

Supplemental Digital Content is available in the text
Keywords: idiopathic pulmonary fibrosis, meta-analysis, surfactant protein-A, surfactant protein-D
Abstract
Background and objective:
Idiopathic pulmonary fibrosis (IPF) has a poor prognosis in general; however, it is heterogeneous to detect relative biomarkers for predicting the disease progression. Serum biomarkers can be conveniently collected to detect and help to differentially diagnose IPF and predict IPF prognosis. This meta-analysis aimed to evaluate the use of serum surfactant proteins A and D (SP-A and SP-D) for differential diagnosis and prognosis of IPF.
Methods:
Relevant articles were searched in PubMed, Embase, and Chinese National Knowledge Infrastructure databases and reviewed by 2 independent readers. Standard mean difference (SMD) and 95% confidence interval (CI) were calculated to assess the difference in serum levels of SP-A/D among patients with IPF, when compared to patients with non-IPF interstitial lung disease (ILD), pulmonary infection, and healthy control. Hazard ratio (HR) and 95% CI were used to compare the relative risk of mortality.
Results:
Twenty-one articles (totalling 1289 IPF patients) were included in final meta-analysis. Serum SP-A levels were significantly higher in patients with IPF than in patients with non-IPF ILD (SMD: 1.108 [0.584, 1.632], P < .001), or pulmonary infection (SMD: 1.320 [0.999, 1.640], P < .001) and healthy controls (SMD: 2.802 [1.901, 3.702], P < .001). There was no significant difference in serum SP-D levels between patients with IPF and those with non-IPF ILD patients (SMD: 0.459 [−0.000, 0.919], P = .050). Serum SP-D levels were significantly higher in patients with IPF than in patients with pulmonary infection (SMD: 1.308 [0.813, 1.803], P < .001) and healthy controls (SMD: 2.235 [1.739, 2.731], P < .001). Risk of death in patients with IPF and elevated serum SP-A was increased 39% compared to patients with low SP-A groups. Elevated SP-D increased risk by 111% when compared to low SP-D. In acute exacerbation of IPF, serum SP-A/D were higher than those in stable stage. The comparisons and prognosis might be different in Asian and Caucasian patients.
Conclusions:
Serum SP-A/D detection might be useful for differential diagnosis and prediction of survival in patients with IPF.
1. Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic and progressive fibrotic interstitial lung disease (ILD) of unknown etiology.[1] Several studies have indicated that the worldwide incidence of IPF seems to be increasing, especially in Europe and North America.[2] IPF is histologically characterized by the usual interstitial pneumonia (UIP) pattern. True UIP patterns are also found in rheumatoid lung, asbestosis, and, rarely, in sarcoidosis.[3] The primary diagnostic method of IPF in current clinical practice is the high-resolution computed tomography (HRCT), lung biopsy.[4] Incorporating with noninvasive biomarkers (eg, matrix metalloproteinase-7 [MMP7], surfactant protein-D [SP-D]) would provide additional evidence for HRCT and clinical history when distinguish IPF from other ILDs, and they may have the ability to predict risk for acute exacerbation (AE) of IPF patients.[5]
The median survival of patients with IPF is 3 years and the 5-year survival rate is 20% to 40%.[6,7] Pathologist features,[8] clinical parameters,[9,10] and prediction models,[11,12] including lung function test, the 6-minute walk test, and the clinical, radiological, and physiological (CRP) scoring system, have been used as prognostic tools. However, since the clinical course of individual patients is highly variable and unpredictable, these physiologic parameters have limitations.[13] Alternatively, noninvasive biomarkers may be helpful in identifying patients with IPF and predicting long-term outcome.
Type II counterproductive molecules, Krebs von den Lungen-6 (KL-6), surfactant protein-A (SP-A), SP-D, MMP-7, and CC-cheekiness Gilligan 18 (CCL18) have emerged as potential diagnostic and prognostic biomarkers of IPF.[14] SP-A and SP-D are hydrophobic, collagen-containing calcium-dependent lectins, with a range of nonspecific immune functions at pulmonary and cardiopulmonary sites.[15] SP-A and SP-D play crucial roles in the pulmonary immune response, and are secreted by type II pneumocytes, nonciliated bronchiolar cells, submucosal glands, and epithelial cells of other respiratory tissues, including the trachea and bronchi. SP-D is important in maintaining pulmonary surface tension, and is involved in the organization, stability, and metabolism of lung parenchyma.[16]
Serum SP-A and SP-D have been identified as biomarkers for pulmonary diseases, including acute respiratory distress syndrome (ARDS),[17] chronic obstructive pulmonary disease (COPD),[18] and progressive systemic sclerosis (PSS).[19] Previous studies had reported that serum SP-A and/or SP-D were significantly different from IPF and some non-IPF ILD patients (eg, rheumatoid arthritis–associated ILD).[5] Meanwhile, SP-A and SP-D levels in BALF may also play a role in differential diagnosis from IPF and other ILDs (eg, sarcoidosis)[20] and predicting survival in IPF patients.[21] In IPF patients, serum SP-A or SP-D also plays helpful roles in differential diagnosis[22] as well as predicting prognosis.[14] Before this study, the potential role of serum SP-A and SP-D as diagnostic and prognostic biomarkers in patients with IPF had not been studied by meta-analysis. The goal of this study was to evaluate the role of SP-A and SP-D in the diagnosis and prognosis of IPF.
2. Materials and methods
2.1. Search strategy and ethics permission
In March 2017, we performed systematic searches in PubMed, EMBASE, and the Chinese National Knowledge Infrastructure (CNKI). We used the following search terms: “Surfactant protein” or “SP” and “Idiopathic pulmonary fibrosis” or “IPF” or “Interstitial lung disease” or “Pulmonary fibrosis.” We also identified relevant publications by reviewing the reference of the search results. The meta-analysis was approved by the Ethics Committee of the Xijing Hospital, the First Affliated Hospital of the Fourth Military Medical University.
2.2. Inclusion and exclusion criteria
Included studies met the following criteria: SP-A and/or SP-D were evaluated in human subjects for the differential diagnosis and prognosis of IPF. In comparison part, patients with IPF were compared to at least one reference group (non-IPF patients or healthy subjects). Studies were written in English or Chinese. A definitive diagnosis of IPF was evident by clinical features, chest HRCT, laboratory findings, and/or surgical lung biopsy. Data, for example, serum levels of SP-A and SP-D, and hazard ratios (HRs), were available from the reviewed articles. In addition, for inclusion in the prognostic evaluation of SP-A/SP-D in patients with IPF, studies had to have a defined patient follow-up schedule, and include death as an endpoint. Studies were excluded if the data were unavailable after attempts to contact author.
