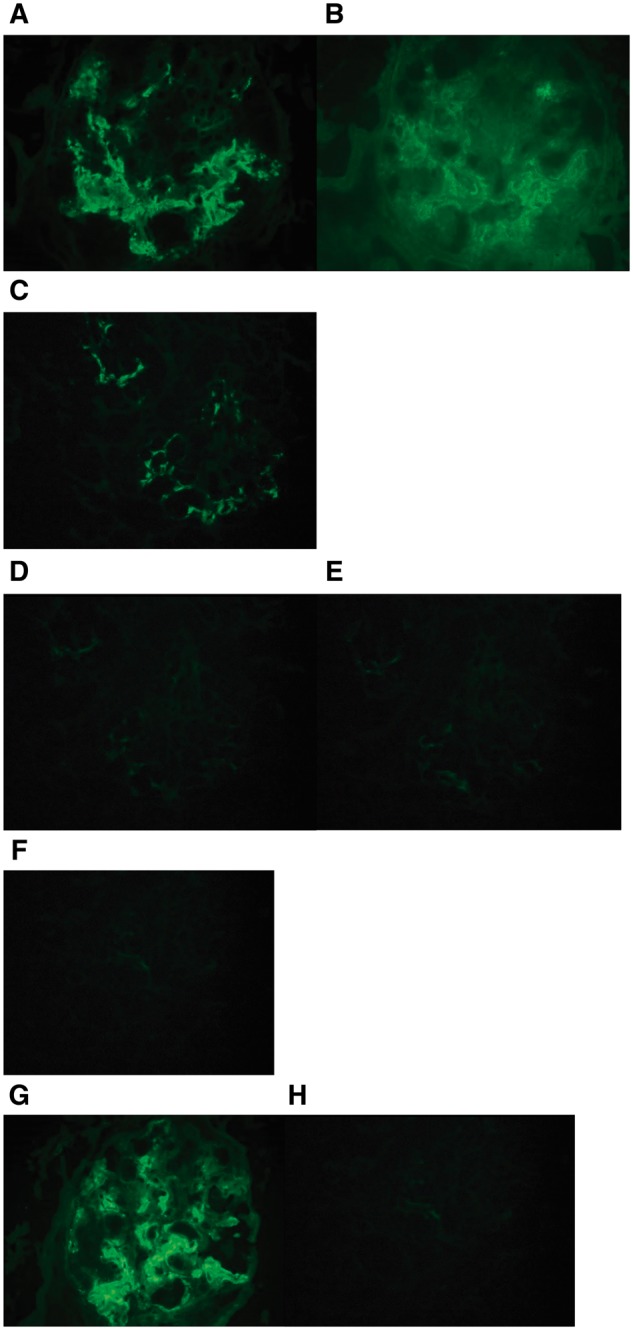
Immunofluorescence microscopy showed granular mesangial and occasional capillary wall staining for (A) IgG and (B) C1q. IgG subclasses showed positive glomerular staining for (C) IgG1 only, whereas the glomeruli were negative for (D) IgG2, (E) IgG3 and (F) IgG4. Additionally, the glomeruli showed positive staining for (G) kappa light chain, but were negative for (H) lambda light chain (original magnification, 400×).
Fig. 3.

Electron microscopy showed numerous mesangial immune-type electron-dense deposits, and rare subendothelial and subepithelial deposits, without organization or substructure (original magnification, 4600×).
Table 1.
