

Abstract
Platensimycin (PTM) and platencin (PTN) are highly functionalized bacterial diterpenoid natural products that target bacterial and mammalian fatty acid synthases. PTM and PTN feature varying diterpene-derived ketolides that are linked to the same 3-amino-2,4-dihydroxybenzoic acid moiety. As a result, PTM is a selective inhibitor for FabF/FabB, while PTN is a dual inhibitor of FabF/FabB and FabH. We previously determined that the PTM cassette, consisting of five genes found in the ptm, but not ptn, gene cluster, partitions the biosynthesis of the PTM and PTN diterpene-derived ketolides. We now report investigation of the PTM cassette through the construction of diterpene production systems in E. coli and genetic manipulation in the PTM–PTN dual overproducer Streptomyces platensis SB12029, revealing two genes, ptmT3 and ptmO5, that are responsible for the biosynthetic divergence between the PTM and PTN diterpene-derived ketolides. PtmT3, a type I diterpene synthase, was determined to be a (16R)-ent-kauran-16-ol synthase, the first of its kind found in bacteria. PtmO5, a cytochrome P450 monooxygenase, is proposed to catalyze the formation of the characteristic 11S,16S-ether ring found in PTM. Inactivation of ptmO5 in SB12029 afforded the ΔptmO5 mutant SB12036 that accumulated nine PTM and PTN congeners, seven of which were new, including seven 11-deoxy-16R-hydroxy-PTM congeners. The two fully processed PTM analogues showed antibacterial activities, albeit lower than that of PTM, indicating that the ether ring, or minimally the stereochemistry of the hydroxyl group at C-16, is crucial for the activity of PTM.
TOC image
INTRODUCTION
Platensimycin (PTM) and platencin (PTN) are highly functionalized bacterial diterpenoid natural products and represent a new class of promising antibiotic and antidiabetic drug leads. PTM and PTN are potent and selective inhibitors of bacterial fatty acid synthase (FASII) and showed efficacy in an in vivo mouse model of a Staphylococcus aureus infection.1,2 PTN dually inhibits the chain-initiation and chain-elongation condensing enzymes FabH and FabF/FabB, respectively,2 while PTM selectively inhibits FabF/FabB.1 PTM was also found to be a potent and selective inhibitor of mammalian fatty acid synthase.3 The structural differences between PTM and PTN account for their biological selectivities.1,2
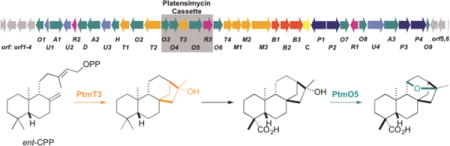
Two distinct moieties, a varying diterpene-derived ketolide and a 3-amino-2,4-dihydroxybenzoic acid (ADHBA), connected via an amide linkage, form the characteristic PTM and PTN scaffolds (Figure 1).4,5 The only structural differences between PTM and PTN reside in their diterpene-derived ketolides. The unique carbon scaffolds of the ketolides support the intermediacy of ent-kaurene and ent-atiserene metabolites in PTM and PTN biosynthesis, respectively.4,5 We recently cloned and sequenced the ptm gene cluster from Streptomyces platensis MA7327 and the ptn gene cluster from Streptomyces platensis MA7339, revealing that (i) MA7327 is a dual PTM-PTN producer and (ii) two distinct diterpene synthases (DTS) control divergence in PTM and PTN biosynthesis.6,7 The ptm and ptn gene clusters are highly homologous in genetic organization and sequence, with the major difference being the inclusion of a 5.4-kb cassette, containing five open reading frames (ORFs) and named the PTM cassette, in the middle of the ptm gene cluster (Figure 2). Introduction of this cassette into the sole PTN producer S. platensis MA7339 afforded a recombinant strain S. platensis SB12604 that produced both PTM and PTN, confirming that the PTM cassette encodes the genes necessary for PTM biosynthesis.6 These data, along with other isolated PTM and PTN congeners,8,9 supported a unified pathway for PTM and PTN biosynthesis where one set of genes – ptm and ptn – encode enzymes that process both PTM and PTN intermediates en route to the final PTM and PTN products.
Figure 1.

Structures of PTM (1), PTN (2), biosynthetic intermediates/congeners and fully processed PTM analogues isolated in this study. (16R)-ent-Kauran-16-ol (3) was isolated from the heterologous diterpene production system E. coli SB12301, and 4–12, featuring the 11-deoxy-16R-hydroxyl functionality, along with 2, were isolated from the ΔptmO5 mutant S. platensis SB12036, with 4, 5, and 8 as the major metabolites.
Figure 2.
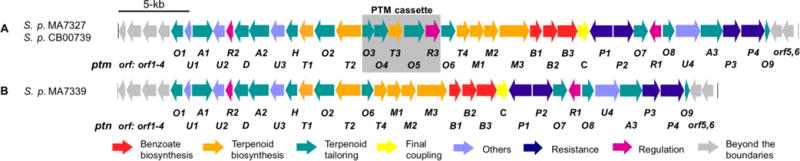
The ptm and ptn biosynthetic gene clusters. (A) Genetic organization of the ptm gene clusters from the dual PTM-PTN producers S. platensis MA7327 and S. platensis CB00739 and (B) the ptn gene cluster from the PTN-only producer S. platensis MA7339. The PTM cassette, which contains five ORFs including ptmT3 and ptmO5 and encodes the genes necessary for PTM biosynthesis, is present in the ptm gene cluster but absent in the ptn gene cluster. The PTM-PTN dual overproducer S. platensis SB12029 used in this study was derived from S. platensis CB00739 by in-frame deletion of the pathway-specific negative regulator ptmR1.9
Using a high-throughput real-time PCR method for strain prioritization and natural products discovery, we discovered six additional dual PTM–PTN producers from the Actinomycetales collection at The Scripps Research Institute.10 Inactivation of the pathway-specific negative regulator ptmR17 in one of these producers, Streptomyces platensis CB00739, afforded a genetically amenable dual PTM–PTN overproducer SB12029 (Figure 2), which has been developed into a model strain to study PTM and PTN biosynthesis and engineering, thereby circumventing technical difficulties encountered with the original MA7327 and MA7339 producers.9 Thus, ptmO4, one of the genes in the PTM cassette that encodes an acyl-CoA dehydrogenase, was inactivated in SB12029 and found to catalyze β-oxidation of both PTM and PTN diterpene intermediates.9 The other four genes within the PTM cassette are therefore likely candidates that encode the enzymes necessary to partition PTM biosynthesis from that of PTN. They are ptmO3, ptmT3, ptmO5, and ptmR3, encoding an α-ketoglutarate-dependent dioxygenase, a DTS, a cytochrome P450 monooxygenase, and a regulatory kinase, respectively, based on bioinformatics analysis.6
One of the key structural features of PTM is the 11S,16S-ether ring found in the diterpene-derived ketolide moiety (Figure 1). The oxygen atom of this O-heterocycle hydrogen bonds to a Thr in FabF, strongly contributing to the potency of PTM11 and the selectivity of PTM for FabF, as evidenced by the IC50 = 48 nM for PTM vs IC50 = 4.6 μM for PTN against S. aureus FabF, respectively.1,2 The biosynthesis of this crucial ether linkage, however, is unknown. The PTM ketolide is initially generated through cyclization of geranylgeranyl diphosphate (GGPP, 13) into ent-copalyl diphosphate (ent-CPP, 14) by the type II DTS PtmT212 and a subsequent cyclization by the type I DTS PtmT3, affording the characteristic ent-kaurene-derived scaffold.6 α-Ketoglutarate-dependent dioxygenases and cytochrome P450 monooxygenases, such as PtmO3 and PtmO5, respectively, are well known candidates to catalyze C–H activation, thereby imbuing oxygen functionalities into the various hydrocarbon scaffolds. In fact, two P450s have been implicated in the formation of exomethylene tetrahydrofuran rings in natural products, AveE in avermectin and AurH in aureothin (Figure S1).13–17 Given that PtmT3, PtmO3, and PtmO5 (i.e., proteins encoded by the PTM cassette) are necessary for PTM biosynthesis, we hypothesized that the tetrahydrofuran-containing ketolide moiety of PTM is constructed by one of, or a combination of, these enzymes. In fact, an ent-kaurene-derived natural product, featuring a similar 11S,16S-ether ring, has been isolated from the gibberellin-producing fungus Gibberella fujikuroi (Figure S1), the biosynthesis of which however is unknown.18 Therefore, studying the biosynthetic origin of the ether ring in PTM will not only reveal novel biochemistries and enzymes found in bacteria, but also provide opportunities to addresses unanswered questions of plant and fungal diterpenoid biosynthesis in a bacterial model system.
Here we report the biosynthetic origin of the 11S,16S-ether ring in PTM. We first constructed the heterologous diterpene production systems E. coli SB12301 and SB12302 and functionally characterized PtmT3 as a (16R)-ent-kauran-16-ol synthase. We then exploited the genetically amenable dual PTM-PTN overproducer SB12029 to construct the ΔptmO5 mutant SB12036. Fermentation of SB12036 led to the isolation of nine PTM and PTN congeners, seven of which were new. Each of the PTM congeners lacked the 11S,16S-ether ring, characteristic of PTM, but featured the 11-deoxy-16R-hydroxyl functionality instead, revealing that PtmO5 is involved in the formation of the ether ring in PTM. Finally, we complemented SB12036, as well as the ΔptmT2 mutant SB12034,19 with strategically selected biosynthetic intermediates, both of which restored PTM production, providing additional support to the function and mechanism of PtmO5. The biosynthetic intermediates/congeners or fully processed PTM analogues isolated in this study continue to reveal new insights into PTM and PTN biosynthesis and their structure-activity relationships.
