

Abstract
Background: Acute kidney injury (AKI) with renal tubular obstruction by red blood cell casts (RBCC) has been described in patients treated with warfarin and is known as warfarin-related nephropathy (WRN).
Methods: To determine whether other vitamin K antagonists (VKA) cause WRN, we retrospectively collected and analyzed the clinical and histological data of 13 patients treated with different VKA (seven with fluindione, four with warfarin and two with acenocoumarol) in seven French hospitals.
Results: They all developed gross hematuria following overanticoagulation complicated by severe AKI (median serum creatinine concentration = 693 μmol/L). Histological analysis of the kidney biopsies highlighted the presence of intratubular RBCC and acute tubular necrosis in all patients and of an underlying kidney disease in 12 patients. WRN was suspected in patients treated with warfarin; however, the initial diagnosis was incorrect in six of the nine patients treated with other VKA. Nine patients progressed to chronic kidney disease, one fully recovered renal function, two died and one still needs dialysis.
Conclusions: This is the first report of AKI caused by fluindione. In agreement with the recent publication on AKI in two patients treated with dabigatran, we suggest that the term ‘anticoagulant-related nephropathy’ is more appropriate than WRN. Gross hematuria in patients with an underlying kidney disease and treated with VKA requires rapid control of the international normalized ratio and renal function monitoring.
Keywords: acute kidney injury, drug nephrotoxicity, renal biopsy, vitamin k antagonists, warfarin-related nephropathy
Introduction
Vitamin K antagonists (VKA) inhibit the synthesis of the coagulation factors II, VII, IX and X, and have been used since the 1950s. VKA are mainly prescribed for the treatment of cardiovascular diseases (arrhythmia, heart valve disease) and venous thromboembolism. They can be divided in two families: (i) coumarin-derived VKA (warfarin, the most prescribed VKA worldwide, and acenocoumarol) and (ii) indanedione-derived VKA (fluindione). The main features of the different VKA are summarized in Table 1. In France, 80% of the 1.1 million people treated with VKA in 2014 received fluindione [1].
Table 1.
Characteristics of the different VKA
| Acenocoumarol | Warfarin | Fluindione | |
|---|---|---|---|
| Trade name | Sintrom® | Coumadin® | Previscan® |
| Family | Coumarin-derived VKA | Coumarin-derived VKA | Indanedione-derived VKA |
| Plasma half-life (h) | 8 | 35 – 45 | 31 |
| Activity duration (h) | 18-24 | 36 | 24 – 48 |
| Cases report of WRN | Yes | Yes | No |
| Marketed in the USA | No | Yes | No |
| AKI description | WRN | WRN | AIN |
AIN, acute interstitial nephritis.
Acute kidney injury (AKI) induced by VKA is due to tubulointerstitial damage. This could be caused by an immuno-allergic reaction or by tubular obstruction with red blood cell casts (RBCC). The former has been observed in patients treated with fluindione, but very rarely in patients treated with coumarin-derived VKA [2, 3]. Conversely, tubular obstruction with RBCC has been mainly reported in patients treated with warfarin. This is characterized by gross hematuria that appears during overanticoagulation [defined by an acute increase of the international normalized ratio (INR) >3] followed by AKI. Based on kidney biopsy findings, AKI can be considered the result of severe glomerular hemorrhage, leading to extensive renal tubular obstruction by RBCC and acute tubular necrosis (ATN). It was described for the first time in a patient with glomerular basement membrane nephropathy [4]. Later, two other cases were reported: a patient with immunoglobulin A (IgA) nephropathy [5] and a patient with inactive systemic lupus erythematosus [6]. Then, Brodsky et al. [7] described nine biopsy-proven cases of AKI by tubular obstruction with RBCC and proposed the term ‘warfarin-related nephropathy’ (WRN).
The prevalence of unexplained increase in serum creatinine (SC) during overanticoagulation (INR >3) was evaluated in two retrospective studies and was 20.5% [8] and 19.3% [9], respectively. In these studies, some patients did not develop gross or microscopic hematuria and the histological analysis of kidney biopsy specimens was not available.
Biopsy-proven cases of AKI with tubular obstruction by RBCC have been mostly reported during treatment with warfarin, the most used VKA in the USA. So far, only one case of AKI during acenocoumarol treatment has been described [10]. Here, we describe 13 biopsy-proven cases of AKI with tubular obstruction by RBCC in patients treated with fluindione, warfarin or acenocoumarol.