2.3. Data extraction and quality assessment
Two reviewers independently screened titles and abstracts for potentially eligible articles. Full texts of articles were obtained, and 2 reviewers independently determined eligibility. Discrepancies were resolved by a discussion or with a third reviewer. For every eligible study, we extracted the following information: first author, year of publication, number and sex of patients with IPF, patient smoking history, study endpoints, study methods, and epidemiological study methods. When the association between SP-A/SP-D and IPF mortality was studied, cutoff values for SP-A and SP-D were also collected. When study data for meta-analysis were not available in the article, we attempted to contact author(s) to obtain original data. Some original data were not available. In this case, we derived mean and standard deviation using median and range from graphs and curve diagrams, according to the method recommended by Hozo et al.[23] In one case, the relative risk of mortality data was presented as Kaplan–Meier curves, not HR and 95% confidence interval (CI).[24] In this case, we calculated the HR and 95% CI according to the method described by Parmar et al.[25] In the analysis of the association of SP-A/SP-D with death of patients with IPF, there was an overlap of subjects in the studies of Greene et al.[26] and Kinder et al.[27]. To avoid sampling bias, we included only the Greene et al.'s[26] study in the analysis.
We used Newcastle–Ottawa quality assessment scale of case control studies to evaluate the quality of these studies.[28] A study could be awarded a maximum of one star for each numbered item within the Selection and Exposure categories. A maximum of 2 stars could be given for comparability. A study with >5 stars was included in the meta-analysis.
2.4. Statistical analysis
Statistical analyses were conducted using STATA software (Stata Corp, TX, version 12.0). All tests were 2-tailed with the significance level set at P < .05. For analyses of SP-A/SP-D as diagnostic and prognostic biomarkers, Cochran's Q test was used to assess between-study heterogeneity. Heterogeneity was presented in the form of the inconsistency index, I2, ranging from 0% to 100%.[29] To assess heterogeneity, the value of I2 was divided into 3 groups: <25%, 25% to 75%, and >75%, corresponding to low, moderate, and high heterogeneity. If statistical heterogeneity existed, the potential causes were estimated using sensitivity and subgroup analysis. A random-effect model was applied to reduce the effect of heterogeneity. To estimate the potential publication bias, we used a funnel plot and Egger's test. An asymmetric funnel plot and a P < .05 on the Egger's test identified the existence of publication bias.
When assessing the diagnostic effect of SP-A/SP-D, we treated the biomarkers as continuous, and gave an estimate of the combined overall effect size utilizing standard mean difference (SMD) in the random-effects model. For the evaluation of SP-A/SP-D as prognostic biomarkers of mortality, we used the Z test to test the pooled HR, and presented both a Z and P value. We compared the weight of the individual article in influencing the pooled HR, and the 95% CI. We also did subgroups analysis, stratified by race, for both diagnostic and prognostic analysis.
3. Results
3.1. Study inclusion, characteristics, and study quality
We identified articles from three databases: PubMed (n = 1353), EMBASE (n = 1230), and CNKI (n = 305). Nine hundred and ninety duplicate articles were removed. We excluded 1877 articles that failed to meet the inclusion criteria. A final group of 21 articles met the eligibility criteria[5,14,22,24,26,30–45] (Fig. 1). Seven of 21 articles evaluated the prognosis of IPF as an outcome and the observed endpoint was death (Table 1). Eighteen studies compared the serum level of SP-A/SP-D in patients with IPF to patients with non-IPF ILD or pulmonary infection, or healthy controls. All 21 articles were retrospective studies, and none of these studies reported blindness. No relevant prospective studies or large-scale meta-analyses were identified. The main characteristics of the 21 articles are summarized in Table 1. A total of 1289 patients with IPF were included for analyses. The majority of patients (>58%) were Asian. The date of publication ranged from 1992 to 2016. Data reflecting severity included FEV1% predicted, FVC% predicted, and DLco% predicted. DLco% predicted values varied from 39.1% to 74.9%. The number of male patients was greater than the number of female patients.
Figure 1.
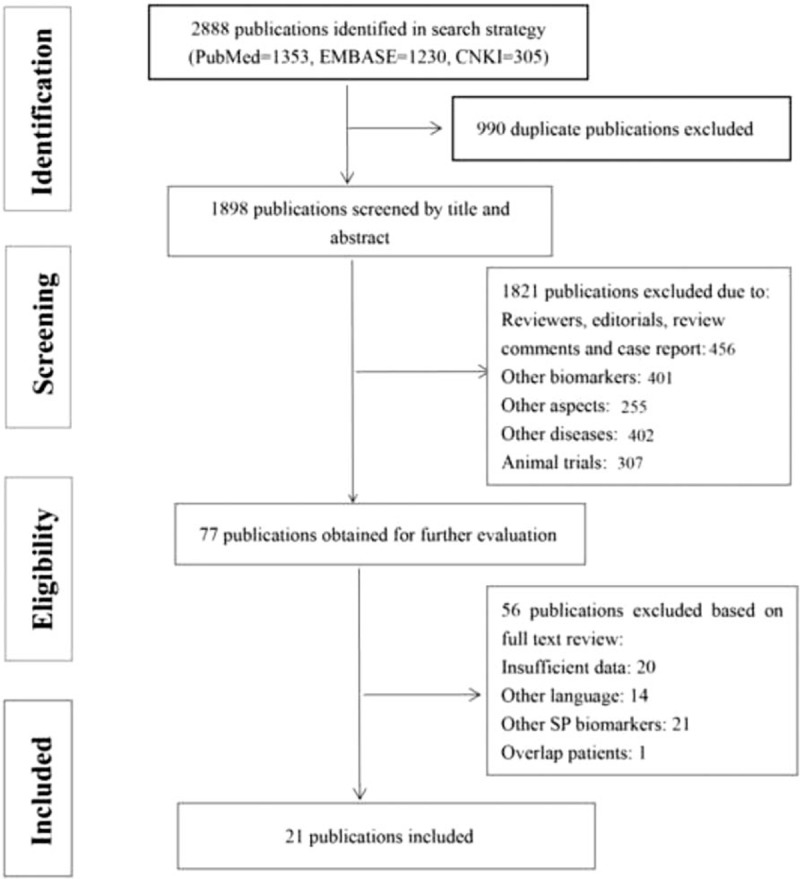
PRISMA processing map: 2888 publications identified in search strategy (PubMed 1353, EMBASE 1230, CNKI 305). After screening and eligibility process, only 21 articles met the including criteria and were included in ultimate analysis.
Table 1.
Baseline information for all articles included.