EXPERIMENTAL SECTION
General experimental procedures
All 1H, 13C, DEPT 135 and 2D NMR (1H-1H COSY, 1H-13C HSQC, 1H-13C HMBC, 1H-1H ROESY) experiments were run on a Bruker Avance III Ultrashield 700 at 700 MHz for 1H and 175 MHz for 13C nuclei. MPLC separation was conducted on a Biotage Isolera One equipped with a Biotage SNAP Cartridge HP-Sil column (60 g). Preparative HPLC was carried out on an Agilent 1260 Infinity LC equipped with an Agilent Eclipse XDB-C18 column (250 mm × 21.2 mm, 7 μm). LC-MS was performed on an Agilent 1260 Infinity LC coupled to a 6230 TOF (HRESI) equipped with an Agilent Poroshell 120 EC-C18 column (50 mm × 4.6 mm, 2.7 μm). For GC-MS analysis, an Agilent 7890A GC 240 MS ion trap system operating in splitless mode and equipped with a VF-5ms capillary column (30 m × 0.25 mm, 5% phenyl-95% methyl polysiloxine, 0.25 μm film thickness; Agilent) was used. Melting points were measured on a Stuart automatic melting point SMP40. Optical rotations were obtained using an AUTOPOL IV automatic polarimeter (Rudolph Research Analytical). UV was measured with a NanoDrop 2000C spectrophotometer (Thermo Scientific). IR spectra were attained using a Spectrum One FT-IR spectrophotometer (PerkinElmer). X-ray crystallographic data were obtained on a Bruker AXS Smart APEX CCD diffractometer using graphite monochromated MoKα radiation. The crystal structure was solved by direct methods (SHELXS97)20 and refined by least-squares calculations. The nonhydrogen atoms were refined anisotropically, and the hydrogen atoms were fixed at calculated positions.
Bacterial strains, plasmids, and chemicals
Strains, plasmids, and PCR primers used in this study are listed in Tables S1–S3, respectively. PCR primers were obtained from Sigma-Aldrich. Q5 high-fidelity DNA polymerase, restriction endonucleases, and T4 DNA ligase were purchased from NEB and used by following the protocols provided by the manufacturers. DNA gel extraction and plasmid preparation kits were purchased from Omega Bio-Tek. DNA sequencing was conducted by Eton Bioscience. The John Innes Center (Norwich, U.K.) provided the REDIRECT Technology kit for PCR-targeting homologous recombination.21 E. coli ET12567/pUZ800222 was used as the host for intergeneric conjugation with S. platensis SB12029.9 Other common chemicals, biochemical, and media components were purchased from standard commercial sources.
Culture conditions
E. coli strains harboring plasmids or cosmids were grown in lysogeny broth (LB) with appropriate antibiotic selection.23 Streptomyces strains were grown on solid ISP4 medium at 28 °C or cultured in liquid tryptic soy broth (TSB) at 28 °C and 250 rpm, with appropriate antibiotic selection, if needed.24 E. coli-Streptomyces conjugations were plated onto ISP4 medium supplemented with 10 mM MgCl2. Fermentation of S. platensis recombinant strains were conducted as described previously.7,9,10 Briefly, fresh spores of Streptomyces strains were inoculated into TSB seed medium and cultured for 2 d. PTM fermentation medium was inoculated with 4% (v/v) seed culture and 3% (w/v) Amberlite XAD-16 resin (Sigma-Aldrich) and incubated for 7 d.
Construction of plasmids pBS12301, pBS12302, pBS12303, and pBS12304 for diterpene production in E. coli
pRSFDuet-1 and pCDFDuet-1 (Novagen) were used as the cloning vectors to construct a diterpene production system in E. coli (Figure S2). The E. coli genes idi, dxs, and dxr were amplified by PCR from E. coli DH5α and individually cloned into pRSFDuet-1. Thus, idi was cloned into the NcoI and BamHI sites of multiple cloning site (MCS)-1, and dxr, cloned into the NdeI and KpnI sites, was placed upstream of dxs, which was ligated into the XhoI and PacI sites of MCS-2. The resulting construct, containing idi-dxr-dxs, was named pBS12301.
The three diterpene synthase-encoding genes from the ptm cluster, ptmT2, ptmT3, and ptmT4 were amplified by PCR from the genomic DNA of S. platensis CB00739 and individually cloned into pCDFDuet-1. Thus, ptmT2 was cloned into the EcoRI and HindIII sites of MCS-1, while ptmT3 was cloned into the NdeI and KpnI sites of MCS-2, affording pBS12302. Two additional plasmids were constructed by cloning ptmT4 into either the SacI and PstI sites (MCS-1) of pBS12301, downstream of idi, to afford pBS12303 or the EcoRI and HindIII sites (MCS-1) of pBS12302, upstream of ptmT2, to afford pBS12304.
Functional characterization of PtmT3 in E. coli SB12301 and SB12302
Plasmids pBS12301 and pBS12304, or pBS12302 and pBS12303, were co-transformed into E. coli BL21(DE3), affording the recombinant E. coli strains SB12301 and SB12302, respectively. SB12301 and SB12302 were cultured in either LB or TB media as described previously for heterologous diterpene production in E. coli.25 Briefly, 3 L of LB or TB were inoculated with overnight cultures of SB12301 or SB12302 and incubated at 37 °C and 250 rpm until an OD600 = 0.6 was reached. The cultures were then cooled on ice, induced with 0.1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG), and further incubated at 16 °C and 250 rpm for an additional 90 h. Following centrifugation, extraction of the supernatant, using 1× volume of hexanes, and removal of excess solvent in vacuo resulted in a colorless, fragrant oil. A small sample of this crude extract was diluted with hexanes and subjected to GC-MS analysis. The GC conditions were: injector 275 °C, initial column temperature 40 °C for 3 min, temperature ramp 20 °C per min to 300 °C, 4 min hold at 300 °C, with a carrier gas flow rate of 1.1 mL min−1. Diterpenes were analyzed in EI mode, and mass spectral data (50–500 m/z) were collected using Agilent MS Workstation software system control version 7.0.0.
The rest of the crude extract was subjected to silica gel flash chromatography using a gradient elution system of hexane/ethyl acetate (from 50:1 to 4:1) to obtain partially pure 3. This material was further chromatographed on a silica gel column using an isocratic elution system of hexane/ethyl acetate (9:1) to afford a white, amorphous solid (6.3 mg), 1H and 13C NMR analysis confirmed its identity as (16R)-ent-kauran-16-ol (3), a known diterpenoid (SI).26
Inactivation of ptmO5 in SB12029 to afford the ΔptmO5 mutant SB12036
Gene replacement of ptmO5 was performed as previously described for other recombinant strains constructed in S. platensis CB00739 and SB12029.9,10,19 Briefly, the ptmO5 gene was replaced with the aac(3)IV + oriT cassette from pIJ773 using λRED-mediated PCR-targeting mutagenesis in E. coli BW25113/pIJ79021 harboring pBS12039, a cosmid containing a partial ptm gene cluster (Table S3).9 The genotype of the resultant ΔptmO5 mutant cosmid, pBS12063, was confirmed by PCR analysis using primers 739O5ID_F and 739O5ID_R (Table S3). pBS12063 was transformed into E. coli ET12567/pUZ800222 and introduced into S. platensis SB12029 by intergeneric conjugation. Isolation of the ΔptmO5 mutant SB12036 by double crossover homologous recombinantion between SB12029 and pBS12063 was selected by the apramycin resistant and kanamycin sensitive phenotype. The genotype of SB12029 was confirmed by PCR analysis (Figure S3) and Southern analysis (Figure S4).
Chemical complementation of the ΔptmT2 mutant SB12034 and the ΔptmO5 mutant SB12036
The ΔptmT2 mutant S. platensis SB12034, constructed previously,19 was chemically complemented with (16R)-hydroxy-ent-kauran-19-oic acid (4), isolated in this study from SB12036. Complementarily, the ΔptmO5 mutant S. platensis SB12036, constructed in this study, was chemically complemented with (11S,16S)-ent-kauran-11,16-epoxy-19-oic acid (18), isolated previously from the ΔptmO4 mutant S. platensis SB12030.9 Each complementation fermentation was carried out, with 1 mg of 4 or 18 added to 50 mL of PTM fermentation media for SB12034 or SB12036, respectively, with the dual PTM-PTN overproducer SB12029 as a positive control.9,10,19
Isolation and structural elucidation of PTM and PTN biosynthetic intermediates/congeners (4–9) and fully processed PTM congeners (10–12) from SB12036
For small-scale fermentations, extraction of natural products from resin followed previously reported procedures.7,9,10,19 Briefly, after harvesting the resin from the fermentation broth, the resin was washed with water and extracted with CH3OH three times. The CH3OH extract was used directly for LC-MS analysis. Chromatography for LC-MS was conducted using an 18 min solvent gradient from 5–100% CH3CN in H2O containing 0.1% formic acid at a flow rate of 0.4 mL min−1.
For a large-scale fermentation (3.2 L) of SB12036, eight 2.0 L baffled flasks, each containing 400 mL of production medium with 4% (v/v) seed culture and 15 g of Amberlite XAD-16 resin, were incubated for 7 d. The harvested and washed resin was extracted three times with ca. 500 mL of CH3OH. Methanol was removed in vacuo and the resulting oil was adsorbed onto C18 reverse-phase resin (Biotage) and fractionated by MPLC with an elution system of CH3OH–H2O (20:80, 40:60, 60:40, 80:20, 100:0) to yield five fractions (Fr01–Fr05). Since the major metabolites of Fr05 were fatty acids, no further purification of Fr05 was carried out.