Materials and methods
This was a French observational retrospective case study on AKI with tubular obstruction by RBCC based on the collection and analysis of clinical and histological data from the various Departments of Nephrology of our network (35 Nephrology Departments, of which 15 are in university hospitals). We have followed the inclusion criteria described in the study by Brodsky etal. [7] where only patients with biopsy-proven AKI were included. Therefore, only patients treated with VKA and who developed (between April 2009 and December 2014) gross hematuria followed by AKI, according to the Kidney Disease: Improving Global Outcomes (KDIGO) criteria [11], with tubular obstruction by RBCC, were retrospectively considered regardless of INR value. This study was conducted by following the Declaration of Helsinki principles and was approved by the ethics committee of Rennes hospital. Where possible, the participants, written informed consent was retrospectively obtained.
For each patient, the following data were collected: VKA molecule, starting date of treatment, delay between overanticoagulation and AKI development, and highest INR value during the overanticoagulation period. The risk factors identified by Brodsky et al. [8] were analyzed: age, history of chronic kidney disease (CKD), hypertension, diabetes and cardiovascular diseases. The initial suspected diagnosis was recorded. Urine sediment was analyzed with a dipstick and a 24-h urine protein test was carried out during AKI. SC levels (in µmol/L) before AKI (the lowest concentration during the 3 months before AKI), during AKI and then at 6 weeks, 3 months, 6 months and 1 year after AKI were collected. The estimated glomerular filtration rate (eGFR) was calculated with the Modification of Diet in Renal Disease (MDRD) formula [12] and the CKD stage according to the Kidney Disease Outcomes Quality Initiative Index (K/DOQI) classification [13]. Recurrence of AKI with tubular obstruction by RBCC during the follow-up period (up to 1 year after the AKI episode) was also recorded. All treatment changes following AKI (especially VKA) were recorded.
Histological analysis of kidney biopsies
Kidney biopsy specimens obtained during the AKI episode were analyzed by staining with the Masson’s trichrome, Marinozzi’s silver, hematoxylin–eosin or periodic acid–Schiff stain (sections from paraffin-embedded tissue blocks) and by immunofluorescence analysis with antibodies against IgA, IgG and IgM, light chain kappa and lambda, fibrin and the complement components C1Q and C3. RBCC semiquantitative quantification in the cortical and medullary tubule segments was done by the same pathologist (N.R.-L., Department of Pathology, Rennes). Electronic microscopy analysis was performed at the Department of Nephrology, Poitiers, when frozen tissue samples from the kidney biopsy were available. Samples were fixed with 4% glutaraldehyde in phosphate buffer (v/v), washed three times in phosphate-buffered saline and post-fixed in phosphate-buffered 1% osmium tetroxide (w/v). After dehydration through a graded series of acetone, embedding in araldite (Fluka, Buchs, Switzerland) and polymerization, ultrathin sections (60 nm), were stained with uranyl acetate and lead citrate, according to the standard techniques, and examined by transmission electron microscopy (JEOL 1010, Jeol Ltd, Tokyo, Japan).
Results
Clinical features
On the basis of the analysis of the clinical and histological data from the different nephrology departments of our network, 13 patients treated with VKA and with biopsy-proven AKI were selected. Five patients were from Rennes, two from the Georges Pompidou European Hospital and one from the Tenon Hospital in Paris, two from Lorient, one from Tours, one from Quimper and one from Saint-Lô.
The median age of the 13 patients (10 men and 3 women) was 71 years (range: 48–90 years) (Table 2). The indications for VKA treatment were arrhythmia (9/13), venous thromboembolism (2/13), heart valve disease (1/13) and nephrotic syndrome with important hypoalbuminemia (1/13). Patients were treated with fluindione (7/13), warfarin (4/13) or acenocoumarol (2/13) with a median interval of 3 years (range: 2 weeks–20 years) between the beginning of the treatment and AKI occurrence. Eleven patients (84.6%) had hypertension, four (30.8%) a vascular disease, two (15.4%) diabetes mellitus and five (38.5%) had stage 3 CKD. A gross hematuria episode was systematically detected before AKI occurrence. When available (three missing data), urinalysis found microscopic hematuria and proteinuria during AKI with a median 24-h proteinuria of 1.62 g/24 h (range: 0.61–5.7 g/24 h). Eleven patients (84%) developed overanticoagulation with a median INR value of 3.8 (range: 2.51–10) during AKI. The median interval between the first INR value >3 and AKI appearance could be calculated only for 4 of these 11 patients (36%) and was 22 days (range: 6 days–5 weeks), and no correlation was found between SC level and INR. All patients had severe AKI (stage 3 according to the KDIGO classification) with a median SC during AKI of 693 µmol/L (range: 311–1654 µmol/L).