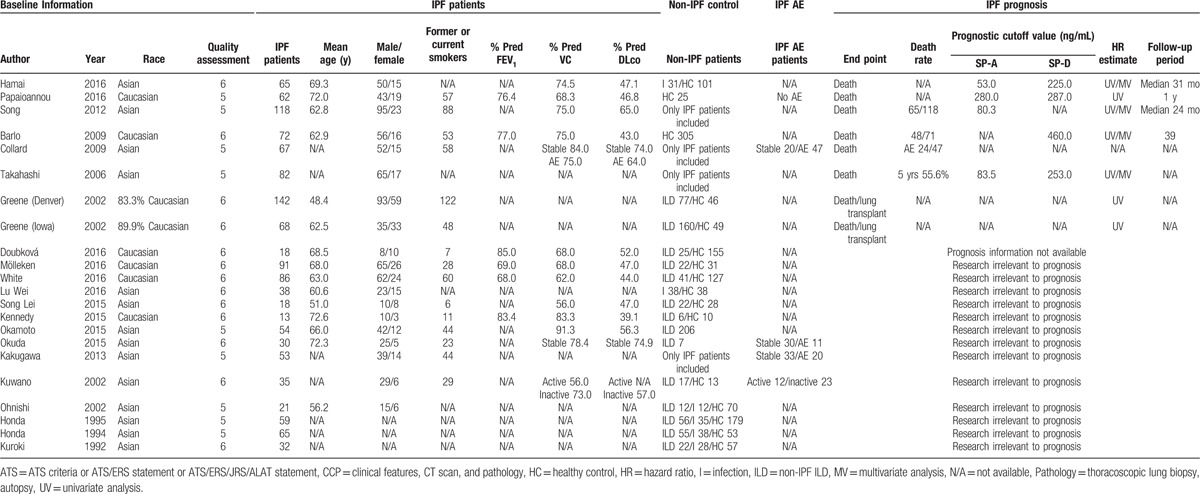
According to study quality assessment criteria (Supplemental Table 1), the study participation was adequate and the baseline study sample was completely delineated in all included articles. Every study that assessed the prognostic effect of SP-A/SP-D included patient follow-up. All studies assessed SP-A and (or) SP-D levels; however, different cutoff values were used, as described in Table 1. SP-A cutoff values ranged from 53 to 83.5 except Papaioannou's (280), and SP-D cutoff values ranged from 225 to 287 except Barlo's (460). Moreover, 5-year survival rates based on these cutoff points were listed in Supplemental Table 2.
3.2. Meta-analysis
3.2.1. Comparative analysis
Eighteen publications that reported serum levels of SP-A and (or) SP-D were included in these analyses. We compared the serum levels of SP-A and (or) SP-D of patients with IPF to patients with non-IPF ILD or pulmonary infection, or healthy controls (Figs. 2 and 3).
Figure 2.
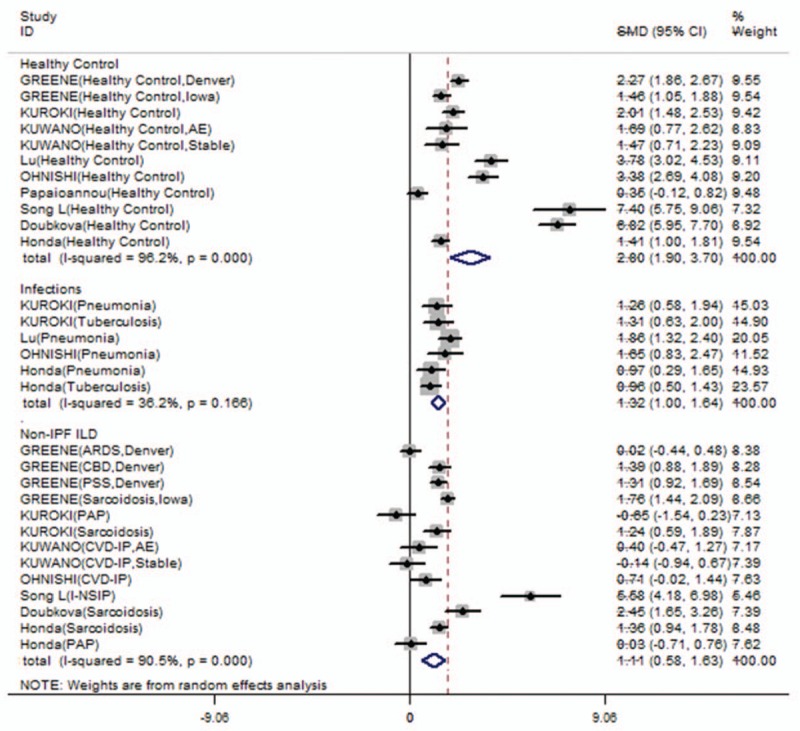
Serum SP-A levels comparisons between IPF and other groups (non-IPF ILD, pulmonary infections or healthy controls). Serum SP-A levels in IPF patients were higher than non-IPF ILD patients (SMD: 1.108 [0.584, 1.632], Z value = 4.15, P < .001), pulmonary infections (SMD: 1.320 [0.999, 1.640], Z value = 8.07, P < .001) or healthy controls (SMD: 2.802 [1.901, 3.702], Z value = 6.10, P < .001).
Figure 3.
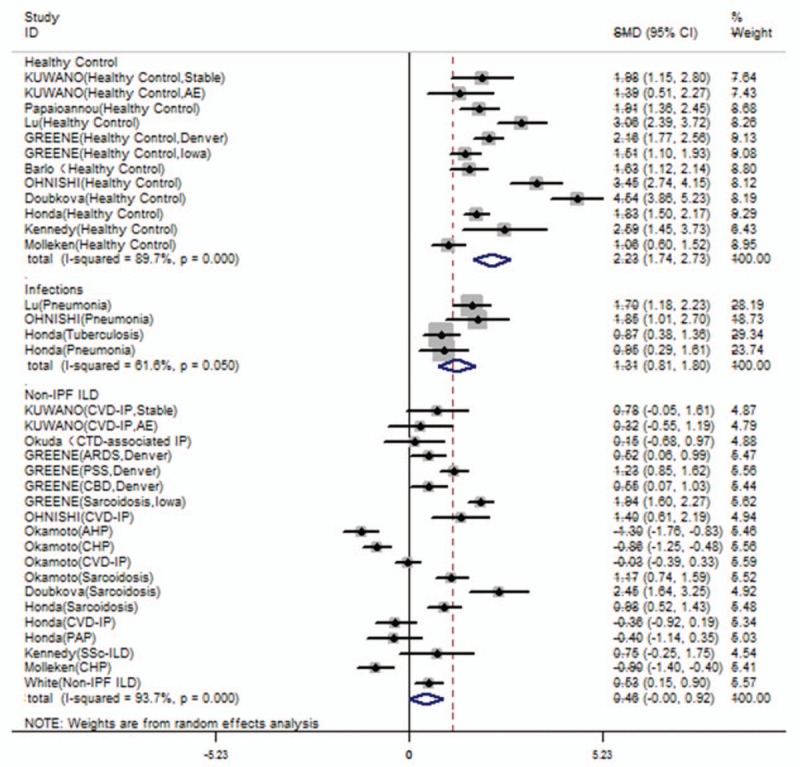
Serum SP-D levels comparisons between IPF and other groups (non-IPF ILD, pulmonary infections or healthy controls). Serum SP-D levels in IPF patients were higher than pulmonary infections (SMD: 1.308 [0.813, 1.803], Z value = 5.18, P < .001) or healthy controls (SMD: 2.235 [1.739, 2.731], Z value = 8.83, P < .001). However, there was no significant difference of serum SP-D levels between IPF and non-IPF ILD patients (SMD: 0.459 [-0.000, 0.919], Z value = 1.96, P = .050).
