

Highlights
-
•
Sensitive DNA markers for identification of plant species are highly desirable.
-
•
Ubiquitin ligase gene region was first found to be useful for DNA marker development.
-
•
Ubiquitin ligase gene region will be another good source for DNA marker development.
-
•
Our methodology will improve evaluation and conservation of plant genetic diversity.
Keywords: Biotechnological development, DNA marker, Genetic diversity detection
Abstract
Development of more sensitive nuclear DNA markers for identification of species, particularly closely allied taxa has been a challenging task that has attracted interest from scientists in fields of biotechnological development and genetic diversity detection. In this study, the sequence of the ubiquitin ligase gene (UBE3) region of nuclear DNA was tested for applicability and efficacy in revealing genetic diversity of walnut resources, with an emphasis on inter- and intra-specific levels. Analysis on genetic relationship among the taxa was conducted with the neighbor-joining (NJ) method. The number of variable bases in the UBE3 region was 20 sites. All nine taxa (species/variety/cultivars) were distinguished using the UBE3 sequence. In addition, each taxon was characterized molecularly with a unique nucleotide molecular formula using ten variable base sites derived from the nuclear DNA UBE3 gene sequence. This study presents a good complementary methodology for developing new DNA markers for identification of genus Juglans.
1. Introduction
The internal transcribed spacer (ITS) sequence of nuclear ribosomal DNA with bi-parental inheritance is currently used widely to determine the genetic diversity of land plants [1], [2], [3], [4], [5], [6], [7], [8], [9], [10]. However, one limitation of species identification using the ITS sequence is that the method has limited resolution in identifying species, especially within closely related taxa [1], [2], [3], [4], [5], [6], [7], [8], [9], [10], [11]. For instance, the discriminating power of the four recommended DNA markers at the species level in Alnus (Betulaceae) was 10% (rbcL), 31.25% (matK), 63.6% (trnH–psbA), and 76.9% (ITS) [3]. Among the four DNA regions (rbcL, matK, trnH–psbA, and ITS), the ITS sequence has the most variable information, and appears to have limited power to discriminate closely related taxa in Juglandaceae [5]. Hanabusaya and Adenophora sect. Remotiflorae of the family Campanulaceae could not be resolved using ITS sequences [6]. As a result of insufficient morphological information and DNA markers, the development of scientific research has been severely hindered in fields such as taxonomy, ecology, and genetic resource evaluation. Development of new and more sensitive nuclear DNA markers for biodiversity detection is highly desirable [11], [12].
The ubiquitin–proteasome system, which plays a key role in degradation of proteins, is imperative for maintaining the cellular homeostasis in eukaryotic cells [13]. Three enzymes are required in the ubiquitination and targeting of proteins for degradation: ubiquitin activating enzyme E1, ubiquitin conjugating enzyme E2, and the highly conserved ubiquitin ligase E3. Ubiquitin ligases are key components of the ubiquitin–proteasome system. After a protein has been ubiquitinated, the substrate protein will be located to the proteasome (a cylindrical complex) to be degraded into smaller polypeptides or other molecules with biological activity for reutilization [13].
Ubiquitin plays a vital role not only in protein degradation but also in many cellular functions including DNA repair processes, cell cycle regulation, cell growth, immune system functionality, and hormone-mediated signaling in plants. In recent years, several types of the ubiquitin ligase E3, such as RING finger-containing E3s (RBR and TRIM families), cullin-containing E3 complexes, Ubox E3s and HECT E3s, were reported [14]. However, few reports are available concerning development of nuclear DNA markers from the genomic regions in relation to the ubiquitin–proteasome system.
In this study, we used plant material from Juglandaceae to develop a new nuclear DNA marker within the ubiquitin ligase gene (UBE3) region to discriminate the representative samples (species/variety/cultivars) of the genus Juglans. Our objectives were: (i) to test the applicability of the nuclear DNA marker from the UBE3 gene region; and (ii) to evaluate the resolution ability of the nuclear DNA marker from the UBE3 gene region. The results of this effort show that UBE3 is sensitive for characterizing genetic diversity in the family Juglandaceae.