Table 2.
Clinical characteristics of patients and AKI outcome
| Patient no. | Age (years) | VKA | INR | Baseline eGFR | Peak SC | Underlying kidney disease | Treatments | Recovery kidney function | AKI recurrence | eGFR at 1-year follow-up |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 76 | F | 2.51 | 83 | 316 | IgA nephropathy | Switch from fluindione to warfarin | Partial | No | 59 |
| 2 | 48 | F | 4.2 | 98 | 473 | Post-infectious glomerulonephritis (Staphylococcus aureus) | Switch from fluindione to warfarin | Partial | No | 42 |
| 3 | 81 | F | 4.85 | 80 | HD | IgG kappa glomerulonephritis | Switch from fluindione to warfarin | Partial | No | 34 |
| 4 | 73 | F | 3.8 | 51 | 311 | IgA nephropathy | Switch from fluindione to warfarin | Partial | Yes | 28 |
| 5 | 66 | F | DM | 89 | 635 | Not found | Switch from fluindione to warfarin | Partial | No | 48 |
| 6 | 80 | F | >10 | 48 | 803 | Diabetic nephropathy | Switch from fluindione to warfarin | Patient died of respiratory distress 3 months after AKI | Patient died | |
| 7 | 90 | F | 3.39 | 47 | 589 | IgA nephropathy | Fluidione was continued | Patient died a few weeks after the diagnosis of congestive heart failure | Patient died | |
| 8 | 49 | A | >10 | 98 | HD | Hypertensive nephrosclerosis | VKA stopped | Partial | No | 31 |
| 9 | 70 | A | >10 | >120 | HD | Hypertensive nephrosclerosis | Acenocoumarol was continued | Partial | No | 29 |
| 10 | 71 | W | 3.3 | 98 | 322 | Hypertensive nephrosclerosis | Warfarin was continued after AKI | Total | Yes | 89 |
| 11 | 51 | W | >10 | 97 | HD | Henoch-Schönlein Purpura | Warfarin stopped for 1 month and then started again following venous thromboembolism | Partial | No | 59 |
| 12 | 78 | W | 4.5 | 56 | HD | IgA nephropathy | Warfarin stopped | Partial | No | 47 |
| 13 | 69 | W | 2.7 | 47 | HD | IgA nephropathy | Warfarin stopped | No recovery | No | HD |
The baseline eGFR was the lowest level found within 3 months before AKI. INR value was the highest value detected before AKI. A, acenocoumarol; AIN, acute interstitial nephritis; DM, data missing; eGFR, estimated glomerular filtration rate (mL/min/1.73 m2); F, fluindione; HD, hemodialysis; IgA, immunoglobulin A; IgG, immunogloulin G; INR, international normalized ratio (IU); SC, serum creatinine (µmol/L); W, warfarin.
Kidney damage
The histological findings are summarized in Table 3. RBCC were systematically found in the tubule lumen; on average, RBCC obstructed 10% of cortical tubule segments (range: 0–100%) in all biopsies and 25% of medullar tubule segments (range: 0–50%) in nine biopsies (Figure 1A). ATN was systematically found. Red blood cells in the Bowman’s space were observed in three patients (23%) (Figure 1B). Histological signs of an underlying kidney disease were found in 12 of the 13 patients: 9 had a glomerulopathy (IgA nephropathy: n = 6; Henoch-Schönlein Purpura: n = 1; IgG kappa glomerular deposits: n = 1; acute post-infectious glomerulonephritis: n = 1; and diabetic glomerulonephritis: n = 1) and 3 patients had hypertensive nephrosclerosis. In nine patients, the underlying kidney disease was discovered during AKI. The others (patient #2 with acute post-infectious glomerulonephritis, and patients #11 and #12 with IgA nephropathy) had a confirmed kidney disease already before starting the VKA-based treatment. In these three patients, renal function declined following the beginning of the VKA treatment due to the appearance of the tubular obstructive lesions. Conversely, the glomerular lesions linked to the underlying kidney disease were improved compared with the previous biopsy findings. In four patients, morphological analysis of the glomerular lesions could be done also by electron microscopy. No common or specific lesion could be found. Glomerular lesions seemed to be present already before AKI. Specifically, mesangial deposits, compatible with IgA nephropathy, were observed in two patients. Important inflammatory lesions, compatible with his post-infectious glomerulonephritis, were detected in another patient. Glomerulitis with accumulation of lymphocytes and macrophages, which was difficult to interpret, was found in the fourth patient. Tubule lesions could not be analyzed by electron microscopy because of the biopsy preparation method.