Serum SP-A levels among patients with IPF were significantly higher than patients with non-IPF ILD (SMD: 1.108 [0.584, 1.632], Z value = 4.15, P < .001, I2 = 90.5%, P < .001); patients with pulmonary infection (SMD: 1.320 [0.999, 1.640], Z value = 8.07, P < .001, I2 = 36.2%, P = .166); and healthy controls (SMD: 2.802 [1.901, 3.702], Z value = 6.10, P < .001, I2 = 96.2%, P < .001; Fig. 2). The I2 values suggest high heterogeneity across healthy and non-IPF ILD comparisons. Serum SP-A levels in patients with ILD and pulmonary infections were significantly higher than controls (SMD: 2.200 [1.627, 2.773]; 1.097 [0.420, 1.774], Supplemental Fig 1). These comparisons suggest a high level of heterogeneity (I2 = 95.8%, 87.0%, respectively, with P < .001, Supplemental Fig 1). To further evaluate the cause of heterogeneity, we repeated the serum SP-A level comparisons in specific diseases. The SP-A levels were significantly higher in patients with IPF than in patients with sarcoidosis and pneumonia, but not collagen vascular disease-associated interstitial pneumonia (SMD: 0.344 [−0.158, 0.846], Z value = 1.34, P = .180; Table 2).
Table 2.
Role of serum SP-A/D levels in specific disease comparisons.
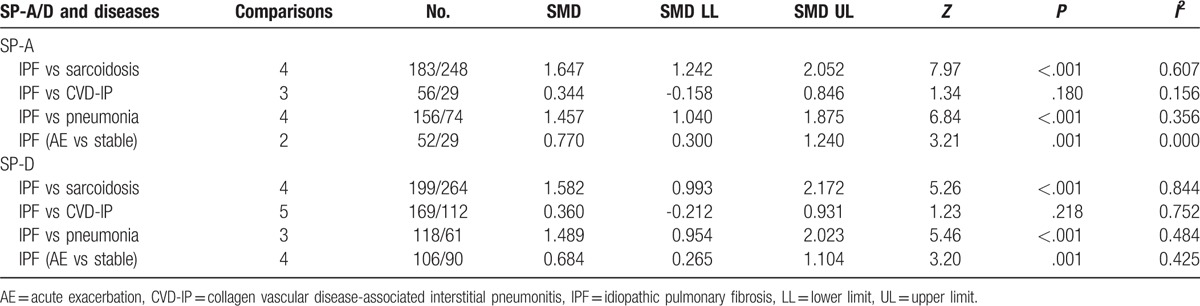
Serum SP-D levels in patients with IPF were significantly higher than SP-D in patients with pulmonary infection (SMD: 1.308 [0.813, 1.803], Z value = 5.18, P < .001; Fig. 3) and healthy controls (SMD: 2.235 [1.739, 2.731], Z value = 8.83, P < .001; Fig. 3). There was no significant difference in SP-D levels between patients with IPF and non-IPF ILD (SMD: 0.459 [−0.000, 0.919], Z value = 1.96, P = .050; Fig. 3). Surfactant protein-D levels are significantly elevated in patients with ILD (SMD: 1.935 [1.504, 2.367], Supplemental Fig 2), as well as pulmonary infections (SMD: 2.466 [0.558, 4.374], Supplemental Fig. 2). Furthermore, we compared the serum SP-D level in specific diseases. The SP-D serum level in patients with IPF patients was significantly higher than in patients with sarcoidosis (SMD = 1.582 [0.993, 2.172], Z value = 5.26, P < .001; Table 2), and the heterogeneity was high (I2 = 84.4%). There was no significant difference between patients with collagen vascular disease-associated interstitial pneumonia and IPF (SMD: 0.360 [−0.212, 0.931], Z value = 1.23, P = .218; Table 2). Moreover, elevated SP-A and SP-D might indicated AE of IPF (SP-A SMD: 0.770 [0.300, 1.240], SP-D SMD: 0.684 [0.265, 1.104]; Table 2). Only Kakugawa's research reported elevated SP-A and potentially elevated SP-D level in AE phase compared to stable phase in the same patients.[35]
3.2.2. Prognostic analysis
Seven publications were included to evaluate the effect of serum levels of SP-A and SP-D on the death of patients with IPF. A higher SP-A level was associated with a significantly higher risk of death, and there was no heterogeneity (pooled HR: 1.39 [1.10, 1.75], Z value = 2.80, P = .005, I2 = 0.0%; Fig. 4A). There was a significant association between higher SP-D level and increased risk of death, without heterogeneity (HR: 2.11 [1.60, 2.78], Z value = 5.31, P < .001, I2 = 0.0%; Fig. 4B). Sensitivity analysis indicated that no specific study influenced the pooled HR (Supplemental Fig. 3).
Figure 4.
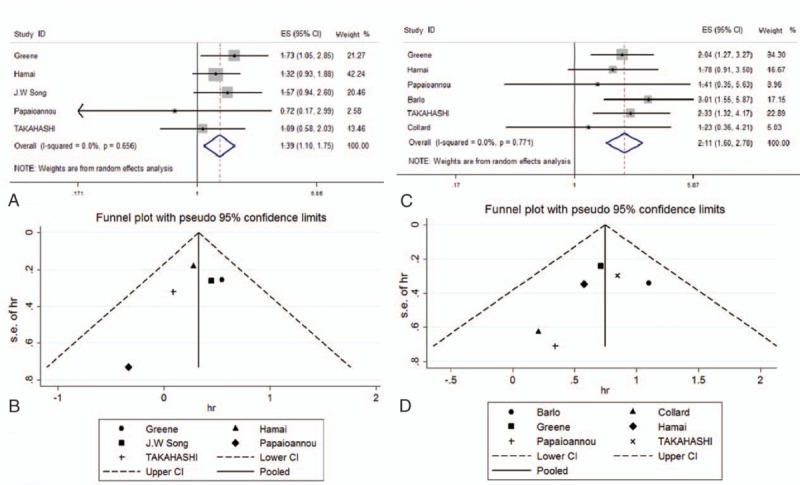
Forest plot and funnel plot for the association between serum SP-A/D levels and IPF patients’ prognosis. (A) Forest plot for the role of serum SP-A levels in IPF prognosis. HR and heterogeneity were shown (HR: 1.39 [1.10–1.75], Z value: 2.80, P = .005, I2 = 0.0%). P value of Q statistic test was also presented (P = .656). (B) Funnel plot for role of serum SP-A levels in IPF prognosis. No obvious asymmetry could be observed (Egger's test: P = .388). (C) Forest plot for the role of serum SP-D levels in IPF prognosis. HR and heterogeneity were shown (HR: 2.11 [1.60–2.78], Z value: 5.31, P < .001, I2 = 0.0%). P value of Q statistic test was also presented (P = .771). 4D: funnel plot for role of serum SP-D levels in IPF prognosis. No obvious asymmetry could be observed (Egger's test: P = .276).