2. Materials and methods
2.1. Plant materials
Nine representative taxa of the genus Juglans and two outgroups (Cyclocarya paliurus and Pterocarya stenoptera in Juglandaceae) were used in this study (Table 1). The eleven taxa were sampled from three places: the resources nursery (N 34°18′, E 111°30′) of Forestry Bureau of Luoning County, Henan Province, China; the Arboretum (N 25°08′, E 102°45′) of Forestry Academy of Yunnan Province, located at Heilongtan in the northern suburbs of Kunming City, Yunnan, China; and Beijing Botanical Garden (N 39°48′, 116°28′) under the Institute of Botany, Chinese Academy of Sciences, Beijing, China. All necessary permits for the collection of fresh leaves from the trees growing at each place were acquired prior to material collection. All collected material was verified by a taxonomic expert. Fresh leaves of each accession were collected in the spring and dried immediately using silica gel for future DNA extraction.
Table 1.
Materials used in this study.
| No. | Taxon | Rank | Section | Place of collection |
|---|---|---|---|---|
| 1 | Juglans regia ‘Zha 343′ | Cultivar | Juglans | * |
| 2 | Juglans sigillata | Species | Juglans | ** |
| 3 | Juglans sigillata ‘Lushui 1Hao’ | Cultivar | Juglans | ** |
| 4 | Juglans cathayensis | Variety | Cardiocaryon | ** |
| 5 | Juglans mandshurica | Species | Cardiocaryon | ** |
| 6 | Juglans hindsii | Species | Rhysocaryon | * |
| 7 | Juglans major | Species | Rhysocaryon | * |
| 8 | Juglans microcarpa | Species | Rhysocaryon | * |
| 9 | Juglans nigra | Species | Rhysocaryon | * |
| 10 | Pterocarya stenoptera | Species | *** | |
| 11 | Cyclocarya paliurus | Species | *** |
Resources Nursery, Forsetry Bureau of Luoning County, Henan, China.
Forestry Academy of Yunnan Province, Kunming City, Yunnan, China
Beijing Botanical Garden, Institute of Botany, Chinese Academy of Sciences, Beijing, China.
2.2. DNA extraction, PCR amplification and sequencing
Total genomic DNA was extracted using the Plant Genomic DNA Kit (DP305) from Tiangen Biotech (Beijing) Co., Ltd. China. The nuclear DNA UBE3 gene locus was amplified using the primer pair H_UBE3_23f (5′-TCGCCTCCAAGTTCAGTG-3′) and H_UBE3_838r (5′-CTCCCATAGGTGTAGTTCCA-3′). Taq DNA polymerase and PCR buffer (TaKaRa Code: DR100B) were from TaKaRa Biotechnology Co., Ltd. (Dalian, China). The PCR protocol were as follows: preheating at 94 °C for 4 min, 34 cycles at 94 °C for 45 s, annealing at 52 °C for 45 s and elongation at 72 °C for 1.2 min, followed by a final extension at 72 °C for 10 min. PCR amplification of the regions of interest was performed in an Applied Biosystems VeritiTM 96-Well Thermal Cycler (Model#: 9902, made in Singapore). The amplicons were resolved simultaneously on 2% agarose gels (Promega, the USA) run in 1 × TAE buffer at 3 V cm−1 for 2.5 h and were stained with ethidium bromide. The fragments (PCR products) were directly sequenced with the same primer pair mentioned above using a 3730xl DNA analyzer (Applied Biosystems, Foster City, CA, USA).
2.3. Data analysis
The DNA sequences were aligned with ClustalX [15] and then were manually confirmed using Sequencher (v4.6) software. Sequence haplotype diversity was calculated using DnaSP (DNA Sequences Polymorphism version 5.10.01) software [16]. Sequence datasets were analyzed using Mega 6 software [17]. Tamura’s 3-parameter model was found to be the best nucleotide substitution model for the UBE3 sequences with Mega 6. The analysis on genetic relationship among the taxa was conducted in Mega 6 using neighbor-joining (NJ) method. Bootstrapping was done with 1000 replicates. All positions containing gaps and missing data were treated with pairwise deletion option. The sequences of the accessions were deposited in GenBank (GenBank accession numbers: KF994007-KF994018).
3. Results
3.1. Identification and confirmation of the DNA sequences
The nuclear DNA UBE3 sequences (Fig. S1) obtained in this study were confirmed to be part of the coding region of the UBE3 gene, as shown using BLAST to compare the sequence to that in the GenBank database. Additional comparisons were made using the E3 ubiquitin-protein ligase sequences of Prunus mume (GenBank accession no. XM_003607148.1) and several other plant species (GenBank accession no. XM_007199611.1, XM_003537761.2, XM_004505735.1, and XM_003607148.1). At least three independent samples from each species or cultivar have been sequenced, identical results were obtained. Therefore, only one sample data was used to represent each taxon.