Table 3.
Histological findings in kidney biopsy specimens
| Patient no. | Glomeruli |
Tubulointerstitium |
Vasculature |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| No. of glomeruli | No. of sclerotic glomeruli | No. of crescents | RBC in Bowman's space (%) | Interstitial inflammation | ATN | Fibrosis | RBC in cortical tubules (%) | RBC in medullar tubules (%) | Arteriolar hyalin | Immunofluorescence | |
| 1 | 7 | 1 | 0 | 0 | 0 | 2 | 1 | 8 | 25 | 2 | Mild mesangial C3 and IgA |
| 2 | 20 | 0 | 1 partial crescent | 0 | 1 | 2 | 1 | 5 | 20 | 1 | Mild mesangial C3, IgG, K and L |
| 3 | 12 | 0 | 1 crescent | 15 | 2 | 0 | 0 | 0 | 30 | 2 | Prominent mesangial C3, IgG and K |
| 4 | 1 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 25 | DM | Moderate mesangial IgA |
| 5 | 12 | 2 | 0 | 0 | 3 | 3 | 2 | 10 | 30 | 0 | Negative |
| 6 | 7 | 0 | 0 | 0 | 3 | 3 | 0 | 10 | 20 | 3 | Moderate GBM IgG |
| 7 | 38 | 17 | 0 | 0 | 1 | 2 | 2 | 10 | 10 | 2 | Moderate mesangial IgA and K and L |
| 8 | 15 | 2 | 0 | 10 | 3 | 3 | 0 | 30 | 50 | 3 | Non-specific |
| 9 | 14 | 1 | 0 | 0 | 2 | 2 | 1 | 100 | 0 | 1 | Negative |
| 10 | 30 | 3 | 0 | 0 | 2 | 2 | 1 | 20 | No | 2 | Moderate cylinders IgA and K and L |
| 11 | 17 | 1 | 3 crescents | 0 | 2 | 2 | 1 | 10 | No | 0 | No immunofluorescence |
| 12 | 14 | 2 | 0 | 25 | 2 | 2 | 2 | 10 | No | 2 | Prominent mesangial C3 and IgA |
| 13 | 8 | 3 | 0 | 0 | 1 | 1 | 2 | 10 | No | 2 | Moderate mesangial IgA and C3 |
Histological findings were scored semiquantitatively by using the following criteria: 0: absent; 1: mild; 2: moderate; and 3: prominent. DM, data missing; GBM, glomerular basal membrane; IgA, immunoglobulin A; IgG, immunoglobulin G; K, light chain Kappa deposits; L, light chain Lambda deposits; RBC, red blood cells.
Fig. 1.