The fully processed PTM analogues (10, 8.6 mg; 11, 4.6 mg) were isolated from Fr01 using preparative reverse-phase HPLC with a gradient elution of 5–100% CH3CN in H2O containing 0.1% formic acid. Fraction Fr02 was fractionated by preparative reverse-phase HPLC using an elution system of 5–70% CH3CN in H2O containing 0.1% formic acid to afford subfraction Fr0201 and PTN (2, 42 mg). The major compound in Fr0201 was 8, which, due to its poor aqueous solubility, easily precipitated out upon slow removal of CH3CN from its aqueous mixture in vacuo. After centrifuging and washing three times with CH3OH, a large quantity of pure 8 (120 mg) was obtained. Fraction Fr03 was fractionated by preparative reverse-phase HPLC using a 30 min elution system of 10–70% CH3CN in H2O containing 0.1% formic acid to afford subfraction Fr0301, from which 9 (15.2 mg) was similarly precipitated out upon slow removal of CH3CN in vacuo, collected by centrifugation, and washed with CH3OH.
Fraction Fr04 was split into four subfractions (Fr0401–Fr0404) by silica gel column chromatography using a gradient elution system of CH2Cl2–CH3OH (20:1 to 2:1). Compound 4 (110 mg) precipitated out from Fr0402 as a white solid. Subfraction Fr0403 was further purified by chromatography using a Sephadex LH-20 column, eluted with CH3OH, to generate two subfractions Fr040301 and Fr040302. Compound 5 (82 mg) crystallized directly from both fractions upon concentration and addition of H2O at room temperature. The mother liquids of Fr040301 and Fr040302 were further purified by preparative reverse-phase HPLC using a 30 min elution system of 30–80% CH3CN in H2O containing 0.1% formic acid. Compound 7 (7.5 mg) was isolated from Fr040301, whereas 6 (1.5 mg) and 12 (7.1 mg), along with an additional 6.0 mg of 7, were isolated from Fr040302.
(16R)-Hydroxy-ent-kauran-19-oic acid (4)
Isolated as a white, amorphous solid, 4 was confirmed to be (16R)-hydroxy-ent-kauran-19-oic acid, a known diterpenoid also known as (16α)-hydroxy-ent-kauran-19-oic acid,27–29 based on combined 1H and 13C NMR and HRESIMS analysis (SI).
Platensimycin ML9 (5)
Colorless needles (CH3OH/H2O); mp 242.9–244.3 °C −55.3 (c 1.15, CH3OH); IR (film) νmax 3379, 2932, 1698, 1463, 1375, 1332, 1241, 1198, 1145, 1059, 1036, 939, 875, 796 cm−1; 1H and 13C NMR data, see Table S4; HRESIMS affording the [M + Na]+ ion at m/z 359.2191 (calcd [M + Na]+ ion for C20H32O4 at m/z 359.2193). Crystallographic data for 5 was deposited in the Cambridge Crystallographic Data Centre (deposition number: CCDC 1476684).
(7S,16R)-Dihydroxy-ent-kauran-19-oic acid (6)
Isolated as a white, amorphous solid, 6 was confirmed to be (7S,16R)-dihydroxy-ent-kauran-19-oic acid, a known diterpenoid also known as (7β,16α)-dihydroxy-ent-kauran-19-oic acid.30 As 6 was not previously characterized in full,30 detailed spectroscopic characterization of 6 is reported here (SI, Table S4).
Platensimycin ML10 (7)
White, amorphous solid; −52.8 (c 0.39, CH3OH); IR (film) νmax 3567, 3405, 2944, 1683, 1464, 1246, 1201, 1054, 1019, 933, 873, 792 cm−1; 1H and 13C NMR data, see Table S5; HRESIMS affording the [M + Na]+ ion at m/z 359.2193 (calcd [M + Na]+ ion for C20H32O4 at m/z 359.2193).
Platensimycin ML11 (8)
Light gray, amorphous solid; −67.1 (c 0.34, DMSO); UV (CH3OH) λmax (log ε) 259 (5.45), 208 (5.60) nm; IR (film) νmax 3334, 2938, 1695, 1416, 1240, 1225, 1075, 1016, 951, 760 cm−1; 1H and 13C NMR data, see Table S4; HRESIMS affording the [M + H]+ ion at m/z 666.2892 (calcd [M + H]+ ion for C30H44N5O10P at m/z 666.2899).
Platencin ML3 (9)
Light gray, amorphous solid; −52.0 (c 0.51, DMSO); UV (CH3OH) λmax (log ε) 261 (5.15), 210 (5.46) nm; IR (film) νmax 3338, 2929, 1693, 1468, 1424, 1234, 1060, 870 cm−1; 1H and 13C NMR data, see Table S5; HRESIMS affording the [M + H]+ ion at m/z 648.2790 (calcd [M + H]+ ion for C30H42N5O9P at m/z 648.2793).
Platensimycin ML12 (10)
White, amorphous solid; −36.7 (c 0.55, CH3OH); UV (CH3OH) λmax (log ε) 296 (4.76), 229 (5.60) nm; IR (film) νmax 2936, 1652, 1534, 1379, 1316, 1251, 1018, 796 cm−1; 1H and 13C NMR data, see Table S6; HRESIMS affording the [M + H]+ ion at m/z 444.2015 (calcd [M + H]+ ion for C24H29NO7 at m/z 444.2017).
Platensimycin ML13 (11)
White, amorphous solid; −40.4 (c 0.46, CH3OH); UV (CH3OH) λmax (log ε) 296 (4.89), 229 (5.64) nm; IR (film) νmax 3316, 2938, 1652, 1441, 1318, 1265, 1064, 789 cm−1; 1H and 13C NMR data, see Table S6; HRESIMS affording the [M + H]+ ion at m/z 458.2172 (calcd [M + H]+ ion for C25H31NO7 at m/z 458.2173).
Platensimycin ML14 (12)
White, amorphous solid; −67.8 (c 0.18, CH3OH/CH2Cl2 1:1); UV (DMSO) λmax (log ε) 303 (4.35), 256 (4.32) nm; IR (film) νmax 3277, 2934, 1652, 1622, 1440, 1308, 1236, 1077, 1014, 672 cm−1; 1H and 13C NMR data, see Table S6; HRESIMS affording the [M + H]+ ion at m/z 919.3508 (calcd [M + H]+ ion for C48H58N2O12S2 at m/z 919.3504).
Antibacterial assay
Compounds 4–12 were tested for antibacterial activities against S. aureus ATCC 25923 and Micrococcus luteus ATCC 9431 using a standard disk diffusion assay.9,19,31 Quantities (20 μg) of compounds in DMSO were applied to 7 mm filter disks (Whatman), dried, and placed onto solid LB agar plates containing dense bacterial liquid cultures (ca. ~18 h). PTM (1) and PTN (2) (5 μg) were used as positive controls. The plates were incubated overnight at 37 °C and zones of inhibition, if present, were observed. For compounds (10 and 12) that showed antibacterial activity, minimum inhibitory concentration (MIC) values were determined using a 96-well plate format with Muller-Hinton (MH) broth, as previously described.32 Briefly, 2 μL of each compound, serially diluted in DMSO, were added to wells containing 98 μL of cell cultures diluted to an OD600 = 0.005. The MIC values, performed in duplicates, were determined after incubation at 37 °C for 18 h. PTM (1) and DMSO were used as positive and negative controls, respectively.
RESULTS
Production of diterpenoids in E. coli establishing PtmT3 as a (16R)-ent-kauran-16-ol synthase
Inspired by previous diterpenoid production in E. coli,25,33 we sought to construct a two-plasmid system for diterpenoid production based entirely on genes of bacterial origin. pBS12301, a pRSFDuet-1-derived plasmid that contained the idi, dxs, and dxr genes from E. coli, encoding isopentenyl diphosphate isomerase (IDI), 1-deoxy-D-xylulose-5-phosphate synthase (DXS), and 1-deoxy-D-xylulose-5-phosphate reductoisomerase (DXR), respectively, was constructed to boost the metabolic pool of diterpene precursors, i.e., dimethylallyl diphosphate (DMAPP, 13) and isopentenyl diphosphate (IPP, 14) (Figure 3), as previously reported.25 pBS12304, a pCDFDuet-1-derived complementary plasmid that contained the ptmT4, ptmT2, and ptmT3 genes from S. platensis CB00739, encoding geranylgeranyl diphosphate synthase, ent-copalyl diphosphate synthase, and the type I DTS responsible for the ent-kaurene-derived PTM-ketolide, respectively, was constructed for modular production of diterpenoids in E. coli (Figure 3). (Two variants, pBS12303 and pBS12302, were also constructed, and together they were functionally equivalent to pBS12301 and pBS12304). pBS12301 and pBS12304 (or pBS12302 and pBS12303) were co-transformed into E. coli BL21(DE3), affording E. coli SB12301 (or SB12302), which was cultured for 90 h at 16 °C after IPTG induction. Since both strains showed similar metabolite profiles, only the characterization of the metabolites from SB12301 was described. Thus, GC-MS analysis of the crude extract of SB12301 revealed three new peaks compared to the negative control, an extract made from the parent E. coli BL21(DE3) strain (Figure S5). Each of the three peaks had MS spectra consistent with those of ent-kaurene or ent-kauran-16-ol scaffolds (Figure S6). It is well known that terpene synthases can exhibit relaxed control over their carbocation-based cyclization cascades, resulting in numerous product outcomes.34 Indeed, the bifunctional ent-kaurene synthase (PpCPS/KS) from the moss Physcomitrella patens catalyzes the cyclization of GGPP into both ent-kaur-16-ene and 16α-hydroxy-ent-kaurane [i.e., (16R)-ent-kauran-16-ol (3)] (Figure S7).35 The three peaks visible by GC-MS analysis suggested that PtmT3 may also display relaxed catalytic control.