3.2. Genetic variation of the UBE3 region among the taxa
According to the UBE3 sequence dataset, eight variable base sites were detected at inter- and intra-specific levels. These variable sites were found at nucleotide positions 46, 125, 205, 227, 459, 562, 595 and 663, and represent 40% of the total variable sites detected. The closely related taxa within each section are successfully discriminated (Table 2; Fig. S1).
Table 2.
The twenty variable base sites detected in the nuclear DNA ubiquitin ligase E3 gene (UBE3) region among the nine taxa in Juglans L.
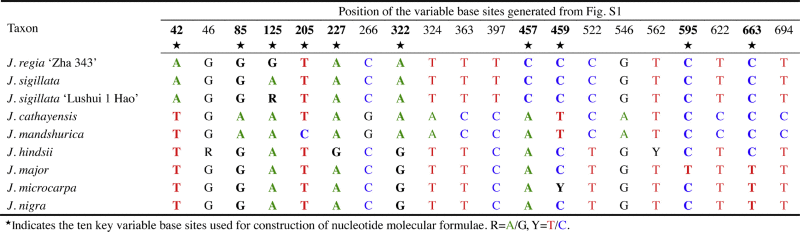 |
The remaining 12 variable sites that were unique at the section level comprise 60% of the total (Table 2; Fig. S1). They can be classified into three categories: (i) two variable sites unique to Juglans sect. Juglans, No. 42 and 397 (10% of the total); (ii) seven variable sites unique to J. sect. Cardiocaryon, No. 85, 266, 324, 363, 546, 622 and 694 (35% of the total); and (iii) two variable sites unique to J. sect. Rhysocaryon, No. 322 and 522 (10% of the total) (Table 2; Fig. S1).
Genetic differentiation was about 55–65% of the total between J. sect. Juglans and sect. Cardiocaryon, 30–45% of the total between J. sect. Juglans and J. sect. Rhysocaryon, 55–70% of the total between J. sect. Cardiocaryon and J. sect. Rhysocaryon (Table S2; Fig. S1). The genetic variation was 5% of the total either within J. sect. Juglans or J. sect. Cardiocaryon, but ranged from 5–70% of the total within J. sect. Rhysocaryon (Table S2; Fig. S1).
3.3. Genetic relationship revealed by the UBE3 sequence
Neighbor-joining (NJ), maximum likelihood (ML) and maximum parsimony (MP) algorithms were tried. However, the NJ method showed the best resolution, and significant differences existed between the methods. Thus, only NJ trees are presented. Twenty variable sites were detected according to the aligned UBE3 sequences (Table 2; Table S1; Fig. S1). Using NJ analysis and UBE3 sequences, all nine taxa (species/variety/cultivars) within the three sections were uniquely identified, and the outgroups were placed at reasonable positions outside the genus Juglans. Bootstrap support values were more than 50% at each clade (Fig. 1). This topology was identical to the classification based on their morphology and habits [10], [18], [19], [20], [21], [22], [23]. Pairwise distances are shown in Table S3.
Fig. 1.

The neighbor-joining (NJ) tree generated based on the UBE3 gene sequences showing a success in discrimination at species, variety and cultivar levels. Numbers above the branches are bootstrap support values (%) for each clade with 1000 replicates.