Clinical outcome 2 years after rituximab in two kidney allografts with PGNMID
| sCr upon presentation | Uric acid upon presentation | UPCR upon presentation | sCr after 2 years of rituximab | Uric acid 2 years after rituximab | UPCR 2 years after rituximab | |
|---|---|---|---|---|---|---|
| Case 1 | 2 mg/dL | 3+ blood, >180 RBC/HPF and >500 protein | 4.4 g/g | 1.08 mg/dL | 1+ blood, 7 RBC/HPF and 100 protein | 0.85 g/g |
| Case 2 | 2.25 mg/dL | 3+ blood, >180 RBC/HPF and >500 protein | 1.5 g/g | 1.2–1.5 mg/dL | 1+ blood, 4 RBC/HPF and 100 protein | 1.5–2 g/g |
Patient 2
A 69-year-old male patient with a history of hypertension and ESRD, presumed secondary to obesity-related glomerular disease (never biopsied), underwent a deceased donor kidney transplant. He initially enjoyed excellent allograft function (sCr 1.2 mg/dL) on an immunosuppressive regimen of prednisone, tacrolimus and mycophenolic acid, and prophylaxis with valganciclovir, trimethaprim-sulfamethoxazole and clotrimazole troche. Posttransplant course, however, was complicated with BK viremia with BK PCR of 20 400 copies/mL and mild worsening of allograft function (sCr of 1.4 mg/dL). His dose of mycophenolic acid was decreased by 50% while tacrolimus was kept as the same dose for a target level of 7 ng/dL with a complete clearance of his viremia. On routine labs, his urinalysis 1 month posttransplant was consistent with positive blood, 30+ protein and >180 RBC/HPF. Repeated urinalysis over the next few months showed persistent hematuria and proteinuria. A kidney biopsy was performed (6 months posttransplant) showing Banff type 1A acute cellular rejection, and an immunohistochemical stain was negative for SV40 large T antigen. There were also glomerular alterations consistent with an early membranoproliferative pattern of injury, and limited mesangial and capillary wall immune complex deposition by IF and electron microscopy that could not be further characterized. These findings were concerning for a recurrent or de novo GN in the allograft. At that time, serologic studies were significant for a low C3 level at 83 mg/dL (normal range: 88–200 mg/dL); additional studies were negative/normal including C4, hepatitis B and C, ANAs, cryoglobulins, SPEP/UPEP and urine immunofixation. Due to persistent hematuria, worsening proteinuria and declining allograft function (sCr 2.27 mg/dL), the patient underwent repeat allograft biopsy (9 months posttransplant), which confirmed the diagnosis of PGNMID with IgG3κ restriction. Specifically, the light microscopy demonstrated a membranoproliferative pattern of glomerular injury with accentuated lobulation of the capillary tufts, mesangial and endocapillary hypercellularity, and thickening and duplication of the glomerular basement membranes (Figure 4). Direct IF showed granular capillary wall and mesangial staining for IgG (3+), C3 (3+), C1q (1+) and kappa light chain (3+), and subclass staining demonstrated monoclonality for IgG3 (Figure 5). Electron microscopy revealed numerous amorphous mesangial, subendothelial and focal subepithelial immune-type electron-dense deposits without substructure, and duplication of the glomerular basement membranes (Figure 6). There was no histologic evidence of rejection, and there was no C4d deposition in the peritubular capillaries by indirect IF microscopy. The DSA were checked each time he underwent an allograft biopsy with negative results. The patient was treated with two courses of intravenous rituximab 375 mg/m2 2 weeks apart. His allograft function improved to sCr of 1.2 mg/dL. He remained on a triple immunosuppressive regimen with prednisone, tacrolimus and low-dose mycophenolate mofetil. His clinical follow up 2 years after therapy with rituximab showed renal function of 1.5 mg/dL with urinalysis showing only 1+ blood (4 RBC/HPF), 100 protein with UPCR of 1.5–2 g/g (Table 1).
Fig. 4.