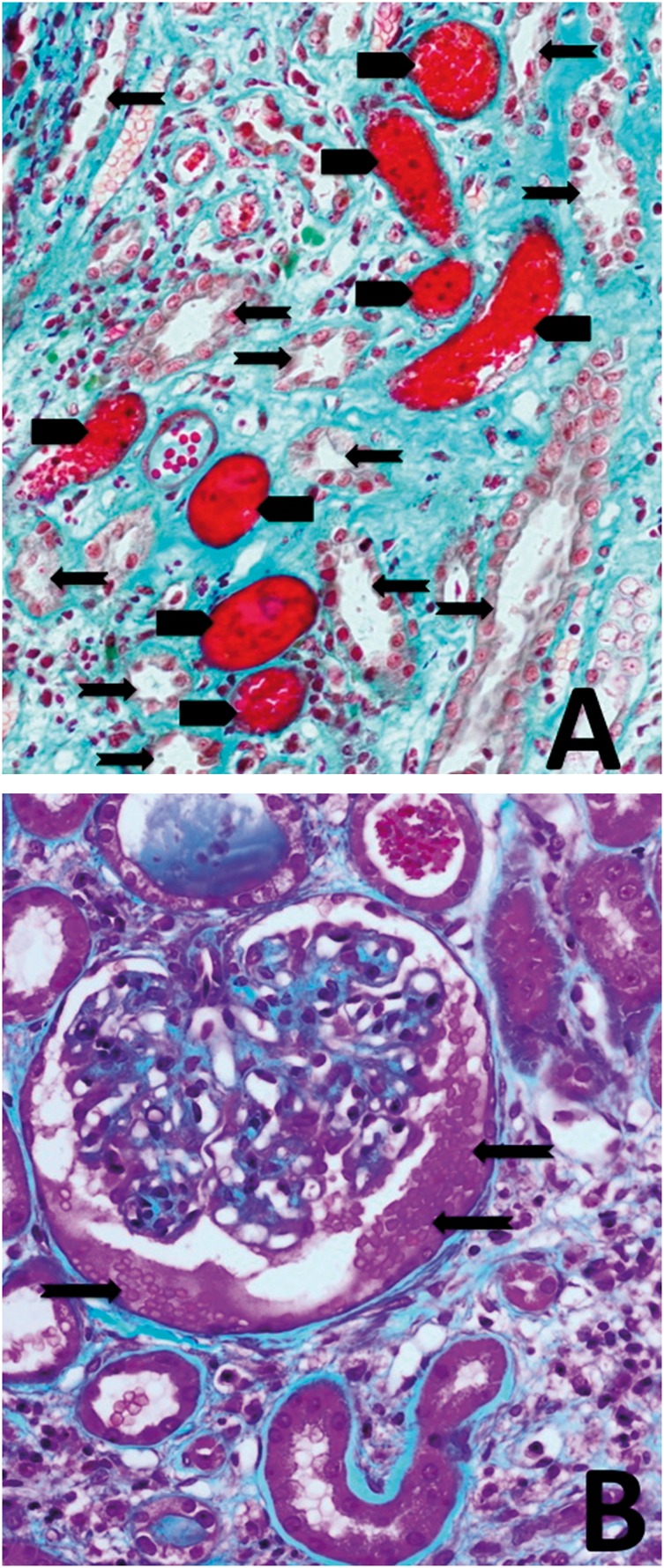
Different lesions observed in kidney biopsies from patients with AKI. (A) Masson’s trichrome staining showing acute tubular injury (arrows) associated with tubular obstruction by red blood cell casts (arrowheads) (magnification ×200). (B) Masson’s trichrome staining showing red blood cells (arrows) in the Bowman’s space (magnification ×200).
Initial diagnosis and treatment
WRN was initially suspected in all patients treated with warfarin (n = 4), in two of the patients treated with fluindione and in one of the patients treated with acenocoumarol. The initial diagnosis was incorrect in six patients treated with fluindione (n = 5) or with acenocoumarol (n = 1). All patients treated with fluindione were switched to warfarin. Of the two patients treated with acenocoumarol, one continued his VKA treatment after AKI, while the second stopped VKA.
Outcomes and follow-up
One patient (7.7%) rapidly recovered renal function, two (15.4%) patients died and one (7.7%) still required dialysis 3 months after AKI (Figure 2). In nine patients (69.2%), recovery was delayed and six of them initially required dialysis therapy with a median of five dialysis sessions. Moreover, one patient recovered renal function 6 months after AKI. At the 1-year follow-up visit, seven patients had stage 3 CKD (median eGFR = 47 mL/min/1.73 m2) and two had stage 4 CKD (median eGFR = 28.5 mL/min/1.73 m2). Two AKI recurrences were reported during a new episode of overanticoagulation (Figure 3). In the first case, the patient was still treated with the same VKA, whereas in the second case, recurrence occurred after switching from fluindione to warfarin.
Fig. 2.

Renal function changes in the 13 patients during the follow-up after AKI.
Fig. 3.

INR value and SC concentration changes over time in the two patients (#4 and #10) with AKI recurrence.
Discussion
Here, we describe 13 cases of biopsy-proven AKI with tubular obstruction by RBCC of which 7 were caused by fluindione and 2 by acenocoumarol. To the best of our knowledge, these are the first reports of AKI with tubular obstruction due to fluindione. AKI with tubular obstruction by RBCC has recently been described in the USA, where warfarin is the only used VKA, and was therefore named WRN.