No obvious asymmetry was observed in the funnel plot regarding the association between SP-A and death (Egger's test: P = .388; Fig. 4C), indicating no evidence of publication bias. Similar asymmetry was observed for the association between SP-D and death (Egger's test: P = .276; Fig. 4D).
3.3. Race subgroup analysis
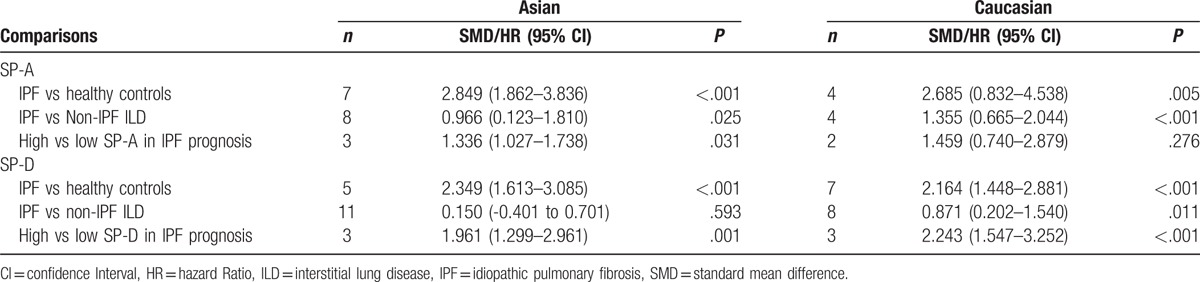
Both Asian (SMD 2.849 [1.862, 3.836], P < .001) and Caucasian (SMD 2.685 [0.832, 4.538], P = .005; Table 3) patients with IPF had significantly higher serum levels of SP-A when compared with healthy controls. Similar results were observed for SP-D (Table 3). Significantly higher SP-A serum levels (SMD 1.355 [0.665, 2.044], P < .001) and SP-D (SMD 0.871 [0.202, 1.540], P = .011) were identified in Caucasian patients with IPF, when compared to Caucasian patients with non-IPF ILD. However, no significant differences in serum levels of SP-D (SMD 0.150 [-0.401, 0.701], P = .593) were identified when Asian patients with IPF were compared to Asian patients with non-IPF ILD (Table 3). Higher levels of SP-A (pooled HR 1.336, 95% CI [1.027, 1.738], P = .031) and SP-D (pooled HR 1.961, 95% CI [1.299, 2.961], P = .001) were associated with increased risk of death among Asian patients with IPF. In Caucasian patients with IPF, a higher level of SP-D (pooled HR 2.243, 95% CI [1.547, 3.252], P < .001) was a risk factor for increased mortality, while a higher SP-A was not associated with increased mortality (pooled HR 1.459, 95% CI [0.740, 2.879], P = .276).
Table 3.
Race subgroups analysis in IPF comparisons and prognosis.
4. Discussion
In this study, serum levels of SP-A could be used to differentiate patients with IPF from patients with non-IPF ILD, pulmonary infection, and healthy controls. Serum SP-D levels could be used to differentiate patients with IPF from those with pulmonary infections and healthy controls, but not from patients with non-IPF ILD. Among Caucasian patients, both SP-A and SP-D levels differentiated patients with IPF from those with non-IPF ILD, and healthy controls. However, among Asian patients, higher level of SP-D differentiated patients with IPF only when compared to healthy controls. SP-A and SP-D could predict prognosis. Patients with IPF and elevated levels of SP-A had a 1.39-fold (95% CI 1.10, 1.75) increased risk of poor prognosis. Patients with IPF and elevated levels of SP-D had 2.11-fold (95% CI 1.60, 2.78) increased risk of poor prognosis. The cutoff points of SP-A (or SP-D) were the same order of magnitude except particular research. Different cutoff points may due to different calculation methods, race, severity, and so on.
Higher SP-A and SP-D serum levels among patients with IPF may be a result of genetic and environmental susceptibility. Pulmonary fibrosis may occur in genetically susceptible individuals after exposure to one of several environmental stressors. These stressors cause abnormal surfactant synthesis, secretion, and recycling, in both children and adults.[46] Serum levels of SP-A and SP-D are significantly elevated in patients with IPF, bacterial pneumonia, and tuberculosis, compared to the healthy controls. Kati et al.[47] suggested that serum SP-D levels were significantly higher in patients with IPF and submassive pulmonary embolism than in controls and SP-D may have a diagnostic role in sub-massive pulmonary embolism.
AE of IPF is characterized by an acute worsening of dyspnea, with the potential of hypoxemic respiratory failure, and new or progressive pulmonary infiltrates on radiologic images.[48] There is a difference in levels of serum SP-A and SP-D between stable and AE of IPF. In this study, we found that serum levels of SP-A and SP-D increased significantly in an AE of IPF, when compared to levels in stable IPF. Pathologically, it may result from functional abnormalities of type II alveolar epithelial cells, including endothelial cells injuries and coagulation abnormalities.[49] Therefore, SP-A and SP-D, reflecting type II alveolar epithelial cell injury and proliferation, will increase with an AE of IPF. However, in clinical practice, the combination of serum SP-A/SP-D with other predictors (eg, symptoms or HRCT) will enhance diagnostic accuracy of AE.
Elevated SP-A and SP-D in patients with IPF is likely the result of increased secretion by type II alveolar epithelial cells. An increase in total number of type II cells, resulting from diffuse hyperplasia, leads to increased leakage from the alveoli to the interstitium, and decreased clearance from vascular compartment. Selman and Pardo[50] reported that IPF was not an inflammatory disorder, and that alveolar epithelial cell damage and activation were a result of constant microscopic injury. Hyperplasia of type II alveolar epithelial cells results in loss of type I alveolar epithelial cells. In patients with IPF, Nishikiori et al.[20] identified thickened matrix, near the alveolar space, and capillary hyperplasia was detected using CD34.[51,52] These capillaries were near the regenerated type II alveolar epithelial cells, and were SP-A and SP-D positive. Surfactant proteins can leak directly into the bloodstream through capillaries instead of lymphatic ducts.