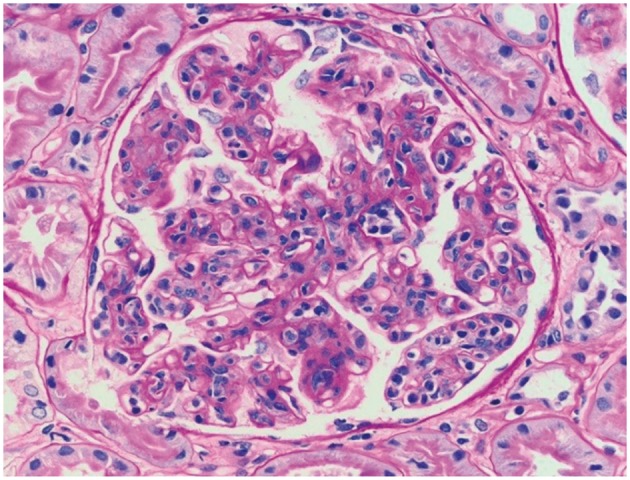
Proliferative GN with monoclonal IgG deposits in the kidney allograft biopsy specimen from patient 2, 9 months posttransplantation. Light microscopy showed features of membranoproliferative GN, including accentuated lobulation of the capillary tufts, mesangial and endocapillary hypercellularity, and thickening and duplication of the glomerular basement membranes (periodic acid–Schiff stain; original magnification, 400×).
Fig. 5.
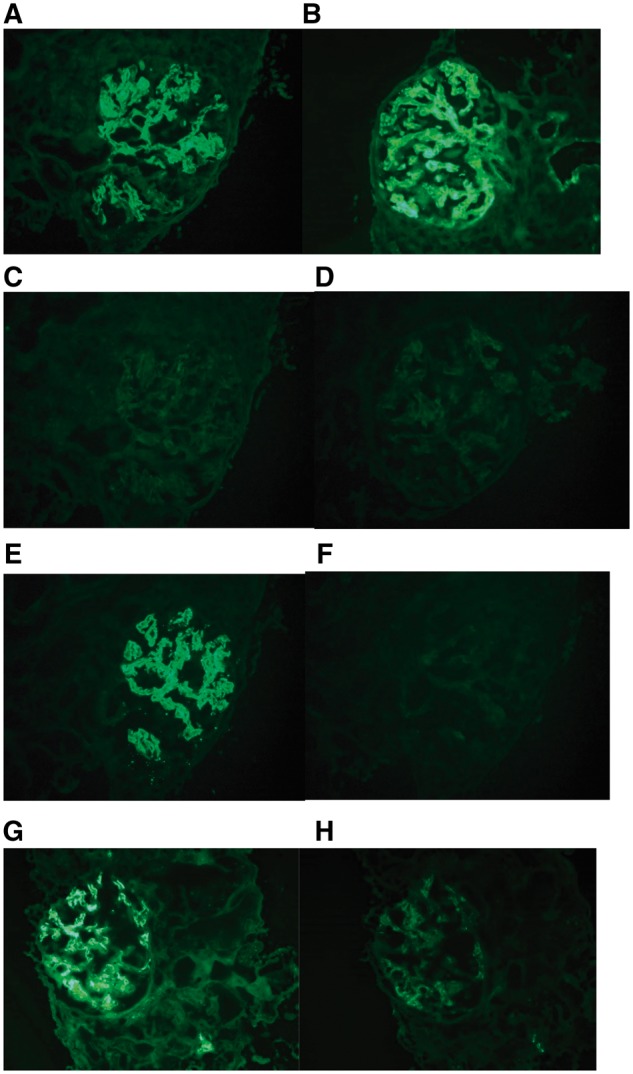
Immunofluorescence microscopy showed granular mesangial and capillary wall staining for (A) IgG and (B) C3. IgG subclasses for (C) IgG1, (D) IgG2, (E) IgG3 and (F) IgG4 showed isolated glomerular staining for IgG3 only. The glomeruli also showed positive staining for (G) kappa light chain, but were negative for (H) lambda light chain (original magnification, 400×).
Fig. 6.

Electron microscopy showed numerous subendothelial immune-type electron-dense deposits with frequent duplication of the glomerular basement membranes, as well as mesangial deposits (original magnification, 3200×).