Figure 3.
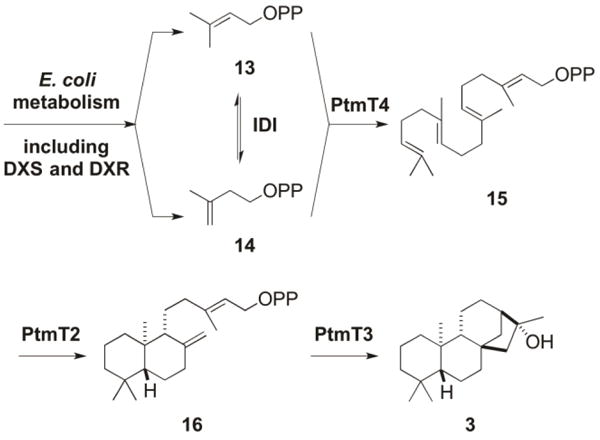
Production of diterpenes in E. coli SB12301 by a two-plasmid system. The terpene building blocks DMAPP (13) and IPP (14) are biosynthesized using the E. coli endogenous pathway supplemented with extra copies of DXS, DXR, and IDI encoded by pBS12301. PtmT4 catalyzing GGPP (15) formation from 13 and 14, the type II DTS PtmT2 cyclizing 15 into ent-CPP (16), and the type I DTS PtmT3 finally converting 16 into (16R)-ent-kauran-16-ol (3), respectively, were encoded by pBS12304.
We next set out to isolate the product(s) of PtmT3 from SB12301 to unambiguously determine the structure(s), which resulted in the isolation of 3 as the only diterpene metabolite upon silica gel chromatography. NMR analysis revealed that 3 had an ent-kaurane scaffold with one oxygenated carbon (δC 79.3) and was identified as the known compound (16R)-ent-kauran-16-ol (or 16α-hydroxy-ent-kaurane).26 GC-MS analysis of pure 3 afforded the same three peaks that were seen in the crude extract (Figure S5), indicating that 3 was the only major diterpene metabolite produced by SB12301; diterpene alcohols are known to undergo dehydration during GC-MS analysis.36 These results, taken together, support the functional assignment of PtmT3 as a product specific (16R)-ent-kauran-16-ol synthase.
Inactivation of ptmO5 in SB12029 revealing that PtmO5 is necessary for PTM, but not PTN, biosynthesis
With PtmT3 functionally characterized as a (16R)-ent-kauran-16-ol synthase, and PtmO4 previously determined as a long-chain acyl-CoA dehydrogenase necessary for β-oxidation of both PTM and PTN,9 ptmO3, ptmO5, and ptmR3, the three remaining genes within the PTM cassette, were next considered as candidates to complete the divergence of PTM biosynthesis from PTN biosynthesis, namely the 11S,16S-ether ring formation for PTM. Close examination of the ptm gene cluster revealed two genes that encode highly homologous α-ketoglutarate-dependent dioxygenases (99% protein sequence identity over 94% coverage): ptmO3, found within the PTM cassette, and ptmO6, the first gene downstream of the 3′-end of the PTM cassette (Figure 2). A homologue of ptmO6, ptnO6, is found in the ptn gene cluster,6 implying a functional role of PtmO6/PtnO6 in the biosynthesis of PTM and PTN. Given the extreme homology between PtmO3 and PtmO6, we reasoned that these enzymes might be functionally redundant, and hence unlikely to be PTM-specific. Likewise, PtmR3, a putative regulator, is unlikely to play a catalytic role in the biosynthesis of PTM. We, therefore, turned our attention to ptmO5, which was predicted to encode a cytochrome P450 monooxygenase.6
Using λRED-mediated PCR-targeting mutagenesis and E. coli–Streptomyces conjugation techniques,21,24 we replaced ptmO5 in the dual PTM-PTN overproducer S. platensis SB120299 with the apramycin resistance cassette aac(3)IV + oriT, to generate the ΔptmO5 mutant SB12036. SB12036 was fermented under the conditions known for both PTM and PTN production, with SB12029 as a positive control (Figure 4, panel I). LC-MS analysis revealed that SB12036 completely lost the production of PTM and PTM S1, the thioacid analogue of PTM (Figure S8),31 but retained the ability to produce PTN and PTN S131 (Figure 4, panels I vs II and III). These findings unambiguously establish that PtmO5 is specific for PTM biosynthesis. Interestingly, concomitant with the loss of PTM biosynthesis in SB12036 was the detection, upon LC-MS analysis, of a large number of new metabolites, none of which had been seen previously in the dual PTM-PTN overproducer SB12029 (Figure 4, panels I vs II).
Figure 4.
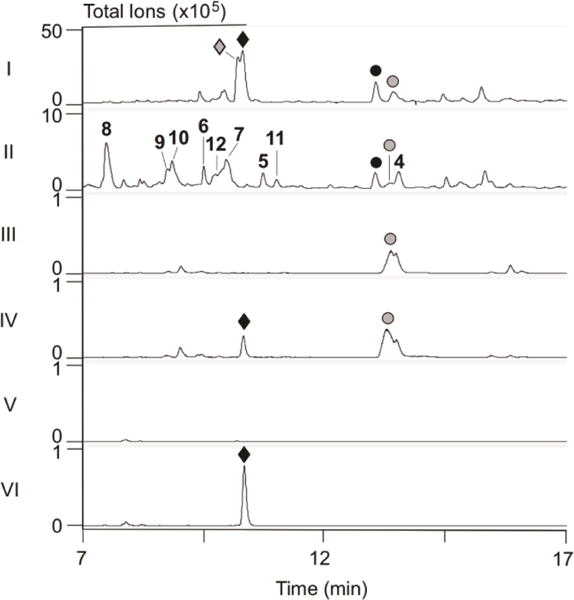
Metabolite profiles of selected S. platensis strains upon LC-MS analysis. Crude extracts were analyzed by (I–II) total ion current (TIC) or (III–VI) extracted ion current (EIC, m/z at 442 for the [PTM + H]+ ion, which coincidentally also extracted the [PTN S1 + H]+ ion with the same m/z value31) chromatograms. (I) SB12029; (II–III) SB12036 (ΔptmO5); (IV) SB12036 + 18; (V) SB12034 (ΔptmT2); (VI) SB12034 + 4. (◆) PTM; (⚫); PTN; (◆) PTM S1; (○) PTN S1; (4–8) PTM and (9) PTN biosynthetic intermediates and (10–12) fully processed PTM analogues isolated from SB12036 (see Figure 1 for structures).
Isolation and structural elucidation of metabolites from the ΔptmO5 mutant SB12036
A large scale fermentation of SB12036 was carried out to isolate the new metabolites for structural elucidation. Following previously reported procedures,9 extraction of a 3.2-L culture and chromatography of the crude extract resulted in the isolation of PTN (2), one PTN (9) and five PTM (4-8) biosynthetic intermediates/congeners, and three fully processed PTM analogues (10-12) (Figures 1 and 4). Of the nine metabolites, excluding PTN, seven (5 and 7–12) are new and two (4 and 6) are known diterpenoids from plants and fungi.27–30 Each new compound was named with an “ML” designation, as described previously,9 to denote that they were isolated from a markerless ΔptmR1 overproducer SB12029.9
(16R)-Hydroxy-ent-kauran-19-oic acid (4) (110 mg isolated) was a major metabolite of SB12036. High-resolution ESIMS (HRESIMS) analysis yielded an [M + Na]+ ion at m/z 343.2241, consistent with a molecular formula of C20H32O3 (calculated [M + Na]+ ion at m/z 343.2244). The 1H and 13C NMR spectra of 4 were consistent with those of the previously reported (16R)-hydroxy-ent-kauran-19-oic acid.27–29
Platensimycin ML9 (5) (82 mg isolated), another major metabolite of SB12036, had a molecular formula of C20H32O4 based on the observed [M + Na]+ ion at m/z 359.2191 (calculated [M + Na]+ at 359.2193). 1D and 2D NMR analysis (SI, Table S4, Figure S50) revealed that 5 had a scaffold similar to 4, with the exception of a hydroxyl group at C-7 (δC 75.5; δH 3.68, dd, J = 11.2, 4.2 Hz). The relative configuration at C-7 was supported to be R by ROESY correlations of H-7 with H-5 (δH 1.27) and H-9 (δH 1.08) (Figure 1). The absolute stereochemistry of 5 was determined by X-ray diffraction using molybdenum radiation (Table S7), together with its biosynthetic relationship with PTM, confirming the 7R,16R-configurations of 5 (Figure 5A).
Figure 5.