3.4. Construction of molecular taxonomic key based on nucleotide molecular formulae
The UBE3 sequence dataset was employed for construction of the nucleotide molecular formulae (NMF). The 724 bp aligned sequence corresponds to the DNA tract from bases 15 to 738 of the entire sequence of the UBE3 fragment from the 5′ end and includes all the variable sites of this region (Table 2; Fig. S1). The position number of each variable site used in the formula was determined according to the newly generated 724 bp-length sequence alignment. The ten polymorphic base sites used in the NMF of the taxa for the genus Juglans are No. 42, 85, 125, 205, 227, 322, 457, 459, 595 and 663 (Table 2; Fig. S1). For instance, “Nuclear_DNA_UBE3_cds” was used to refer to the coding region of the nuclear UBE3 gene employed in the NMF and “aln_724 bp” refers to the aligned sequence length (724 bp) of the nine representative species/variety/cultivars in Juglans L. As a result, “Nuclear_DNA_UBE3_cds_aln_724bp_ 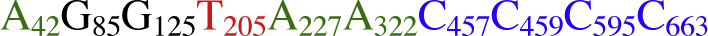 ” can be constructed as an NMF for molecularly characterizing the cultivar Juglans regia ‘Zha 343’, with the figure following the nucleotide character indicating the position of the corresponding polymorphic base site from the 5′ end of the aligned sequence [24]. The NMF can be constructed in a similar way for the rest of the samples of the genus Juglans and the outgroups. “Nuclear DNA_UBE3_cds_aln_724bp_” is omitted to save space in the description below. “Type
” can be constructed as an NMF for molecularly characterizing the cultivar Juglans regia ‘Zha 343’, with the figure following the nucleotide character indicating the position of the corresponding polymorphic base site from the 5′ end of the aligned sequence [24]. The NMF can be constructed in a similar way for the rest of the samples of the genus Juglans and the outgroups. “Nuclear DNA_UBE3_cds_aln_724bp_” is omitted to save space in the description below. “Type  ”, for example, in the following taxonomic key, refers to the taxon/taxa with
”, for example, in the following taxonomic key, refers to the taxon/taxa with  –typed base mutation, i.e., nucleotide A can be detected at base position 42 from the 5′ end in the UBE3 region. Other types of base mutation are indicated in the same way.
–typed base mutation, i.e., nucleotide A can be detected at base position 42 from the 5′ end in the UBE3 region. Other types of base mutation are indicated in the same way.
As shown in Fig. 2, a novel taxonomic key based on nucleotide molecular formulae is constructed by which the molecular feature of each taxon is given.
Fig. 2.

Molecular taxonomic key compiled based on nucleotide molecular formulae derived from the UBE3 sequence.
4. Discussion
Plants of Juglans sect. Cardiocaryon are precious tree species for high quality wood production. J. mandshurica and J. cathayensis are closely related taxa in Juglans sect. Cardiocaryon. J. mandshurica is mostly distributed in provinces of North and Northeast China, where the climate is colder. J. cathayensis is mainly distributed in warmer provinces of South and Southwest China [19], [20], [23]. The four black walnut species of Juglans sect. Rhysocaryon are closely related with each other, with some presence in North America as well [18], [19], [20], [21], [22], [23].
Members of Juglans sect. Juglans are economically important tree species for edible walnut production. The distribution of Juglans sigillata and J. sigillata ‘Lushui 1Hao’ is limited to Southwest China (mainly Yunnan Province) [19], [20], [23]. J. sigillata ‘Lushui 1 Hao’ is a traditional local cultivar with an annual nut production of more than 1.0 × 108 kg. In contrast, the annual nut production of J. sigillata is around 4.0 × 108 kg. They are the major walnut trees cultivated in Yunnan Province, China. J. sigillata ‘Lushui 1Hao’ prefers a warmer climate with higher humidity for normal growth compared to J. sigillata. Fruit maturation time of J. sigillata ‘Lushui 1Hao’ is about 15 days earlier than that of J. sigillata. There is almost no difference in floral morphology between them. J. sigillata ‘Lushui 1Hao’ possesses 9–11 leaflets in the odd-pinnate leaf without obvious degradation of the terminal leaflet, whereas J. sigillata has 9–13 leaflets in its odd-pinnate leaf whose terminal leaflet degraded significantly [19], [20], [23].
Nearly 2.0 × 109 kg of the annual walnut production in China is provided by J. regia. In fact, J. regia ‘Zha 343’ is a major walnut cultivar in Xinjiang Uygur Autonomous Region, China. In the Yunnan Province, the growth of J. regia gradually becomes weaker after planting because the local climate averages lower temperature and higher humidity than what is required by the species. Thus, in China, J. regia is mainly cultivated in the walnut distribution area outside the Southwest, although plants of J. regia can be seen in Yunnan Province.
Generally, the greater the number of informative base sites available, the higher discrimination efficiency should be achieved during genetic diversity detection. One of the important tasks in DNA marker development is to seek DNA regions with a large number of variable base sites [19], [20], [23]. However, when compared to researches on genetic variations at the family, genus, or section level, development of nuclear DNA marker covering lower taxa is time consuming and expensive [19], [20], [23].
The key to increasing the discrimination ability of a locus is commonly to obtain more variable sites that contribute genetic variations at inter- and intra-specific levels. Here, the three taxa of Juglans sect. Juglans were chosen to represent the genetic variation between closely related species (J. sigillata and J. regia) and between cultivars (J. sigillata ‘Lushui 1Hao’ and J. regia ‘Zha 343’) and to test the ability of the variable genomic region to correctly discriminate between them.