The blood-air barrier of lung comprises the alveolar epithelium, alveolar epithelial basal layer, substantia propria, capillary basement membrane, and capillary endothelial cells. Injury to any part of the barrier will increase permeability, resulting in leakage of SP-A and SP-D from the alveolus to the capillaries. Barbas-Filho et al.[53] conducted a retrospective study including 55 patients with histologically confirmed IPF. Using electron microscopy, they detected apoptosis in 89% of the patients with IPF/UIP.
In animal experiments, alveolar epithelial cell death was observed, and bleomycin was used to induce pulmonary fibrosis.[54,55] Repeated injuries of alveolar epithelial cells resulted in cell death, but the exact mechanism is unclear. Angiotensin II and Fas signaling were associated with alveolar epithelial cell death in bleomycin-induced lung injury.[56,57]
The inflammatory reaction may be involved in the pathogenesis of IPF. There is evidence of immune cell infiltration and inflammatory cytokine production in patients with IPF. Carre et al.[58] reported that the levels of interleukin-8 (IL-8) mRNA in alveolar macrophages, and the level of IL-8 protein in bronchoalveolar lavage, were significantly higher in patients with IPF than in healthy subjects. Both IL-8 mRNA and IL-8 protein were associated with disease severity. Other cytokines and chemokines, including tumor necrosis factor alpha (TNF-α), IL-1β, IL-4, and IL-5, are overexpressed in patients with IPF.[59,60] In an inflammatory environment, capillary permeability increases, and SP-A and SP-D leakage from the alveolus to the capillary increases. Mechanisms include damage of the junction of capillary endothelial cells, dysfunction of endothelial cell signal transduction, and release of inflammatory cytokines.
In addition, the concentration difference between alveolar airspace and the blood and reduction of SP clearance contribute to the elevation of the serum levels of SP-A and SP-D. IPF and non-IPF ILD have a similar pathogenesis. We found no significant differences in the serum concentration of SP-A and SP-D between patients with IPF and non-IPF ILD (including progressive systemic sclerosis, pulmonary alveolar proteinosis, idiopathic nonspecific interstitial pneumonia, and sarcoidosis). When comparing patients with IPF and sarcoidosis, there is no difference in SP-A, but there is a difference in SP-D, and the reason is unclear.
The median survival of patients with IPF patients is three years, indicating a poor prognosis.[61] Despite the potential use of SP-A/SP-D as diagnostic aids, the diagnosis of IPF is confirmed by surgical biopsy or HRCT. According to guidelines, serum SP-A/SP-D cannot replace biopsy or HRCT.[4] However, SP-A/SP-D has the following advantages. First, SP-A/SP-D serum levels could be preliminarily evaluated in patients apprehensive of traumatic examination and surgery. Second, image findings in HRCT are not typical, which complicate the diagnosis, and may delay early diagnosis and treatment. Other physiological measurements have been suggested for predicting the severity and prognosis of patients with IPF, including the GAP index[62] (gender, age, and 2 lung physiology variables, FVC and DLco) and the CPI[13] (composite physiologic index). Ley et al.[62] reported that the GAP index and staging system and the GAP calculator were better predictors of morality in patients with IPF than previously developed prediction models. This method has important utility for both clinical practice and trials. Our analyses indicate that the use of serum biomarkers should not replace the existing physiological measurements for predicting prognosis, as each method has its own limitation. Combining these methods (eg, GAP models, HRCT, and serum biomarkers) could increase the accuracy and sensitivity in determining the prognosis of patients with IPF. Several studies suggested that serum SP-A and/or SP-D can combine with existing physiologic parameters to enhance the ability of predicting survival. Those clinical variables include age, smoking status, FVC, Dlco, and so on.[27,44]
Our study still has several limitations. Although we tried to control the study quality by the Newcastle–Ottawa quality assessment scale, the included studies were all retrospective, which intrinsically presents difficulty in causality inference. Only Chinese and English articles were included in this analysis. Statistical heterogeneity was prevalent among our included studies in the overall analyses. The cutoff of high versus low serum levels was inconsistent among the included publications. In the articles included, 3 did not provide a specific cutoff value, and we were unable to obtain the original data. Among the cutoff values provided by the author, the numbers in each article were different.
Acknowledgments
We thank all authors and patients from the included articles. Especially we thank Andriana I. Papaioannou for providing her processed data and analysis.
Supplementary Material
Supplementary Material
Footnotes
Abbreviations: 95% CI = 95% confidence interval, AE = acute exacerbation, AHP = acute hypersensitivity pneumonitis, ARDS = acute respiratory distress syndrome, ATS = American Thoracic Society, CBD = chronic beryllium disease, CCL18 = CC-chemokine ligand 18, CHP = chronic hypersensitivity pneumonitis, CNKI = Chinese National Knowledge Infrastructure, COPD = chronic obstructive pulmonary disease, CPI = composite physiologic index, CRP = clinical, radiological, and physiological, CTD-IP = connective tissue disease-associated interstitial pneumonia, CVD-IP = collagen vascular disease-associated interstitial pneumonia, Dlco = diffusing capacity of the lung for carbon monoxide, FEV1 = forced expiratory volume in 1 second, FVC = forced vital capacity, GAP = gender, age, and two lung physiology variables (FVC and Dlco), HR = hazard ratio, HRCT = high-resolution computed tomography, ILD = interstitial lung disease, I-NSIP = idiopathic non-specific interstitial pneumonia, IPF = idiopathic pulmonary fibrosis, KL-6 = Krebs von den Lungen-6, MMP-7 = matrix metalloproteinase-7, PAP = pulmonary alveolar proteinosis, PSS = progressive systemic sclerosis, SMD = standard mean difference, SP-A = surfactant protein-A, SP-D = surfactant protein-D, SSc-ILD = scleroderma-associated interstitial lung disease, UIP = usual interstitial pneumonia.
KW and QJ are co-first authors.
Supplemental Digital Content is available for this article.
The authors report no conflicts of interest.