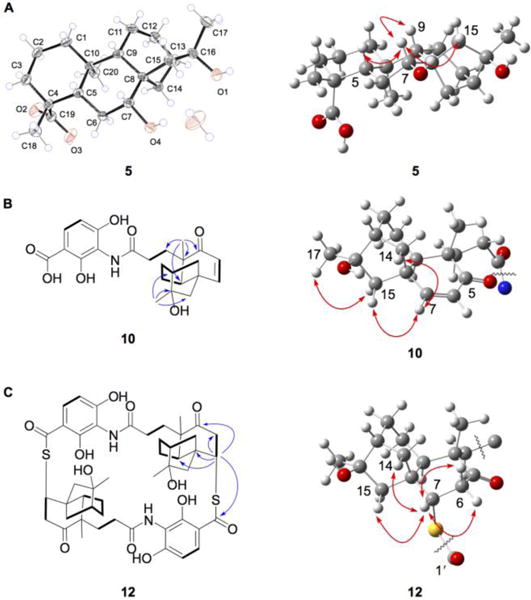
Spectroscopic data supporting the structures of (A) 5, with ORTEP drawing of its crystal structure, confirming the R-configurations at C-7 and C-16, (B) 10, confirming the R-configuration at C-16, and (C) 12, confirming the S-configurations at C-7 of both monomers. Selected key correlations: COSY, bold lines; HMBC, blue arrows; and ROESY, red arrows.
(7S,16R)-Dihydroxy-ent-kauran-19-oic acid (6) (1.5 mg isolated), a known diterpenoid,30 was determined to be a diastereomer of 5 based on its HRESIMS and NMR data (SI, Table S4). The hydroxyl group at C-7 in 6 was assigned the 7S configuration (Figure 1) based on the ROESY correlations between H-7 (δH 4.15) and H-6a (δH 2.64), H-6b (δH 2.46), H-14a (δH 2.22), and H-15a (δH 2.65).
Platensimycin ML10 (7) (13.5 mg isolated) had an identical molecular formula with that of 5 and 6. It was evident from the 1D and 2D NMR (SI, Table S4, Figure S50) that the hydroxyl group at C-7 in 5 and 6 was absent (δC 42.2; δH 1.87 and 1.57), and a hydroxyl group was at C-12 (δC 71.6; δH 4.31, ddd, J = 11.9, 6.3, 3.5 Hz). Based on ROESY correlations of H-12 (δH 4.31) with H3-20 (δH 1.22) and H-14b (δH 2.03), and H3-20 with H-14b, the hydroxyl group at C-12 was assigned the 12R configuration (Figure 1).
Platensimycin ML11 (8) (120 mg isolated), another major metabolite of SB12036, and platencin ML3 (9) (15.2 mg isolated) were found to be ent-kauranol- and ent-atiserene-adenylates, respectively. HRESIMS of 8 afforded the [M + H]+ ion at m/z 666.2892, supporting a molecular formula of C30H44N5O10P (calculated [M + H]+ ion at m/z 666.2899), which is consistent with a structure of 5 linked to an adenosine monophosphate (AMP) moiety. 1D and 2D NMR analysis (SI, Table S5, Figure S50) confirmed the presence of the diterpene scaffold of 5 and an AMP moiety. The 13C NMR signals for C-4 (δC 45.3, d, JP→C = 5.3 Hz) and C-19 (δC 175.2, d, JP→C = 8.8 Hz) were split into doublets due to the long-range coupling with phosphorus, supporting the AMP moiety is linked to C-19. Compound 9 also had a molecular formula (C30H42N5O9P) that was consistent with a diterpene and an AMP moiety. Its 1D and 2D NMR (SI, Table S5, Figure S50) verified an ent-atiserene skeleton linked to an AMP moiety at C-19, again due to the presence of split signals for C-4 (δC 45.3, d, JP→C = 4.6 Hz) and C-19 (δC 175.2, d, JP→C = 9.3 Hz). As with 5, 6, and 8, the ent-atiserene scaffold of 9 also possessed a hydroxyl group at C-7 (δC 78.3; δH 3.50, d, J = 7.7 Hz). ROESY correlations between H-7 and H-5 (δH 1.19), H-6a (δH 2.52), H-9 (δH 1.11), and H-15b (δH 1.96) supported the assignment of the hydroxyl group at C-7 as 7R configuration for 9, the same stereochemistry as that of 5 and 8 (Figure 1).
Platensimycin ML12 (10) (8.6 mg isolated) had a molecular formula of C24H29NO7 based on the observed [M + H]+ ion at m/z 444.2015 (calculated [M + H]+ at 444.2017). The 1H and 13C NMR of 10 (SI, Table S6, Figure S50) was very similar to that of PTM, with one major difference. The 11S,16S-ether linkage between C-11 (δC 18.5; δH 1.61, m) and C-16 (δC 79.0) was absent; only C-16 was oxygenated, indicating a hydroxyl group at C-16 as in 3–8 (Figure 1). ROESY correlations between H3-17 (δH 1.58) and H-15b (δH 1.83), and between H-7 (δH 6.72), H-14a (δH 2.41), and H-15a (δH 2.22) enabled the assignment of 16R configuration for the hydroxyl group at C-16 of 10 (Figure 5B), the same configuration as that for 3–8 but opposite of that in PTM (Figure 1). Platensimycin ML13 (11) (4.6 mg isolated) was easily determined to be the methyl ester of 10 by its almost identical NMR spectra (SI, Table S6), with the one exception being a –OCH3 group (δC 52.4; δH 3.74, s) attached to C-1´ (δC 171.3) (Figure 1).
Finally, platensimycin ML14 (12) (7.1 mg isolated) is an intriguing macrocyclic, sulfur-containing dimer of 10 (Figure 1). HRESIMS of 12 afforded the [M + H]+ ion at m/z 919.3508 supporting a molecular formula of C48H58N2O12S2 (calcd [M + H]+ at m/z 919.3504). As 10 is comprised of 24 carbons, and the 1H and 13C NMR spectra of 12 showed resonances that were consistent with those of 10 (SI, Table S6, Figure S50), it was clear that a dimeric compound had formed. Similarly to the previously isolated sulfur-containing pseudodimer platensimycin D1,31 the enone moieties of both monomers of 10 were lost [C-5 (δC 212.2), C-6 (δC 127.2; δH 5.95, d, J = 9.8 Hz), C-7 (δC 159.4; δH 6.72, d, J = 9.8 Hz)] and the C-1´ carboxylic acid carbonyls of the ADHBA moieties (δC 175.1) were significantly shifted downfield, suggestive of thiocarboxlic acids or esters. The 13C chemical shift of C-7, along with an HMBC correlation between H-7 of one monomer and C-1´ of the other, supported sulfur attachment at C-7. ROESY correlations between H-7 and H-6a (δH 3.40), H-6b (δH 2.46), H-14a (δH 2.49), and H-15a (δH 2.23) finally established the 7S and 7'S configurations of 12 (Figure 5C).
The PtmO5 cytochrome P450 monooxygenase catalyzing 11S,16S-ether ring formation in PTM biosynthesis
With the exception of the PTN congener 9, eight of the nine metabolites (4-8 and 10-12) isolated from SB12036 are derived from 4, lacking the 11S,16S-ether ring characteristic of PTM and instead featuring the (16R)-hydroxyl group at C-16 that is installed by PtmT3 (Figures 1 and 3). Accumulation of 4 as the major biosynthetic intermediate, as well as its hydroxylated (5–7) or adenylated (8) congeners, in SB12036 revealed that PtmO5 must play a role in catalyzing 11S,16S-ether ring formation in PTM biosynthesis, acting early in the pathway before A-ring cleavage and β-oxidation en route to the PTM-ketolide (Figure 6A). This proposal is also consistent with the isolation of the three fully processed PTM analogues (10-12) (Figure 1). Platensimycin ML12 (10) is identical to PTM with the exception that it lacks the 11S,16S-ether ring in the ketolide moiety, with 11 and 12 identified as the corresponding methyl ester and a sulfur-containing cyclic dimer, respectively (Figure 1). The accumulation of these fully processed 11-deoxy-16R-hydroxy-PTM analogues supports the inherent substrate promiscuity of the downstream enzymes in PTM and PTN biosynthesis (Figure 6), which has been recently exploited to construct a focused PTM and PTN library by mutasynthesis.19
Figure 6.
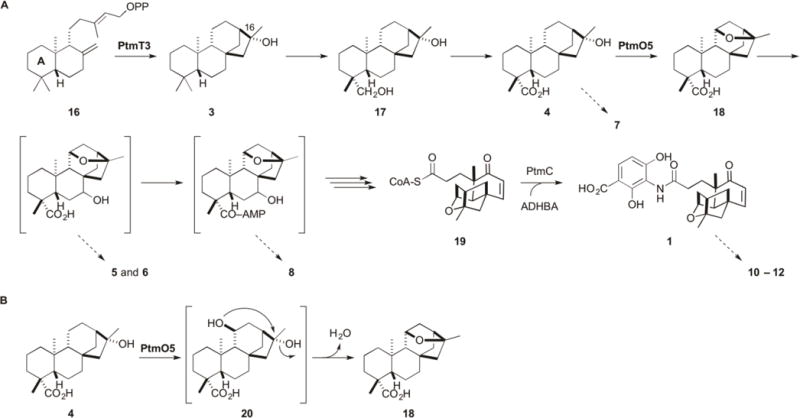
Proposed biosynthesis of PTM supported by the isolation of two intermediates (3 and 4), four congeners (5–8), as well as three fully processed PTM analogues (10–12) from S. platensis SB12036, and intermediates (17 and 18) previously isolated from SB12030.9 (A) ent-CPP (16) is cyclized into (16R)-ent-kauran-16-ol (3) by PtmT3. Oxidation at C-19 likely occurs before 11S,16S-ether ring formation, which is catalyzed by PtmO5. Hydroxylation at C-7 and activation by adenylation at C-19, occurs before A-ring cleavage and β-oxidation. The A-ring is shown in 16. Platensicyl-CoA (19) is coupled to ADHBA by PtmC in the final step of PTM biosynthesis.19 Proposed intermediates in brackets have not been isolated or experimentally confirmed, but are supported by the congeners isolated and indicated with the dotted arrows. Isolation of the adenylated ent-atiserene congener (9) (see Figure 1) also supports parallel processing of the ent-atiserene-derived intermediates in PTN biosynthesis. Congeners 10–12 highlight the flexible nature of the processing enzymes. (B) Proposed model for 11S,16S-ether ring formation by PtmO5. Due to the necessary inversion of stereochemistry at C-16 from 4 to the ether product, an 11S,16R-bis-hydroxylated intermediate (20) is predicted. The final 11S,16S-ether ring formation step may proceed enzymatically as a part of the PtmO5 activity or nonenzymatically as a spontaneous nucleophilic substitution.