The clinical characteristics of our patients matched the WRN description. Specifically, gross hematuria was rapidly followed by AKI in a context of overanticoagulation in patients with an underlying kidney disease. The clinical features of patients treated with warfarin, fluindione or acenocoumarol were comparable. This suggests that AKI could be caused by different VKA molecules and not only by warfarin. In our series, one patient who switched from fluindione to warfarin after AKI developed a second AKI during an episode of overanticoagulation with warfarin. It is important to suspect this AKI etiology also in patients treated with fluindione or acenocoumarol, because AKI was severe and 46.2% of patients required dialysis. AKI prognosis also was severe. Indeed, two patients died and one patient still required dialysis 3 months after AKI. Recovery was delayed in one patient. In the last biopsy-proven study, prognosis was more severe than in our study with one death and three of their nine patients still undergoing dialysis [7]. The follow-up duration was not mentioned in this previous study and recovery could also have been delayed.
The diagnosis of AKI with tubular obstruction by RBCC is based on the kidney biopsy findings. This diagnosis was suspected in patients treated with warfarin. Conversely, it was retrospective in most patients who received fluindione and acenocoumarol. Indeed, diagnosis was immuno-allergic acute interstitial nephritis or ATN caused by treatment with a VKA before biopsy reviewing; however, the presence of gross hematuria and the discrepancy between AKI severity and the moderate interstitial lesions made this diagnosis uncertain. The review of the kidney biopsy results highlighted the presence of cortical and medullary tubular segment obstruction with RBCC (the hallmark of WRN), and the diagnosis was rectified. Histological analysis of the kidney biopsies also systematically found moderate ATN and these lesions were comparable in patients treated with warfarin, fluindione or acenocoumarol. Like in previous case reports, it is important to note that an underlying kidney disease, mainly IgA nephropathy [4–7], was found in almost all patients (except in one patient without any histological sign of kidney disease by optical microscopy analysis). Glomerular hemorrhage (AKI-initiating mechanism) would result from the association of glomerular permeability alterations (due to the underlying kidney disease) with overanticoagulation. The presence of an underlying kidney dysfunction is crucial. Indeed, in two animal models in which acute overanticoagulation was induced by overdose of brodifacoum [14] and warfarin [15], AKI only appeared in rats with 5/6 nephrectomy (inducing focal segmental glomerulosclerosis by nephron reduction), but not in the control group. We did not find any significant correlation between SC changes and the extent of INR increase. Moreover, two of our patients developed AKI by tubular obstruction with RBCC in the absence of overanticoagulation (the maximal INR values were 2.51 and 2.7, respectively). This could simply be due to the absence/loss of blood testing results when INR >3. However, it can be hypothesized that glomerular hemorrhage might appear independently of the anticoagulation level because of the glomerular barrier permeability alterations due to the underlying kidney disease. This hypothesis is supported by a rat model in which RBCC developed even when warfarin was used within the ‘therapeutic’ range (INR between 2 and 3) [15].
Two hypotheses had been proposed to explain the AKI mechanism. The first one is intratubular obstruction by RBCC. However, this mechanism has been questioned [16] based on a study in patients with glomerular diseases (IgA nephropathy, acute post-infectious glomerulonephritis, pauci-immune focal necrotizing glomerulonephritis) who developed gross hematuria and AKI. Analysis of the kidney biopsies revealed no retro-diffusion of the Tamm–Horsfall protein, which is secreted in the large ascendant loop of Henle, in the Bowman’s glomerular space. This is an indirect sign that intratubular obstruction was incomplete and that, therefore, it could not explain AKI on its own [17]. Heme toxicity was then proposed due to the systematic presence of ATN in histological findings. Hemoglobin released by intratubular degradation of red blood cells or hemoglobin directly filtered by the glomerulus may be incorporated in the proximal tubules through the megalin–cubilin receptor system or degraded in the tubular lumen, releasing heme-containing molecules and free iron. Cell-free hemoglobin promotes lipid peroxidation in the tubular lumen. Hemoglobin/heme/iron accumulation within tubular cells generates reactive oxygen species, leading to caspase activation and consequently to apoptosis, mitochondrial damage, up-regulation of vascular adhesion molecules and of pro-inflammatory/pro-fibrotic cytokines [18]. Tubular injury can be increased by hypoxic damage from intrarenal vasoconstriction due to the binding of heme-containing molecules to nitric oxide [19]. An experimental study [20] supports the model in which tubular toxicity by heme-induced oxidative stress is the main mechanism of AKI. Indeed, in rats with 5/6 nephrectomy and warfarin-induced overanticoagulation, treatment with the antioxidant N-AcetylCysteine (NAC) prevents AKI without any effect on hematuria or tubular RBCC development. It is unlikely that free iron plays a significant role in the pathogenesis of acute tubular injury because treatment with iron chelators does not improve renal function in these rats.
