

Abstract
Bleomycin (BLM) is used clinically in combination with a number of other agents for the treatment of several types of tumours. Members of the BLM family of drugs include zorbamycin (ZBM), phleomycin D1, BLM A2 and BLM B2. By manipulating the BLM biosynthetic machinery, we have produced two new BLM analogues, BLM Z and 6′-deoxy-BLM Z, with the latter exhibiting significantly improved DNA cleavage activity. Here we determined the DNA sequence specificity of BLM Z, 6′-deoxy-BLM Z and ZBM, in comparison with BLM, with high precision using purified plasmid DNA and our recently developed technique. It was found that ZBM had a different DNA sequence specificity compared with BLM and the BLM analogues. While BLM and the BLM analogues showed a similar DNA sequence specificity, with TGTA sequences as the main site of cleavage, ZBM exhibited a distinct DNA sequence specificity, with both TGTA and TGTG as the predominant cleavage sites. These differences in DNA sequence specificity are discussed in relation to the structures of ZBM, BLM and the BLM analogues. Our findings support the strategy of manipulating the BLM biosynthetic machinery for the production of novel BLM analogues, difficult to prepare by total synthesis; some of which could have beneficial cancer chemotherapeutic properties.
Keywords: Anti-tumour agent, DNA cleavage, DNA sequence specificity, bleomycin analogue, zorbamycin
1. INTRODUCTION
The bleomycins (BLMs) are a family of glycopeptide antibiotics that were originally isolated by Umezama and colleagues from Streptomyces verticillis [1]. BLM is utilised as part of regimens to treat testicular, head and neck cancers, as well as Hodgkin’s lymphoma [2–4]. After intravenous administration of BLM in a metal-free form, Cu(II) binds to form BLM-Cu(II) that is transported into the cell [5, 6]. The BLM-Cu(II) is intracellularly reduced to BLM-Cu(I), followed by replacement by Fe(II). In a series of reactions, this BLM-Fe(II) complex removes a hydrogen atom from the C4′ deoxyribose sugar to produce a 4′-radical intermediate which in the presence of O2 leads to the generation of a gapped DNA site with 5′-phosphate and 3′-phosphoglycolate ends. This gapped DNA may undergo further interaction with BLM to form a double-strand DNA break [7, 8]. Alternatively, oxidation of the 4′-radical intermediate followed by addition of H2O generates a 4′-oxidized abasic site [9]. The cytotoxic effect of BLM is thought to arise from its ability to damage DNA [5].
Utilising purified plasmid DNA sequences, the primary site of BLM cleavage was previously found to be 5′-GT-3′ and 5′-GC-3′ sequences [10–18]. BLM cleavage of hairpin DNAs has been investigated and resulted in a different spectrum of BLM binding sites [7, 8, 19, 20]. The DNA sequence specificity of BLM has been determined in human cells [21–27]. Our group has elucidated the DNA sequence specificity of BLM in the entire human genome using next-generation sequencing; and an analysis of over 200 million double-strand break sites in human cells found that BLM preferentially cleaves at RTGT*AY where R is a purine (G or A), Y is a pyrimidine (T or C), and T* is the cleavage site [28]. Chromatin structure has also been shown to affect BLM cleavage since BLM preferentially cleaves in the linker region of the nucleosome [21, 29–32].
The DNA sequence specificity of BLM in purified DNA can be determined using fluorescently end-labelled DNA sequences followed by capillary electrophoresis with laser-induced fluorescence detection (CE-LIF) [33, 34]. By use of DNA sequencing reactions, the DNA sequence specificity can be determined to the precise base pair [35, 36]. The CE-LIF procedure enables precise measurement of cleavage efficiencies at different sites on DNA and hence an accurate DNA sequence specificity can be obtained [35]. A double-fluorescent label CE-LIF technique was used in this study that has previously been shown to be the most accurate method available for DNA sequence specificity measurements [37].
BLM is typically produced and isolated from fermentation of S. verticillus. Although numerous BLM analogues have been synthesized, allowing important structure-activity relationship studies, total synthesis of complex natural products such as BLM is difficult. BLM consists of four components – the metal binding region, disaccharide, linker region, and a bithiazole tail (Fig. 1); and is synthesised via a large 120 kb gene cluster encoding non-ribosomal peptide synthetases and polyketide synthetases. Other members of the BLM family include phleomycin (PLM) D1, and zorbamycin (ZBM) (Fig. 1). We have recently cloned the gene clusters and characterized the biosynthetic machineries for BLM from S. verticillus [38–42] and for ZBM from Streptomyces flavoviridis [42, 43]. While manipulation of the BLM biosynthetic machinery in S. verticillus turned out to be extremely difficult, forfeiting most attempts to engineer BLM analogues in the BLM native producer S. verticillus [41, 42], we have circumvented the genetic recalcitrance of S. verticuillus by expressing the entire BLM biosynthetic gene cluster in the ZBM producer S. flavoviridis. We have recently demonstrated the feasibility of engineered production of novel BLM analogues in S. flavoviridis, which is pliable genetically, and successfully produced two BLM analogues, BLM Z and 6′-deoxy-BLM Z, together with ZBM [43, 44] (Fig. 1). These new BLM analogues, together with ZBM, featured unique structural characteristics and showed varying DNA cleavage activities, thereby providing an outstanding opportunity to study the structure-activity relationship for the BLM family of anticancer drugs.