Chemical complementation of the ΔptmT2 mutant SB12034 and the ΔptmO5 mutant SB12036 supporting the function and mechanism of PtmO5
Chemical complementation of the ΔptmT2 mutant SB12034 and the ΔptmO5 mutant SB12036 was carried out with strategically selected biosynthetic intermediates to provide additional evidence supporting the role PtmO5 plays in 11S,16S-ether ring formation in PTM biosynthesis (Figure 6). Thus, fermentation of SB12036 in the presence of (11S,16S)-ent-kauran-11,16-epoxy-19-oic acid (18),9 the proposed 11S,16S-ether product of PtmO5 in PTM biosynthesis, restored PTM production (Figure 4, panels III vs IV). Complementarily, fermentation of SB12034, the ΔptmT2 mutant that is incapable of biosynthesizing 16, and therefore any diterpenoids,19 in the presence of 4, similarly restored PTM production (Figure 4, panels V vs VI). These data, together with the other metabolites isolated from SB12036, strongly support that PtmO5, a cytochrome P450 monooxygenase, catalyzes the 11S,16S-ether ring formation in PTM biosynthesis, most likely acting on 4 as a substrate (Figure 6).
Antibacterial activities of fully processed PTM analogues
All metabolites (4–12) isolated from SB12036 were tested for antibacterial activity against S. aureus ATCC 25923 and M. luteus ATCC 9431 using a standard disk diffusion assay.9,19,31 Only the fully processed PTM analogue 10 and the sulfur-containing cyclic dimer 12 showed bacterial growth inhibition. The MICs of 10 and 12 were subsequently determined using the broth dilution method26 with PTM (1) as a positive control. While 10 showed a 128-fold decrease in MIC compared to that of PTM, 12 was marginally (2–8-fold) more active than 10 but still significantly less active than PTM (16–64-fold) (Table 1). Due to differences in molecular weights, the molar MICs of 12 against S. aureus and M. luteus were 34.8 and 8.7 μM, respectively, compared to 1.13 μM for PTM.
Table 1.
Antibacterial activities of 10–12 in comparison to PTM (1) as measured by MICs (μg mL−1)a
| strain | 1 | 10 | 11 | 12 |
|---|---|---|---|---|
| S. aureus ATCC 25923b | 0.5 | 64 | >64 | 32 |
| M. luteus ATCC 9431 | 0.5 | 64 | >64 | 8 |
MIC determinations were performed in duplicate.
Methicillin sensitive
DISCUSSION
PtmT3 and PtmO5 are responsible for the 11S,16S-ether ring formation in PTM biosynthesis
When the ptm and ptn gene clusters were cloned and sequenced, it was evident that a five gene cassette, aptly named the PTM cassette, was responsible for partitioning the biosynthesis of PTM from that of PTN.6 Two genes, ptmT1 and ptmT3, with the latter found within the PTM cassette (Figure 2), were established to encode two dedicated type I DTSs that channel ent-CPP (16) into the characteristic ent-kaurene and ent-atiserene scaffolds of PTM and PTN, respectively. How the additional modifications to the ent-kaurene scaffold that are specific to PTM biosynthesis, i.e., formation of the 11S,16S-ether ring, were accomplished, however, remained elusive. Functional characterization of PtmT3 as an (16R)-ent-kauran-16-ol synthase (Figures 3 and 6A), instead of an ent-kaur-16-ene synthase as initially postulated,6 revealed that PtmT3 quenches its carbocation cyclization cascade (i.e., the ent-kauran-16-yl cation) with a water molecule (Figure S7).
Initially, we presumed that the C-16 hydroxyl group installed by PtmT3 was a likely candidate for the 11S,16S-ether ring oxygen of PTM. However, upon full structure determination of (16R)-ent-kauran-16-ol, as well as the other PTM biosynthetic intermediate/congeners and the fully processed PTM analogues, isolated from SB12036, the C-16 hydroxyl group of each compound was found to be R-configuration, opposite to the S-configuration for the 11S,16S-ether ring of PTM. Successful chemical complementation of the ΔptmT2 mutant SB12034, which harbored a functional copy of ptmO5 mutant,19 with 4, which features a 16R-hydroxyl group, resulting in the production of PTM, provided direct evidence that the 16R-hydroxyl group of 4 can in fact be transformed into the 11S,16S-ether ring of PTM. This implies that the R-hydroxyl group at C-16 must either invert its configuration before ether ring formation, or a second oxygen is incorporated into 4 to install the S-configuration at C-16. A potential 11S,16R-diol intermediate (20) can be easily envisaged, which may then undergo 11S,16S-ether ring formation with concomitant dehydration (Figure 6B). In the total synthesis of PTM, the 11S,16S-ether ring of PTM was constructed using an intermediate similar to that of 20, albeit with an alkyl halide (Br on C-11) and 16S-hydroxyl group in strongly basic conditions.37
Isolation and structural elucidation of the metabolites accumulated in the ΔptmO5 mutant SB12036 unambiguously established the necessary role PtmO5 played in 11S,16S-ether ring formation in PTM biosynthesis. PtmO5, a cytochrome P450 monooxygenase, therefore serves as an outstanding candidate to catalyze C-11 hydroxylation to install the 11S,16R-diol as in the proposed intermediate 20. We can not rule out at this time, however, if dehydration of 20 to afford a product such as 18, featuring the characteristic 11S,16S-ether ring of PTM, proceeds enzymatically as a part of the PtmO5 activity or results from a nonenzymatic reaction (Figure 6B).
PtmT3, the first (16R)-ent-kauran-16-ol synthase in bacteria, and PtmO5, an ether ring-forming cytochrome P450 monooxygenase, as models for enzymology
ent-Kaurene synthase catalyzes a key step in gibberellin biosynthesis and plant, fungal, and bacterial versions of this enzymes are known.35,38–41 The bifunctional PpCPS/KS from P. patens produces both ent-kaurene and (16R)-ent-kauran-16-ol in a 1:4 ratio (Figure S7).35 Mutagenesis of A710 in PpCPS/KS to either Met or Phe altered its product specificity to 100% ent-kaurene, suggesting that H2O-quenching of the ent-kauran-16-yl cation can be prevented by a steric block.42 A similar product specificity switch was seen in PtTPS20 from Populus trichocarpa when a T607M (analogous to A710 in PpCPS/KS) mutation altered the typically (16R)-ent-kauran-16-ol dominant (~86%) product profile to 100% ent-kaurene.43 The monofunctional BjKS from the Proteobacterium Bradyrhizobium japonicum solely produces ent-kaurene (Figure S7), and its crystal structure revealed that two bulky and hydrophobic residues, Y136 and L140 (analogous to A710 in PpCPS/KS), likely block water from accessing the cyclization intermediate.44 PtmT3 is the first (16R)-ent-kauran-16-ol synthase from bacteria, providing an excellent opportunity to study diterpene cyclization and product determination from a bacterial DTS.45 Two questions immediately stand out: (i) are L142 and V146, the residues in the Y136 and L140 positions of BjKS, responsible for product determination by allowing a water molecule access to the ent-kauran-16-yl carbocation and (ii) what are the implications of the lack of a classical type I terpene synthase DDxxD motif? In fact, PtmT3 has an apparent DDxE motif that aligns with the DDxxD motifs of other ent-KSs as determined by sequence alignment (Figure S9) and structural homology models (Figure S10).
Likewise, the PTM biosynthetic pathway offers an excellent opportunity to study ether formation in natural product biosynthesis. Our data suggest that PtmO5 catalyzes a regio- and stereospecific formation of an 11S,16S-ether bond from (16R)-ent-kauran-16-ol, possibly via an 11S,16R-diol intermediate (20) due to the necessary inversion of stereochemistry at C-16 (Figure 6B). The stereochemical drive of 11S,16S-ether ring formation in PTM biosynthesis is inherently different from that of AveE in avermectin and AurH in aureothin, where only one of the ether carbons possesses stereochemistry (Figure S1).13–17 The mechanism of ether ring formation in PTM may also differ from that in aureothin biosynthesis as the second and final C–O bond formation occurs at a carbon (C-16) with a pre-installed oxygen, as opposed to a transient oxidation; the mechanism of ether formation in avermectin is still unknown. Continued biochemical and structural investigation of PtmO5 and its catalyzed mechanism will undoubtedly contribute to the understanding of this regio- and stereospecific enzymatic reaction and cytochrome P450 monooxygenases in general.
An ent-kaurene natural product, featuring a similar 11S,16S-ether ring, was previously isolated in the gibberellin-producing fungus G. fujikuroi, but the enzyme(s) responsible for this transformation remains unknown (Figure S1).18 The similarities between the fungal diterpenoid and intermediates in PTM biosynthesis are undeniable. Thus, the study of PtmO5 and the 11S,16S-ether ring formation in PTM may also shed light into unanswered questions in fungal diterpenoid biosynthesis.