Discussion
PGNMID is an increasingly recognized glomerular disease in both native and allograft kidney biopsies. As described in the literature, the diagnosis of PGNMID requires specific clinical and pathological criteria. The pathological diagnosis requires that renal biopsy shows evidence of GN with IF and electron microcopy showing (i) glomerular monoclonal IgG deposits restricted to a single IgG subclass and a single light chain isotype, with IgG3κ subtype being the most commonly recognized, (ii) presence of endocapillary proliferative, membranoproliferative or membranous features and (iii) the detection of immune complex deposits by electron microcopy. Clinically, the exclusion of cryoglobulinemia by laboratory and clinical evidence is required [2]. Although the etiology of PGNMID is not well known, case reports of PGNMID were associated with hematological and infectious causes such as chronic lymphocytic leukemia (CLL), parvovirus B19 and hepatitis C infection [4–6]. The pathogenesis, outcome and management PGNMID still remain controversial [1, 7], with low incidence in native kidneys (0.17%) and unclear incidence in kidney transplant patients [5]. This condition appears to be more common in older Caucasian women [3]. Patients present with nephrotic syndrome (50% of cases), hematuria (77%) and/or decreased kidney function (60%) [3]. The prognosis of PGNMID in native kidneys is overall poor, with 25% of the patients progressing to dialysis within 3 years of the diagnosis [1, 8]. There is no definite treatment for this condition, with the largest series reporting 37 patients with PGNMID in native kidneys and different treatment regimens used [1]. Four of these patients were treated with rituximab: two of them received rituximab in combination with mycophenolate mofetil and/or cyclosporine and the other two received rituximab alone. Their clinical outcome after 30.3 months of follow up showed partial remission (reduction of proteinuria by 50% and stable kidney function <2 mg/dL) in two patients and the other two failed to meet the criteria of complete remission (remission of proteinuria to <500 mg/day with normal renal function) or partial remission. In other cases, PGNMID in native kidneys was treated successfully with steroids resulting in complete remission of proteinuria [7, 9]. Two reported cases of PGNMID associated with CLL were treated with rituximab and showed significant reduction in proteinuria and improving kidney function with parallel control of the underlying CLL [4].
In the kidney transplant population, PGNMID is less well described, with IgG3κ subtype being the most common in published reports [10, 11]. The rate of recurrence of PGNMID in the kidney allograft is either undefined or underdiagnosed since allograft biopsy can be misinterpreted as transplant glomerulopathy, unless IF or electron microscopy are performed [1]. Recurrent PGNMID usually develops within the first 2 years post-kidney transplant, whereas de novo PGNMID usually appears several years after transplantation (13 years in one case report) [3, 12]. The recurrence in the allograft favors the persistence of circulating factors in the recipient as a cause of this condition. The average age of the patients reported in the literature is 57 years old, with female predominance. There were no protocol biopsies on these patients, making it impossible to identify the risk of recurrence. No standardized treatment protocol exists yet for PGNMID after transplantation due to the small number of patients [3]. Multiple regimens have been used for the treatment of PGNMID after renal transplant, including renin angiotensin system blocking agents, high-dose steroids, rituximab with or without steroids, bortezomib and even plasmapheresis (Table 2). The proposition of rituximab as the potential treatment of PGNMID comes from the hypothesis that clonal proliferation of B lymphocytes or plasma cells in PGNMID hypersecrete abnormal IgG capable of self-aggregation and deposit in the glomerulus. Rituximab, a chimeric anti-CD20 monoclonal antibody, was initially approved for the treatment of B-cell