References
- [1].Crystal RG, Fulmer JD, Roberts WC, et al. Idiopathic pulmonary fibrosis. Clinical, histologic, radiographic, physiologic, scintigraphic, cytologic, and biochemical aspects. Ann Intern Med 1976;85:769–88. [DOI] [PubMed] [Google Scholar]
- [2].Hutchinson J, Fogarty A, Hubbard R, et al. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J 2015;46:795–806. [DOI] [PubMed] [Google Scholar]
- [3].Soo E, Adamali H, Edey AJ. Idiopathic pulmonary fibrosis: current and future directions. Clin Radiol 2017. [DOI] [PubMed] [Google Scholar]
- [4].Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011;183:788–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].White ES, Xia M, Murray S, et al. Plasma surfactant protein-D, matrix metalloproteinase-7, and osteopontin index distinguishes idiopathic pulmonary fibrosis from other idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2016;194:1242–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bjoraker JA, Ryu JH, Edwin MK, et al. Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1998;157:199–203. [DOI] [PubMed] [Google Scholar]
- [7].Latsi PI, du Bois RM, Nicholson AG, et al. Fibrotic idiopathic interstitial pneumonia: the prognostic value of longitudinal functional trends. Am J Respir Crit Care Med 2003;168:531–7. [DOI] [PubMed] [Google Scholar]
- [8].King TE, Jr, Schwarz MI, Brown K, et al. Idiopathic pulmonary fibrosis: relationship between histopathologic features and mortality. Am J Respir Crit Care Med 2001;164:1025–32. [DOI] [PubMed] [Google Scholar]
- [9].Flaherty KR, Andrei AC, Murray S, et al. Idiopathic pulmonary fibrosis: prognostic value of changes in physiology and six-minute-walk test. Am J Respir Crit Care Med 2006;174:803–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Flaherty KR, Mumford JA, Murray S, et al. Prognostic implications of physiologic and radiographic changes in idiopathic interstitial pneumonia. Am J Respir Crit Care Med 2003;168:543–8. [DOI] [PubMed] [Google Scholar]
- [11].King TE, Jr, Tooze JA, Schwarz MI, et al. Predicting survival in idiopathic pulmonary fibrosis: scoring system and survival model. Am J Respir Crit Care Med 2001;164:1171–81. [DOI] [PubMed] [Google Scholar]
- [12].Wells AU, Desai SR, Rubens MB, et al. Idiopathic pulmonary fibrosis: a composite physiologic index derived from disease extent observed by computed tomography. Am J Respir Crit Care Med 2003;167:962–9. [DOI] [PubMed] [Google Scholar]
- [13].Kim DS, Collard HR, King TE., Jr Classification and natural history of the idiopathic interstitial pneumonias. Proc Am Thorac Soc 2006;3:285–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hamai K, Iwamoto H, Ishikawa N, et al. Comparative study of circulating MMP-7, CCL18, KL-6, SP-A, and SP-D as disease markers of idiopathic pulmonary fibrosis. Dis Markers 2016;2016:4759040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nayak A, Dodagatta-Marri E, Tsolaki AG, et al. An Insight into the diverse roles of surfactant proteins, SP-A and SP-D in innate and adaptive immunity. Front Immunol 2012;3:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Carreto-Binaghi LE, Aliouat el M, Taylor ML. Surfactant proteins, SP-A and SP-D, in respiratory fungal infections: their role in the inflammatory response. Respir Res 2016;17:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Greene KE, Wright JR, Steinberg KP, et al. Serial changes in surfactant-associated proteins in lung and serum before and after onset of ARDS. Am J Respir Crit Care Med 1999;160:1843–50. [DOI] [PubMed] [Google Scholar]
- [18].El-Deek SE, Makhlouf HA, Saleem TH, et al. Surfactant protein D, soluble intercellular adhesion molecule-1 and high-sensitivity C-reactive protein as biomarkers of chronic obstructive pulmonary disease. Med Princ Pract 2013;22:469–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Takahashi H, Kuroki Y, Tanaka H, et al. Serum levels of surfactant proteins A and D are useful biomarkers for interstitial lung disease in patients with progressive systemic sclerosis. Am J Respir Crit Care Med 2000;162:258–63. [DOI] [PubMed] [Google Scholar]
- [20].Nishikiori H, Chiba H, Ariki S, et al. Distinct compartmentalization of SP-A and SP-D in the vasculature and lungs of patients with idiopathic pulmonary fibrosis. BMC Pulm Med 2014;14:196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].McFadden RG. Surfactant protein A predicts survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1996;154(3 Pt. 1):825–6. [DOI] [PubMed] [Google Scholar]
- [22].Lu WZH, Wei H. The diagnostic significance of KL-6, SP-A, SP-D and MMP-7 in IPF and its relationship with pulmonary function. Acta Univ Med Anhui 2016;51:868–72. [Google Scholar]
- [23].Hozo SP, Djulbegovic B, Hozo I. Estimating the mean and variance from the median, range, and the size of a sample. BMC Med Res Methodol 2005;5:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Takahashi H, Shiratori M, Kanai A, et al. Monitoring markers of disease activity for interstitial lung diseases with serum surfactant proteins A and D. Respirology (Carlton, Vic) 2006;11(Suppl.):S51–4. [DOI] [PubMed] [Google Scholar]
- [25].Parmar MK, Torri V, Stewart L. Extracting summary statistics to perform meta-analyses of the published literature for survival endpoints. Stat Med 1998;17:2815–34. [DOI] [PubMed] [Google Scholar]
- [26].Greene KE, King TE, Jr, Kuroki Y, et al. Serum surfactant proteins-A and -D as biomarkers in idiopathic pulmonary fibrosis. Eur Respir J 2002;19:439–46. [DOI] [PubMed] [Google Scholar]
- [27].Kinder BW, Brown KK, McCormack FX, et al. Serum surfactant protein-A is a strong predictor of early mortality in idiopathic pulmonary fibrosis. Chest 2009;135:1557–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Stang A. Critical evaluation of the Newcastle-Ottawa scale for the assessment of the quality of nonrandomized studies in meta-analyses. Eur J Epidemiol 2010;25:603–5. [DOI] [PubMed] [Google Scholar]
- [29].Higgins JP, Thompson SG, Deeks JJ, et al. Measuring inconsistency in meta-analyses. BMJ 2003;327:557–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Barlo NP, van Moorsel CH, Ruven HJ, et al. Surfactant protein-D predicts survival in patients with idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 2009;26:155–61. [PubMed] [Google Scholar]
- [31].Collard HR, Calfee CS, Wolters PJ, et al. Plasma biomarker profiles in acute exacerbation of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 2010;299:L3–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Doubkova M, Karpisek M, Mazoch J, et al. Prognostic significance of surfactant protein A, surfactant protein D, Clara cell protein 16, S100 protein, trefoil factor 3, and prostatic secretory protein 94 in idiopathic pulmonary fibrosis, sarcoidosis, and chronic pulmonary obstructive disease. Sarcoidosis Vasc Diffuse Lung Dis 2016;33:224–34. [PubMed] [Google Scholar]