The stereochemistry at C-16 of PTM affecting its antibacterial activity
The 11-deoxy-16R-hydroxy-PTM analogue 10 has an ADHBA moiety, amide linkage, and enone functional group of the ketolide moiety, each of which is a vital part for the impressive biological activity of PTM.1,11 The only structural difference of 10 to PTM is the lack of the characteristic 11S,16S-ether ring; instead, 10 is a 11-deoxy-16R-hydroxy congener. PTM, with an 11S,16S-ether ring, makes a key hydrogen bond between the ether oxygen and a Thr side chain in FabF.1 It is likely that 10 is unable to form this crucial hydrogen bond, due to the R-configuration of the hydroxyl group at C-16, projecting it away from the Thr side chain, hence resulting in a significant (128-fold) decrease in activity. It is tempting to predict that PTM analogues with either a 16S- or 11S-hydroxyl group, therefore retaining the crucial hydrogen bond to the Thr residue, would be more active than 10. Nevertheless, it is evident that the 11S,16S-ether ring, or minimally the stereochemistry of the oxygen at C-16 and/or C-11, affects the biological activity of PTM. This is consistent with the sulfur-containing dimer 12, which, featuring the same 16R-hydroxyl group at C-16, is also less active than PTM, albeit marginally more active than 10.
The metabolites isolated from SB12301 and SB12036 continuing to reveal new insights into the PTM and PTN biosynthetic pathway
The model proposed previously for PTM and PTN biosynthesis consisted of a unified pathway where (i) ent-CPP (16) is the final common intermediate in the biosynthesis of both PTM and PTN, (ii) dedicated DTSs partition ent-CPP into the ent-kaurene and ent-atiserene scaffolds, (iii) a group of enzymes, likely encoded by genes within the PTM cassette, reacts specifically on the ent-kaurene scaffold to form the characteristic 11S,16S-ether ring of PTM, (iv) a group of enzymes reacts with both scaffolds of PTM and PTN to process the diterpene intermediates into the PTM- and PTN-ketolides, activated in the form of platensicyl-CoA (19) and platencinyl-CoA, respectively, and (v) a final amide coupling reaction with ADHBA to form PTM (1) and PTN (2).6,8,9,19 The isolation of 3 from E. coli SB12301 and 4–12 from S. platensis SB12036 not only supports the unified pathway for PTM and PTN biosynthesis but also reveals new insights into the complex nature of the biosynthetic pathway for the PTM and PTN family natural products (Figure 6A).
The isolation of 3 from the heterologous expression of ptmT2, ptmT3, and ptmT4 in E. coli SB12301 concludes that (16R)-ent-kauran-16-ol, and not ent-kaurene, is the first pathway specific diterpene intermediate in PTM biosynthesis. As previously observed,8,9 and reminiscent of gibberellin biosynthesis,38 oxidation at C-19 occurs early in PTM biosynthesis as evidenced by carboxylic acid moieties at C-19 in the proposed biosynthetic intermediate 4 and congeners 5-7 (Figure 6A). Oxidation at C-19 appears to set up CoA activation for downstream enzymes and is supported by the isolation of adenylated congeners 8 and 9. CoA synthetases are initially activated by ATP, forming adenylate intermediates that are then converted into acyl-CoA products.46 Accumulation of C-7 hydroxylated congeners, such as 5–9, and as previously reported in S. platensis SB12030 and the heterologous PTN-producing Streptomyces lividans SB12606,8,9 continues to support that hydroxylation at C-7 occurs before A-ring cleavage. However, the isolation of congeners with both configurations at C-7, such as 5 and 6, would suggest that either this hydroxylation was non-stereospecific or one of the diasteromers resulted from adventitious hydroxylation.
The timing of 11S,16S-ether ring formation is proposed to occur after oxidation of C-19 to the corresponding carboxylic acid and before hydroxylation at C-7 (Figure 6). This is supported by the isolation of 4 from the ΔptmO5 mutant SB12036 in this study, and of (16R)-ent-kauran-16,19-diol (17) and (11S,16S)-ent-kauran-11,16-epoxy-19-oic acid (18) from the ΔptmO4 mutant SB12030 previously,9 as well as chemical complementation of the ΔptmT2 mutant SB12034 by 4 in this study. The exact timing, however, would require direct in vitro characterization of PtmO5 with the predicted substrate 4. Isolation of 7, with its C-12 hydroxyl group, is attributed to adventitious oxidation of 4, which was accumulated as a major metabolite in SB12036.
Finally, complete processing of the (16R)-ent-kauran-16-ol scaffold to 10, as opposed to the natural (11S,16S)-ent-kauran-11,16-epoxy-19-ol scaffold to PTM, is evidence that the PTM and PTN biosynthetic machinery possesses remarkable substrate promiscuity, efficiently processing unnatural diterpene congeners to completion as in PTM and PTN biosynthesis. The (16R)-ent-kauran-16-ol and ent-atiserene scaffolds are the only two known natural diterpene scaffolds that are incorporated into the PTM and PTN family of antibiotics known to date. The inherent substrate promiscuity of the PTM and PTN biosynthetic pathway would suggest that alternative diterpene scaffolds, such as ent-kaurene, ent-pimaradiene, or other ent-CPP-derived scaffolds may also be processed to PTM and PTN analogues. Combinatorial biosynthesis, exploiting substrate promiscuity of the PTM and PTN biosynthetic pathway, has indeed been achieved recently, with great success, by a mutasynthesis strategy, yielding a focused library of PTM and PTN that featured varying aminobenzoic acid moieties.19 An excellent opportunity has now emerged to test the limits of diterpene-based combinatorial biosynthesis for PTM and PTN structural diversity featuring varying ketolide moieties within an outstanding model system.
CONCLUSIONS
PTM and PTN are the most highly functionalized bacterial diterpenoid natural products. Their biosynthetic pathways, therefore, provide outstanding opportunities to discover novel chemistry and biology and apply biotechnological strategies to generate structural diversity for privileged natural product scaffolds. In this study, we discovered the biosynthetic origin of the 11S,16S-ether ring in PTM. First, PtmT3 cyclizes ent-CPP into (16R)-ent-kauran-16-ol and is the first (16R)-ent-kauran-16-ol synthase characterized from bacteria. Second, PtmO5 is a cytochrome P450 monooxygenase that catalyzes the formation of the 11S,16S-ether ring in PTM from (16R)-ent-kauran-16-ol. Pending in vitro confirmation, in vivo experiments support the proposal that PtmO5 catalyzes the transformation of 4 to 18, most likely via the 11S,16R-diol intermediate 20. Third, the rarity of bacterial diterpenoids makes PTM and PTN excellent models for studying novel tailoring reactions in diterpene biosynthesis, while similarities between diterpenoid production in bacteria, plants, and fungi (e.g., gibberellins) allows comparison of their biosynthetic pathways and machineries and exposes insights into their possible evolutionary relationships. Finally, the genetically amenable PTM–PTN dual overproducer SB12029, coupled with the remarkable inherent substrate promiscuity of the PTM and PTN biosynthetic machinery, continues to prove its value as a powerful tool for bioengineering, substrate generation, and combinatorial chemistry.
Supplementary Material
Acknowledgments
We thank Dr. Matt Gill at The Scripps Research Institute, Florida for the use of the GC-MS, Dr. HaJeung Park of the X-ray Crystallography Core Facility at The Scripps Research Institute, Florida, and the John Innes Center, Norwich, U.K., for providing the REDIRECT Technology kit. JDR is supported in part by an Arnold O. Beckman Postdoctoral Fellowship. This work is supported in part by the National Institutes of Health Grant GM114353.