WRN had been reported also in two case reports of patients treated with dabigatran (a direct thrombin inhibitor that is increasingly used as an alternative to warfarin) [21, 22]. Moreover, SC increase, hematuria and obstructive tubular RBCC are observed in 5/6 nephrectomy rats treated with dabigatran and an inhibitor of proteinase-activated receptor-1 (PAR-1; a thrombin receptor expressed in endothelial cells) [23]. The authors hypothesized a common physiopathology pathway between WRN and AKI caused by dabigatran. PAR-1 inhibition, due to thrombin activity decrease, could play an important role in glomerular permeability and could be responsible for the glomerular hemorrhage that seems to be the AKI-initiating mechanism. In agreement with these recent publications, Wheeler etal. have described AKI induced by VKA and thrombin inhibitor as ‘anticoagulant-related nephropathy’ [24].
New therapeutic approaches need to be considered because of AKI severity. Vitamin K treatment prevents intratubular RBCC development and AKI occurrence in a WRN rat model with 5/6 nephrectomy [15]. Vitamin K decreases the gross hematuria duration by rapid INR reduction. Gross hematuria duration had been identified as a prognostic factor of AKI recovery in patients with IgA nephropathy without VKA treatment [25]. Moreover, by decreasing oxidative stress induced by hemoglobin toxicity, NAC prevents AKI development in 5/6 nephrectomy rats after over-anticoagulation with warfarin [20]. It is now important to test these treatment options in pre-clinical/clinical trials.
Conclusion
We describe here the largest biopsy-proven case series of AKI by tubular obstruction with RBCC induced by VKA, particularly the first cases of AKI by fluindione. Clinical and histological data were not different in patients treated with warfarin or fluindione. In agreement with a recent publication [24], the term ‘anticoagulant-related nephropathy’ appears to be more appropriate than WRN. Moreover, the appearance of gross hematuria during treatment with any VKA needs rapid INR reduction and close monitoring of the renal function.
Acknowledgements
The authors thank Elisabetta Andermarcher for English reviewing, and Laurent Doucet, François Comoz, Marie-Christine Machet, Dominique Nochy and Isabelle Brocheriou for sharing kidney biopsies from our centers.
Conflict of interest statement
None declared.
References
- 1.Rapport sur les anticoagulants en France en 2014: Etat des lieux, synthèse et surveillance. http://ansm.sante.fr/content/download/61981/795269/version/2/file/ANSM-rapport_NACOs-avril+2014.pdf (22 April 2014, date last accessed)
- 2. Reynaud F, Giraud P, Cisterne J-M. et al. Acute immuno-allergic interstitial nephritis after treatment with fluindione. Seven cases. Néphrol Thér 2009; 5: 292–298 [DOI] [PubMed] [Google Scholar]
- 3. Cam G, Kwetcheu AT, Vigneau C. et al. Acute and chronic nephropathy induced by fluindione must be addressed. Nephrol Dial Transplant 2012; 27: 1554–1558 [DOI] [PubMed] [Google Scholar]
- 4. Abt AB, Carroll LE, Mohler JH.. Thin basement membrane disease and acute renal failure secondary to gross hematuria and tubular necrosis. Am J Kidney Dis 2000; 35: 533–536 [DOI] [PubMed] [Google Scholar]
- 5. August C, Atzeni A, Köster L. et al. Acute renal failure in IgA nephropathy: aggravation by gross hematuria due to anticoagulant treatment. J Nephrol 2002; 15: 709–712 [PubMed] [Google Scholar]
- 6. Kabir A, Nadasdy T, Nadasdy G. et al. An unusual cause of gross hematuria and transient ARF in an SLE patient with warfarin coagulopathy. Am J Kidney Dis 2004; 43: 757–760 [DOI] [PubMed] [Google Scholar]
- 7. Brodsky SV, Satoskar A, Chen J. et al. Acute kidney injury during warfarin therapy associated with obstructive tubular red blood cell casts: a report of 9 cases. Am J Kidney Dis 2009; 54: 1121–1126 [DOI] [PubMed] [Google Scholar]