Only half (10 sites) of the variable sites from the UBE3 region were needed to uniquely identify all the nine taxa of Juglans (Table 2, Fig. S1), showing a high efficacy in revealing genetic diversity of walnut resources. Our results suggest that the UBE3 sequence is good and useful in both discrimination ability and revealing genetic relationship (Fig. 1). Interestingly, our results suggested that the discrimination ability does not directly correlate with the number of variable sites or informative sites.
The UBE3 DNA marker discovered in this study is easy to amplify and sequence. Additionally, insertion and deletions are rare in this locus because it is a coding region. In this study, Juglans sect. Juglans was determined to be basal, while Juglans sect. Rhysocaryon was a more advanced section of the genus Juglans, based on the UBE3 sequence data (Fig. 1). These results are identical to those acquired by the classification based on morphology and previous studies [10], [11], [12], [13], [19], [20], [22], [23].
The length of the UBE3 gene related DNA region is at least 5905 bp in Prunus persica (GenBank accession no. XM_007199611.1), 5955 bp in Medicago truncatula (GenBank accession no. XM_003607148.1), 6473 bp in Glycine max (GenBank accession no. XM_003537761.2), 6488 bp in Prunus mume (GenBank accession no. XM_008237787.1), and 6622 bp in Cicer arietinum (GenBank accession no. XM_004505735.1). The UBE3 gene related DNA sequence data of plant species is growing rapidly in GenBank. There is a great potential for developing more DNA markers with high sensitivity from the UBE3 gene related DNA region for the global detection of genetic diversity in walnut resources.
The identification method using nucleotide molecular formulae, as used here, is simple for widespread use. Because the ubiquitin–proteasome system and its associated DNA regions are present in all eukaryotes, these findings represent a good complementary source for development of nuclear DNA markers for genetic diversity detection, covering both inter-specific and intra-specific levels, and will promote evaluation, conservation, and utilization of plant resources and other organisms.
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgements
This study was financially supported by the Chinese Special Fund Project for the Scientific Research of the Forest Public Welfare Industry (Project No. 201004048) from the State Forestry Administration of China and by the National Natural Science Foundation of China (Grant No. 30972412). The expert who instructed the identification of samples used in this study is Prof. Runquan Dong and Yu Zhang of the Forestry Academy of Yunnan Province, Huzhi Xu, professor and former director of Forsetry Bureau of Luoning County, Henan, China. The authors thanks Wenyu Ma, Chengqian Wang, Fengmin Li, Peng Wang, Zhiguo Li, Zhihong Ding, Weiwei Gao, Hao Liu, Qingguo Ma, Xianlan Li, Bin Lu and Ping Zhao for their kind help in field investigations, material collections and discussions. We are sincerely grateful to three anynomous referees for their thoughtful and meaningful comments.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.btre.2014.11.003.
Contributor Information
Zhili Suo, Email: zlsuo@ibcas.ac.cn, zlsuobj@126.com.
Dong Pei, Email: 1365388727@qq.com.