Footnotes
Supporting Information
Supporting experimental procedures; strains, plasmids, and primers used in this study (Tables S1–S3); summary of NMR data for compounds 5–12 (Tables S4–S6); crystal data and structure refinement for compound 5 (Table S7); relevant tetrahydrofuran ring-containing natural products and the enzymes implicated in ether ring formation (Figure S1); design and construction of the E. coli diterpene production system (Figure S2); design and verification of the ΔptmO5 mutant SB12036 (Figure S3); Southern analysis of the SB12036 mutant (Figure S4); GC-MS analysis of the E. coli diterpene production system (Figure S5); MS spectra of the three diterpene peaks from E. coli SB12301 (Figure S6); proposed cyclization cascade of ent-kaur-16-ene or (16R)-ent-kauran-16-ol formation from ent-CPP (Figure S7); structures of PTM S1 and PTN S1 (Figure S8); sequence alignment of PtmT3 and selected ent-kaurene synthases (Figure S9); active site comparisons of PtmT3, PpCPS/KS, and BjKS (Figure S10); NMR spectra of compounds 5–12 (Figures S11–S49); key 2D NMR correlations of compounds 5–10, and 12 (Figure S50). This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
References
- 1.Wang J, Soisson SM, Young K, Shoop W, Kodali S, Galgoci A, Painter R, Parthasarathy G, Tang YS, Cummings R, Ha S, Dorso K, Motyl M, Jayasuriya H, Ondeyka J, Herath K, Zhang C, Hernandez L, Allocco J, Basilio A, Tormo JR, Genilloud O, Vicente F, Pelaez F, Colwell L, Lee SH, Michael B, Felcetto T, Gill C, Silver LL, Hermes JD, Bartizal K, Barrett J, Schmatz D, Becker JW, Cully D, Singh SB. Nature. 2006;441:358. doi: 10.1038/nature04784. [DOI] [PubMed] [Google Scholar]
- 2.Wang J, Kodali S, Lee SH, Galgoci A, Painter R, Dorso K, Racine F, Motyl M, Hernandez L, Tinney E, Colletti SL, Herath K, Cummings R, Salazar O, Gonzalez I, Basilio A, Vicente F, Genilloud O, Pelaez F, Jayasuriya H, Young K, Cully DF, Singh SB. Proc Natl Acad Sci USA. 2007;104:7612. doi: 10.1073/pnas.0700746104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu M, Singh SB, Wang J, Chung CC, Salituro G, Karanam BV, Lee SH, Powles M, Ellsworth KP, Lassman ME, Miller C, Myers RW, Tota MR, Zhang BB, Li C. Proc Natl Acad Sci USA. 2011;108:5378. doi: 10.1073/pnas.1002588108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh SB, Jayasuriya H, Ondeyka JG, Herath KB, Zhang C, Zink DL, Tsou NN, Ball RG, Basilio A, Genilloud O, Diez MT, Vicente F, Pelaez F, Young K, Wang J. J Am Chem Soc. 2006;128:11916. doi: 10.1021/ja062232p. [DOI] [PubMed] [Google Scholar]
- 5.Jayasuriya H, Herath KB, Zhang C, Zink DL, Basilio A, Genilloud O, Diez MT, Vicente F, Gonzalez I, Salazar O, Pelaez F, Cummings R, Ha S, Wang J, Singh SB. Angew Chem, Int Ed. 2007;46:4684. doi: 10.1002/anie.200701058. [DOI] [PubMed] [Google Scholar]
- 6.Smanski MJ, Yu Z, Casper J, Lin S, Peterson RM, Chen Y, Wendt-Pienkowski E, Rajski SR, Shen B. Proc Natl Acad Sci USA. 2011;108:13498. doi: 10.1073/pnas.1106919108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smanski MJ, Peterson RM, Rajski SR, Shen B. Antimicrob Agents Chemother. 2009;53:1299. doi: 10.1128/AAC.01358-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smanski MJ, Casper J, Peterson RM, Yu Z, Rajski SR, Shen B. J Nat Prod. 2012;75:2158. doi: 10.1021/np3005985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rudolf JD, Dong L-B, Huang T, Shen B. Mol BioSyst. 2015;11:2717. doi: 10.1039/c5mb00303b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hindra, Huang T, Yang D, Rudolf JD, Xie P, Xie G, Teng Q, Lohman JR, Zhu X, Huang Y, Zhao L-X, Jiang Y, Duan Y, Shen B. J Nat Prod. 2014;77:2296. doi: 10.1021/np5006168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nicolaou KC, Stepan AF, Lister T, Li A, Montero A, Tria GS, Turner CI, Tang Y, Wang J, Denton RM, Edmonds DJ. J Am Chem Soc. 2008;130:13110. doi: 10.1021/ja8044376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rudolf JD, Dong L-B, Cao H, Hatzos-Skintges C, Osipiuk J, Endres M, Chang C-Y, Ma M, Babnigg G, Joachimiak A, Phillips George N, Jr, Shen B. J Am Chem Soc. 2016;138:10905. doi: 10.1021/jacs.6b04317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pang C-H, Matsuzaki K, Ikeda H, Tanaka H, Omura SJ. Antibiot. 1995;48:59. doi: 10.7164/antibiotics.48.59. [DOI] [PubMed] [Google Scholar]
- 14.Ikeda H, Nonomiya T, Usami M, Ohta T, Omura S. Proc Natl Acad Sci USA. 1999;96:9509. doi: 10.1073/pnas.96.17.9509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He J, Mueller M, Hertweck C. J Am Chem Soc. 2004;126:16742. doi: 10.1021/ja046104h. [DOI] [PubMed] [Google Scholar]
- 16.Richter MEA, Traitcheva N, Knuepfer U, Hertweck C. Angew Chem, Int Ed. 2008;47:8872. doi: 10.1002/anie.200803714. [DOI] [PubMed] [Google Scholar]
- 17.Zocher G, Richter MEA, Mueller U, Hertweck CJ. Am Chem Soc. 2011;133:2292. doi: 10.1021/ja110146z. [DOI] [PubMed] [Google Scholar]
- 18.Fraga BM, Gonzalez P, Guillermo R, Hernandez MG. Nat Prod Lett. 1996;8:257. [Google Scholar]
- 19.Dong L-B, Rudolf JD, Shen B. Org Lett. 2016;18:4606. doi: 10.1021/acs.orglett.6b02248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sheldrick GM. Acta Crystallogr, Sect A Found Crystallogr. 2008;64:112. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 21.Gust B, Challis GL, Fowler K, Kieser T, Chater KF. Proc Natl Acad Sci USA. 2003;100:1541. doi: 10.1073/pnas.0337542100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.MacNeil DJ, Gewain KM, Ruby CL, Dezeny G, Gibbons PH, MacNeil T. Gene. 1992;111:61. doi: 10.1016/0378-1119(92)90603-m. [DOI] [PubMed] [Google Scholar]
- 23.Sambrook J, Russel D. Molecular cloning: A Laboratory Manual. 3rd. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 2001. [Google Scholar]
- 24.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. Practical Streptomyces Genetics. The John Innes Foundation; Norwich, UK: 2000. [Google Scholar]
- 25.Morrone D, Lowry L, Determan MK, Hershey DM, Xu M, Peters RJ. Appl Microbiol Biotechnol. 2010;85:1893. doi: 10.1007/s00253-009-2219-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanson JR, Siverns M, Piozzi F, Savona G. J Chem Soc, Perkin Trans. 1976;1:114. [Google Scholar]
- 27.Duan H, Takaishi Y, Momota H, Ohmoto Y, Taki T, Jia Y, Li D. J Nat Prod. 1999;62:1522. doi: 10.1021/np9902183. [DOI] [PubMed] [Google Scholar]
- 28.Chen C-Y, Chang F-R, Cho C-P, Wu Y-C. J Nat Prod. 2000;63:1000. doi: 10.1021/np0000320. [DOI] [PubMed] [Google Scholar]
- 29.Serebryakov EP, Simolin AV, Kucherov VF, Rozynov BV. Tetrahedron. 1970;26:5215. [Google Scholar]
- 30.El-Emary NA, Kusano G, Takemoto T. Chem Pharm Bull. 1976;24:1664. doi: 10.1248/cpb.24.661. [DOI] [PubMed] [Google Scholar]
- 31.Dong L-B, Rudolf JD, Shen B. Bioorg Med Chem. 2016;24:6348. doi: 10.1016/j.bmc.2016.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wiegand I, Hilpert K, Hancock REW. Nat Protoc. 2008;3:163. doi: 10.1038/nprot.2007.521. [DOI] [PubMed] [Google Scholar]
- 33.Cyr A, Wilderman PR, Determan M, Peters RJ. J Am Chem Soc. 2007;129:6684. doi: 10.1021/ja071158n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Steele CL, Crock J, Bohlmann J, Croteau R. J Biol Chem. 1998;273:2078. doi: 10.1074/jbc.273.4.2078. [DOI] [PubMed] [Google Scholar]
- 35.Hayashi K-i, Kawaide H, Notomi M, Sakigi Y, Matsuo A, Nozaki H. FEBS Lett. 2006;580:6175. doi: 10.1016/j.febslet.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 36.Keeling CI, Madilao LL, Zerbe P, Dullat HK, Bohlmann J. J Biol Chem. 2011;286:21145. doi: 10.1074/jbc.M111.245951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mulzer J, Tiefenbacher K. Angew Chem Int Ed. 2008;47:2548. doi: 10.1002/anie.200705303. [DOI] [PubMed] [Google Scholar]
- 38.Hedden P, Kamiya Y. Annu Rev Plant Physiol Plant Mol Biol. 1997;48:431. doi: 10.1146/annurev.arplant.48.1.431. [DOI] [PubMed] [Google Scholar]
- 39.Keeling CI, Dullat HK, Yuen M, Ralph SG, Jancsik S, Bohlmann J. Plant Physiol. 2010;152:1197. doi: 10.1104/pp.109.151456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kawaide H, Imai R, Sassa T, Kamiya Y. J Biol Chem. 1997;272:21706. doi: 10.1074/jbc.272.35.21706. [DOI] [PubMed] [Google Scholar]
- 41.Morrone D, Chambers J, Lowry L, Kim G, Anterola A, Bender K, Peters R. J FEBS Lett. 2009;583:475. doi: 10.1016/j.febslet.2008.12.052. [DOI] [PubMed] [Google Scholar]
- 42.Kawaide H, Hayashi K-i, Kawanabe R, Sakigi Y, Matsuo A, Natsume M, Nozaki H. FEBS J. 2011;278:123. doi: 10.1111/j.1742-4658.2010.07938.x. [DOI] [PubMed] [Google Scholar]
- 43.Irmisch S, Muller AT, Schmidt L, Gunther J, Gershenzon J, Kollner TG. BMC Plant Biol. 2015;15:262. doi: 10.1186/s12870-015-0647-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu W, Zheng Y, Feng X, Huang C-H, Chen C-C, Cui Y, Li J, Guo R-T, Nakano C, Hoshino T, Bogue S, Oldfield E, Ko T-P, Wang I, Hsu Shang-Te D. Sci Rep. 2014;4:6214. doi: 10.1038/srep06214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smanski MJ, Peterson RM, Huang S-X, Shen B. Curr Opin Chem Biol. 2012;16:132. doi: 10.1016/j.cbpa.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gulick AM. ACS Chem Biol. 2009;4:811. doi: 10.1021/cb900156h. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.