- 8. Brodsky SV, Nadasdy T, Rovin BH. et al. Warfarin-related nephropathy occurs in patients with and without chronic kidney disease and is associated with an increased mortality rate. Kidney Int 2011; 80: 181–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. An JN, Ahn SY, Yoon C-H. et al. The occurrence of warfarin-related nephropathy and effects on renal and patient outcomes in Korean patients. PloS One 2013; 8: e57661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Martín Cleary C, Moreno JA, Fernández B. et al. Glomerular haematuria, renal interstitial haemorrhage and acute kidney injury. Nephrol Dial Transplant 2010; 25: 4103–4106 [DOI] [PubMed] [Google Scholar]
- 11. Kellum JA, Lameire N, Aspelin P. et al. KDIGO Clinical Practice Guideline for acute kidney injury 2012. Kidney Int Suppl 2012; 2: 1–138 [Google Scholar]
- 12. Levey AS, Bosch JP, Lewis JB. et al. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease Study Group. Ann Intern Med 1999; 130: 461–470 [DOI] [PubMed] [Google Scholar]
- 13. National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis 2002; 39: S1–S266 [PubMed] [Google Scholar]
- 14. Ware K, Brodsky P, Satoskar AA. et al. Warfarin-related nephropathy modeled by nephron reduction and excessive anticoagulation. J Am Soc Nephrol 2011; 22: 1856–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ozcan A, Ware K, Calomeni E. et al. 5/6 nephrectomy as a validated rat model mimicking human warfarin-related nephropathy. Am J Nephrol 2012; 35: 356–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Logan IR, Sheerin NS.. Anticoagulation and kidney injury: rare observation or common problem? J Nephrol 2013; 26: 603–605 [DOI] [PubMed] [Google Scholar]
- 17. Fogazzi GB, Imbasciati E, Moroni G. et al. Reversible acute renal failure from gross haematuria due to glomerulonephritis: not only in IgA nephropathy and not associated with intratubular obstruction. Nephrol Dial Transplant 1995; 10: 624–629 [PubMed] [Google Scholar]
- 18. Moreno JA, Martín-Cleary C, Gutiérrez E. et al. AKI associated with macroscopic glomerular hematuria: clinical and pathophysiologic consequences. Clin J Am Soc Nephrol 2012; 7: 175–184 [DOI] [PubMed] [Google Scholar]
- 19. Heyman SN, Brezis M.. Acute renal failure in glomerular bleeding: a puzzling phenomenon. Nephrol Dial Transplant 1995; 10: 591–593 [PubMed] [Google Scholar]
- 20. Ware K, Qamri Z, Ozcan A. et al. N-acetylcysteine ameliorates acute kidney injury but not glomerular hemorrhage in an animal model of warfarin-related nephropathy. Am J Physiol Renal Physiol 2013; 304: F1421–F1427 [DOI] [PubMed] [Google Scholar]
- 21. Moeckel GW, Luciano RL, Brewster UC.. Warfarin-related nephropathy in a patient with mild IgA nephropathy on dabigatran and aspirin. Clin Kidney J 2013; 6: 507–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Escoli R, Santos P, Andrade S. et al. Dabigatran-related nephropathy in a patient with undiagnosed IgA nephropathy. Case Rep Nephrol 2015; 2015: 298261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ryan M, Ware K, Qamri Z. et al. Warfarin-related nephropathy is the tip of the iceberg: direct thrombin inhibitor dabigatran induces glomerular hemorrhage with acute kidney injury in rats. Nephrol Dial Transplant 2013; 29: 2228–2234 [DOI] [PubMed] [Google Scholar]
- 24. Wheeler DS, Giugliano RP, Rangaswami JR.. Anticoagulation-related nephropathy. J Thromb Haemost 2015; 14: 461–467 [DOI] [PubMed] [Google Scholar]
- 25. Gutiérrez E, González E, Hernández E. et al. Factors that determine an incomplete recovery of renal function in macrohematuria-induced acute renal failure of IgA nephropathy. Clin J Am Soc Nephrol 2007; 2: 51–57 [DOI] [PubMed] [Google Scholar]