lymphoma. Lately, its use has been wide and extensive in the transplant population, especially in the treatment of refractory and steroid-resistant acute antibody-mediated rejection (AMR), posttransplant lymphoproliferative disorder, recurrent glomerular disease after kidney transplantation, an alternative to splenectomy as part of the desensitization protocol in ABOi renal transplantation and in preventing chronic AMR by controlling B-cell immunity and decreasing antibody production [13–20]. After reviewing the literature in PGNMID after kidney transplantation, the patients treated with rituximab showed better outcome and improving allograft function compared with the other regimen used, although the number of patients reported is very limited [3, 8, 11, 12]. The patients treated with rituximab were followed for up to 43 months and they achieved reduction in proteinuria and reduction in creatinine, with repeat biopsies showing reduced histologic activity after treatment [3]. From these limited cases that responded to rituximab and from the hypothesis on the pathophysiology of this condition came the rationale of using rituximab in our patients. Our two male patients, a case of PGNMID with IgG1κ restriction that was likely recurrent, and a case of a de novo PGNMID with IgG3κ restriction occurring early after transplantation, were both treated with rituximab and responded well, with marked improvement of the allograft function and almost back to baseline sCr not only in the short follow-up period, but also after 2 years of follow-up (Table 1), which makes our study unique. In addition, we have performed repeated follow-up biopsy 1 year after treatment in one of the two patients with evidence of extensive decrease in the glomerular immune complex deposits after rituximab therapy. The excellent response of PGNMID posttransplant to rituximab in our two cases is encouraging as it showed persistent benefit clinically and pathologically. More work and larger studies are needed to focus on the pathogenesis of PGNMID, and randomized trials can help investigate the role of rituximab in this condition. It is still unclear if protocol biopsy in the allograft is the standard of care in patients with PGNMID affecting their native kidneys in detecting early recurrence in the allograft, not forgetting that this population is already on triple immunosuppressive regimen after kidney transplantation, which may delay the early manifestations of the disease recurrence. Prospective, multicenter, controlled study on a larger number of patients will be helpful as until now, treatment choice has been based on clinical experience with a small number of patients.
Table 2.
Treatment review of PGNMID in renal allograft
| Number of patients | Patient | De novo versus recurrent disease | Cr (mg/dL) upon presentation | Treatment | Cr (mg/dL) after treatment | |
|---|---|---|---|---|---|---|
| 1 | Recurrent | 2.8 | Steroids, PO cytoxan for 6 months | 2.3 | ||
| Nasr et al. [3] | 4 | 2 | Recurrent | 3.7 | Steroids for 8 months, rituximab × 2, lisinopril for 6 months | 1.1 |
| 3 | Recurrent | 4.8 | Steroids, rituximab × 1, PLX × 4, and HD × 3 | 1.3 | ||
| 4 | Recurrent | 1.2 | Steroids for ACR, rituximab × 2 | 2.3 | ||
| 1 | Recurrent | 2.7 | No change in immunosuppression | 1.7–2.5; patient died from fatal myocardial infarct | ||
| Albawardi et al. [12] | 4 | 2 | Recurrent | 3.9 | Adjusted immunosuppression from MFA and sirolimus to MFA and steroids | 3.3; patient died from fatal cryptococcal meningitis |
| 3 | De novo | 2.6 | Prednisone and cyclosporine | ESRD | ||
| 4 | De novo | 2.8 | No mention | No mention | ||
| Rhangino et al. [8] | 1 | 1 | Recurrent | 2.0 | PLX, IVIG, steroids | 1.1 |
| Al-Rabadi [11] | 3 | 1 | De novo | 1.2 | Bortezomib and dexamethasone × 3 doses, then switched to carfilzomib | 1.70 |
| 2 | Recurrent | 4.48 | Patient opted for no treatment | ESRD | ||
| 3 | De novo | 1.0 | No change in immunosuppression: tacrolimus and azathioprine | 1.28 |
ACR, acute cellular rejection; HD, hemodialysis; IVIG, intravenous immunoglobulin; MFA, mycophenolic acid; PLX, plasmapharesis; PO, oral.