- [33].Honda Y, Kuroki Y, Matsuura E, et al. Pulmonary surfactant protein D in sera and bronchoalveolar lavage fluids. Am J Respir Crit Care Med 1995;152(6 Pt. 1):1860–6. [DOI] [PubMed] [Google Scholar]
- [34].Honda Y, Kuroki Y, Shijubo N, et al. Aberrant appearance of lung surfactant protein A in sera of patients with idiopathic pulmonary fibrosis and its clinical significance. Respiration 1995;62:64–9. [DOI] [PubMed] [Google Scholar]
- [35].Kakugawa T, Yokota S, Ishimatsu Y, et al. Serum heat shock protein 47 levels are elevated in acute exacerbation of idiopathic pulmonary fibrosis. Cell Stress Chaperones 2013;18:581–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kennedy B, Branagan P, Moloney F, et al. Biomarkers to identify ILD and predict lung function decline in scleroderma lung disease or idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 2015;32:228–36. [PubMed] [Google Scholar]
- [37].Kuroki Y, Tsutahara S, Shijubo N, et al. Elevated levels of lung surfactant protein A in sera from patients with idiopathic pulmonary fibrosis and pulmonary alveolar proteinosis. Am Rev Respir Dis 1993;147:723–9. [DOI] [PubMed] [Google Scholar]
- [38].Kuwano K, Maeyama T, Inoshima I, et al. Increased circulating levels of soluble Fas ligand are correlated with disease activity in patients with fibrosing lung diseases. Respirology 2002;7:15–21. [DOI] [PubMed] [Google Scholar]
- [39].Molleken C, Poschmann G, Bonella F, et al. MFAP4: a candidate biomarker for hepatic and pulmonary fibrosis? Sarcoidosis Vasc Diffuse Lung Dis 2016;33:41–50. [PubMed] [Google Scholar]
- [40].Ohnishi H, Yokoyama A, Kondo K, et al. Comparative study of KL-6, surfactant protein-A, surfactant protein-D, and monocyte chemoattractant protein-1 as serum markers for interstitial lung diseases. Am J Respir Crit Care Med 2002;165:378–81. [DOI] [PubMed] [Google Scholar]
- [41].Okamoto T, Fujii M, Furusawa H, et al. The usefulness of KL-6 and SP-D for the diagnosis and management of chronic hypersensitivity pneumonitis. Respir Med 2015;109:1576–81. [DOI] [PubMed] [Google Scholar]
- [42].Okuda R, Matsushima H, Aoshiba K, et al. Soluble intercellular adhesion molecule-1 for stable and acute phases of idiopathic pulmonary fibrosis. SpringerPlus 2015;4:657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Papaioannou AI, Kostikas K, Manali ED, et al. Serum levels of surfactant proteins in patients with combined pulmonary fibrosis and emphysema (CPFE). PloS One 2016;11:e0157789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Song JW, Do KH, Jang SJ, et al. Blood biomarkers MMP-7 and SP-A: predictors of outcome in idiopathic pulmonary fibrosis. Chest 2013;143:1422–9. [DOI] [PubMed] [Google Scholar]
- [45].Song L, Zhao JJ, Hua D, et al. The expression of serum SP-A in idiopathic pulmonary fibrosis and its clinical value. Chin J Gerontol 2015;35:6465–6. [Google Scholar]
- [46].Whitsett JA, Wert SE, Weaver TE. Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease. Annu Rev Med 2010;61:105–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kati C, Alacam H, Duran L, et al. The effectiveness of the serum surfactant protein D (Sp-D) level to indicate lung injury in pulmonary embolism. Clin Lab 2014;60:1457–64. [DOI] [PubMed] [Google Scholar]
- [48].Kim DS, Park JH, Park BK, et al. Acute exacerbation of idiopathic pulmonary fibrosis: frequency and clinical features. Eur Respir J 2006;27:143–50. [DOI] [PubMed] [Google Scholar]
- [49].Selman M, Pardo A. Role of epithelial cells in idiopathic pulmonary fibrosis: from innocent targets to serial killers. Proc Am Thorac Soc 2006;3:364–72. [DOI] [PubMed] [Google Scholar]
- [50].Selman M, Pardo A. Idiopathic pulmonary fibrosis: an epithelial/fibroblastic cross-talk disorder. Respir Res 2002;3:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ebina M, Shimizukawa M, Shibata N, et al. Heterogeneous increase in CD34-positive alveolar capillaries in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2004;169:1203–8. [DOI] [PubMed] [Google Scholar]
- [52].Schlingemann RO, Rietveld FJ, de Waal RM, et al. Leukocyte antigen CD34 is expressed by a subset of cultured endothelial cells and on endothelial abluminal microprocesses in the tumor stroma. Lab InvestV 62 1990;690–6. [PubMed] [Google Scholar]
- [53].Barbas-Filho JV, Ferreira MA, Sesso A, et al. Evidence of type II pneumocyte apoptosis in the pathogenesis of idiopathic pulmonary fibrosis (IFP)/usual interstitial pneumonia (UIP). J Clin Pathol 2001;54:132–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Izbicki G, Segel MJ, Christensen TG, et al. Time course of bleomycin-induced lung fibrosis. Int J Exp Pathol 2002;83:111–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kumar RK, Watkins SG, Lykke AW. Pulmonary responses to bleomycin-induced injury: an immunomorphologic and electron microscopic study. Exp Pathol 1985;28:33–43. [DOI] [PubMed] [Google Scholar]
- [56].Hagimoto N, Kuwano K, Nomoto Y, et al. Apoptosis and expression of Fas/Fas ligand mRNA in bleomycin-induced pulmonary fibrosis in mice. Am J Respir Cell Mol Biol 1997;16:91–101. [DOI] [PubMed] [Google Scholar]
- [57].Wang R, Zagariya A, Ibarra-Sunga O, et al. Angiotensin II induces apoptosis in human and rat alveolar epithelial cells. Am J Physiol 1999;276(5 Pt. 1):L885–9. [DOI] [PubMed] [Google Scholar]
- [58].Carre PC, Mortenson RL, King TE, Jr, et al. Increased expression of the interleukin-8 gene by alveolar macrophages in idiopathic pulmonary fibrosis. A potential mechanism for the recruitment and activation of neutrophils in lung fibrosis. J Clin Invest 1991;88:1802–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Furuie H, Yamasaki H, Suga M, et al. Altered accessory cell function of alveolar macrophages: a possible mechanism for induction of Th2 secretory profile in idiopathic pulmonary fibrosis. Eur Respir J 1997;10:787–94. [PubMed] [Google Scholar]
- [60].Pan LH, Ohtani H, Yamauchi K, et al. Co-expression of TNF alpha and IL-1 beta in human acute pulmonary fibrotic diseases: an immunohistochemical analysis. Pathol Int 1996;46:91–9. [DOI] [PubMed] [Google Scholar]
- [61].Navaratnam V, Fleming KM, West J, et al. The rising incidence of idiopathic pulmonary fibrosis in the U.K. Thorax 2011;66:462–7. [DOI] [PubMed] [Google Scholar]
- [62].Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med 2012;156:684–91. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.