Appendix A. Supplementary data
The following are Supplementary data to this article:
References
- 1.Kress W.J., Wurdack K.J., Zimmer E.A., Weigt L.A., Janzen D.H. Use of DNA barcodes to identify flowering plants. Proc. Natl. Acad Sci. U. S. A. 2005;102:8369–8374. doi: 10.1073/pnas.0503123102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sass C., Little D.P., Stevenson D.W., Specht C.D. DNA barcoding in the cycadales: Testing the potential of proposed barcoding markers for species identification of cycads. PLoS One. 2007;2:e1154. doi: 10.1371/journal.pone.0001154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ren B.Q., Xiang X.G., Chen Z.D. Species identification of Alnus (Betulaceae) using nrDNA and cpDNA genetic markers. Mol. Ecol. Resour. 2010;10:594–605. doi: 10.1111/j.1755-0998.2009.02815.x. [DOI] [PubMed] [Google Scholar]
- 4.Yao H., Song J.Y., Liu C., Luo K., Han J.P., Li Y., Pang X., Xu H., Zhu Y., Xiao P., Chen S. Use of ITS2 region as the universal DNA barcode for plants and animals. PLoS One. 2010;5:e13102. doi: 10.1371/journal.pone.0013102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xiang X.G., Zhang J.B., Lu A.M., Li R.Q. Molecular identification on species in Juglandaceae: a tiered method. J. Syst. Evol. 2011;49(3):252–260. [Google Scholar]
- 6.Cheon K.S., Yoo K.O. Phylogeny of Hanabusaya (Campanulaceae), a Korean endemic, based on ITS sequences of nuclear ribosomal DNA. J. Syst. Evol. 2013;51(6):704–714. [Google Scholar]
- 7.Tripathi A.M., Tyagi A., Kumar A., Singh A., Singh S., Chaudhary L.B., Roy S. The internal transcribed spacer (ITS) region and trnH–psbA are suitable candidate loci for DNA barcoding of tropical tree species of India. PLoS One. 2013;8(2):e57934. doi: 10.1371/journal.pone.0057934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stanford A.M., Harden R., Parks C.R. Phylogeny and biogeography of Juglans (Juglandaceae) based on matK and ITS sequence data. Am. J. Bot. 2000;87:872–882. [PubMed] [Google Scholar]
- 9.Potter D., Gao R.Y., Baggett S., Mckenna J.R., McGranahan G.H. Defining the sources of paradox: DNA sequence markers for North American walnut (Juglans L.) species and hybrids. Sci. Hort. 2002;94:157–170. [Google Scholar]
- 10.Stone D.E., Oh S.H., Tripp E.A., Rios G.L.E., Manos P.S. Natural history distribution, phylogenetic relationships, and conservation of Central American black walnuts (Juglans sect. Rhysocaryon) J. Torrey Bot. Soc. 2009;136:1–25. [Google Scholar]
- 11.Ciarmiello L.F., Pontecorvo G., Piccirillo P., Luca A.D., Carillo P., Kafantaris I., Woodrow P. Use of nuclear and mitochondrial single nucleotide polymorphisms to characterize English walnut (Juglans regia L.) genotypes. Plant Mol Biol Rep. 2013;31:1116–1130. [Google Scholar]
- 12.Cosmulescu S., Botu M. Walnut biodiversity in south-western Romania-resource for perspective cultivars. Pak. J. Bot. 2012;44(1):307–311. [Google Scholar]
- 13.Ganoth A., Tsfadia Y., Wiener R. Ubiquitin: molecular modeling and simulations. J. Mol. Graphics Modell. 2013;46:29–40. doi: 10.1016/j.jmgm.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 14.Marin I. Evolution of plant hect ubiquitin ligases. PLoS One. 2013;8(7):e68536. doi: 10.1371/journal.pone.0068536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Larkin M.A., Blackshields G., Brown N.P., Chenna R., McGettigan P.A., McWilliam H., Valentin F., Wallace I.M., Wilm A., Lopez R., Thompson J.D., Gibson T.J., Higgins D.G. Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 16.Librado, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 17.Tamura K., Stecher G., Peterson D., Filipski A., Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gunter L.E., Kochert G., Giannasi D.E. Phylogenetic relationships of the Juglandaceae. Plant Syst. Evol. 1994;192:11–29. [Google Scholar]
- 19.Lu A.M. The geographical dispersal of Juglandaceae. Acta Phytotaxon. Sin. 1982;20:257–271. [Google Scholar]
- 20.Lu A.M., Stone D.E., Grauke L.J. Juglandaceae. In: Wu Z.Y., Raven P.H., editors. Flora of China. Science Press, Beijing, and Missori Botanical Garden Press; St. Louis, Missori, USA: 1999. pp. 277–285. [Google Scholar]
- 21.Aradhya M.K., Potter D., Gao F., Cimon C.J. Molecular phylogeny of Juglans (Juglandaceae): abiogeographic perspective. Tree Genet Genomes. 2007;3:363–378. [Google Scholar]
- 22.Manning W.E. The classification within the Juglandaceae. Ann. Mo. Bot. Gard. 1978;65:1058–1087. [Google Scholar]
- 23.Pei D., Lu X.Z. China Forestry Publishing House; Beijing: 2011. Walnut Germplasm Resources In China (In Chinese) pp. 1–208. [Google Scholar]
- 24.Suo Z.L., Zhang C.H., Zheng Y.Q., He L.X., Jin X.B., Hou B.X., Li J.J. Revealing genetic diversity of tree peonies at micro-evolution level with hyper-variable chloroplast markers and floral traits. Plant Cell Rep. 2012;31:2199–2213. doi: 10.1007/s00299-012-1330-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.