References
- 1. Nasr SH, Satoskar A, Markowitz GS. et al. Proliferative glomerulonephritis with monoclonal IgG deposits. J Am Soc Nephrol 2009; 20: 2055–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nasr SH, Markowitz GS, Stokes MB. et al. Proliferative glomerulonephritis with monoclonal IgG deposits: a distinct entity mimicking immune-complex glomerulonephritis. Kidney Int 2004; 65: 85–96 [DOI] [PubMed] [Google Scholar]
- 3. Nasr et al. Proliferative glomerulonephritis with monoclonal IgG deposits recurs in the allograft. Clin J Am Soc Nephrol 2011; 6: 122–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barbour SJ, Beaulieu MC, Zalunardo NY. et al. Proliferative glomerulonephritis with monoclonal IgG deposits secondary to chronic lymphocytic leukemia. Report of two cases. Nephrol Dial Transplant 2011; 26: 2712–2714 [DOI] [PubMed] [Google Scholar]
- 5. Fujita E, Shimizu A, Kaneko T. et al. Proliferative glomerulonephritis with monoclonal immunoglobulin G3κ deposits in association with parvovirus B19 infection. Hum Pathol 2012; 43: 2326–2333 [DOI] [PubMed] [Google Scholar]
- 6. Yamada T, Arakawa Y, Mii A. et al. A case of monoclonal immunoglobulin G1-lambda deposition associated with membranous feature in a patient with hepatitis C viral infection. Clin Exp Nephrol 2012; 16: 468–472 [DOI] [PubMed] [Google Scholar]
- 7. Ohashi et al. Proliferative glomerulonephritis with monoclonal IgG2k deposits successfully treated with steroids: a case report and review of the literature. CEN Case Rep 2013; 2: 197–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rhangino et al. A case of recurrent proliferative glomerulonephritis with monoclonal IgG deposits after kidney transplantation. Case Rep Nephrol Urol 2012; 2: 46–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Komatsuda et al. Steroid responsive nephrotic syndrome in a patient with proliferative glomerulonephritis with monoclonal IgG deposits with pure mesangial proliferative features. Clin Kidney J 2010; 3: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Batal et al. Proliferative glomerulonephritis with monoclonal immunoglobulin deposits in a kidney allograft. Am J Kidney Dis 2014 [DOI] [PubMed]
- 11. Al-Rabadi L, Francis S, Henderson J. et al. Proliferative glomerulonephritis with monoclonal immunoglobulin in renal allografts. Clin Kidney J 2015: 8:722–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Albawardi et al. Proliferative glomerulonephritis with monoclonal IgG deposits recurs or may develop de novo in kidney allografts. Am J Kidney Dis 2011; 58: 276–281 [DOI] [PubMed] [Google Scholar]
- 13. Faguer S, Kamar N, Guilbeaud-Frugier C. et al. Rituximab therapy for acute humoral rejection after kidney transplantation. Transplantation 2007; 83:1277–1280 [DOI] [PubMed] [Google Scholar]
- 14. Kaposztas Z, Podder H, Mauiyyedi S. et al. Impact of rituximab therapy for treatment of acute humoral rejection. Clin Transplant 2009; 23:63–73 [DOI] [PubMed] [Google Scholar]
- 15. Ponticelli C, Glassock RJ. Posttransplant recurrence of primary glomerulonephritis. Clin J Am Soc Nephrol 2010; 5: 2363–2372 [DOI] [PubMed] [Google Scholar]
- 16. Zimmermann H, Ulrich Trappe R. Therapeutic options in post-transplant lymphoproliferative disorders. Ther Adv Hematol 2011; 2: 393–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tyden G, Kumlien G, Fehrman I.. Successful ABO-incompatible kidney transplantations without splenectomy using antigen-specific immunoadsorption and rituximab. Transplantation 2003; 76: 730–731 [DOI] [PubMed] [Google Scholar]
- 18. Kohei N, Hirai T, Omoto K. et al. Chronic antibody-mediated rejection is reduced by targeting B-cell immunity during an introductory period. Am J Transplant 2012; 12: 469–476 [DOI] [PubMed] [Google Scholar]
- 19. Loupy A, Suberbielle-Boissel C, Zuber J. et al. Combined posttransplant prophylactic IVIg/anti-CD 20/plasmapheresis in kidney recipients with preformed donor-specific antibodies: a pilot study. Transplantation 2010; 89: 1403–1410 [DOI] [PubMed] [Google Scholar]
- 20. Becker YT, Becker BN, Pirsch JD. et al. Rituximab as treatment for refractory kidney transplant rejection. Am J Transplant 2004; 4: 996–1001 [DOI] [PubMed] [Google Scholar]