Figure 1. The structures of BLM A2, BLM B2, 6′-deoxy-BLM Z, BLM Z, ZBM and PLM.
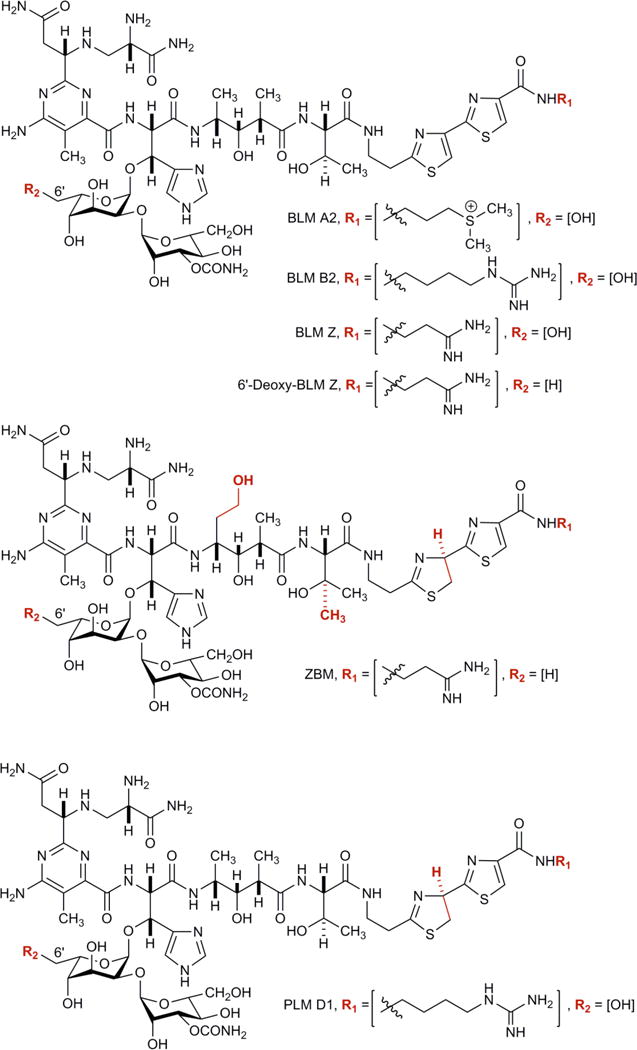
Differences in chemical structure are shown in red.
The bacterium S. flavoviridis naturally produces ZBM that has an alternate C-terminal tail compared with BLM (Fig. 1) [43]. When the BLM gene cluster is expressed in S. flavoviridis, the C-terminal tail is derived from the S. flavoviridis biosynthetic apparatus and hence the C-terminal tails in BLM Z, and 6′-deoxy-BLM Z are similar to ZBM [44]. BLM Z and BLM have identical chemical structures except for the C terminal tail.
In this paper we report the DNA sequence specificity of three BLM analogues, BLM Z, 6′-deoxy-BLM Z and ZBM, that were produced in S. flavoviridis and compared their properties with BLM produced in S. verticillis. The RTGTAY plasmid was used as the DNA target for these sequence specificity studies and this plasmid was designed to contain BLM cleavage sites with systematic variations in the flanking DNA sequences [28]. The double-fluorescent label CE-LIF technique was used since it is the most accurate DNA sequence specificity technology available [37].
2. MATERIALS AND METHODS
BLM was obtained from Bristol Laboratories under the trade name Blenoxane and consists of BLM A2 (70%) and B2 (30%). The BLM analogues, 6-deoxy-BLM Z, BLM Z, and ZBM, were produced in the laboratory of Ben Shen [44].
The RTGTAY plasmid used in this study was derived from pUC19 and included an insert region that consisted of a series of TGTA-related sequences expected to be BLM cleavage sites (Fig. 2). It also contained a telomeric repeat region (T7), comprising of seven consecutive GGGTTA tandem repeats, and a ten consecutive guanine sequence (G10). The plasmid was produced by GenScript.
Figure 2. The sequence of the RTGTAY construct used in this study.

This DNA construct was derived from a pUC19-based plasmid. It contained seven telomeric sequence tandem repeats (GGGTTA) (labelled T7); and also a region of ten consecutive guanines (G10). The region from position 55–427 (underlined in grey) contained 5′-TGTA-3′ BLM cleavage sites with systematic variations in the flanking nucleotides. The Seq and the Rev2 oligonucleotide primers are underlined in bold. The upper strand contained the main BLM cleavage sites and was the focus of this study. Blue arrows indicate the sites where BLM and the analogues were observed to cleave.
The oligonucleotides Rev2 (5′-ATTGTGAGCGGATAAC) and Seq (5′-TCCCAGTCACGACGT) were HPLC-purified by the manufacturer (Invitrogen). The PET-labelled version of Seq was obtained from Applied Biosystems.
The primers PET-Seq and Rev2 were used to generate the 5′-PET-end labelled PCR product. The PCR reaction consisted of approximately 3.5 ng of RTGTAY plasmid DNA template; 5 pmol of each primer; 16.6 mM (NH4)2SO4; 67 mM Tris-HCl, pH 8.8; 6.7 mM MgCl2; and 1 U of AmpliTaq DNA polymerase (Life Technologies). The reaction mixtures were subjected to thermal cycling at 95°C for 5 min; followed by 25 cycles of 95°C for 45 s, 55°C for 1 min, 72°C for 2 min; and finally a single cycle of 72°C for 10 min. Thermal cycling was performed in a Bio-Rad DNA Engine Dyad® Peltier thermal cycler. The products were then purified by 5% (w/v) polyacrylamide gel electrophoresis and dissolved in 10 mM Tris-HCl, pH 8; 0.1 mM EDTA.
For 3′-FAM-end labelling [36, 45], a PCR was performed as above but with the primers Rev2 and Seq. The PCR products were treated with ExoSAP to remove unincorporated nucleotides and primers. To every 10 μL of PCR product, 2 μL of ExoSAP mix (67 mM glycine-KOH, pH 9.5; 6.7 mM MgCl2; 10 mM B-mercaptoethanol; and enzymes 2 U exonuclease I and 0.1 U shrimp alkaline phosphatase) was added, the samples incubated at 30°C for 30 min, followed by heat inactivation at 80°C for 15 min. To the ExoSAP-treated sample, 10 μL of the labelling reaction mix (25 mM Tris-HCl, pH 7.9; 125 mM NaCl; 25 mM MgCl2; 2.5 mM DTT; 100 μM deoxynucleotide triphosphates-A, -G and -C; 2.5 μM FAM-labelled dUTP (Jena Biosciences); and 0.5 U Klenow fragment DNA polymerase) was added. The labelling reaction was incubated at 37°C for 20 min, followed by heat inactivation at 75°C for 20 min. The resulting 3′-FAM-end labelled PCR product was then purified by 5% (w/v) polyacrylamide gel electrophoresis and dissolved in 10 mM Tris-HCl, pH 8; 0.1 mM EDTA.
The cleavage assay for BLM (and analogues) was performed simultaneously on 5′-PET-end labelled and 3′-FAM-end labelled PCR products. In each 10 μL reaction, the cleavage assay contained approximately 12 ng of the 5′-PET-end labelled PCR product and 12 ng of the 3′-FAM-end labelled PCR product with 720 ng purified chicken DNA as carrier. BLM (or BLM analogue) was added to the DNA to final concentrations ranging from 0.03–0.24 mM, supplemented with equimolar concentrations of freshly prepared FeSO4. The cleavage reactions were allowed to proceed at 37°C for 30 min. The treated DNA was then ethanol precipitated and redissolved in 5–10 μL of 10 mM Tris-HCl, pH 8; 0.1 mM EDTA.
BLM-damaged samples were further treated with endonuclease IV to replace the 3′-phosphoglycolate ends with 3′-hydroxyl ends. This is required because of the anomalous migration of 3′-phosphoglycolate ends in capillary electrophoresis that interfere with the determination of the DNA sequence specificity [36]. Approximately 6 ng of end-labelled DNA that had previously been treated with BLM, was used for the endonuclease IV reaction. The 10 μL reaction comprised of the BLM-damaged DNA; 50 mM Tris-HCl, pH 7.9; 100 mM NaCl; 10 mM MgCl2; 1 mM DTT; and 5 U endonuclease IV (New England Biolabs). The reaction was allowed to proceed at 37°C for 1 h, followed by ethanol precipitation and the pellet was redissolved in 5 μL 10 mM Tris-HCl, pH 8; 0.1 mM EDTA. As a size standard, Maxam-Gilbert G+A sequencing reactions were prepared [36, 46].
Approximately 1–2 μL was analysed by capillary electrophoresis on an ABI 3730 DNA Analyser (Applied Biosystems) at the Ramaciotti Centre for Gene Function Analysis (University of NSW). For samples that contained both the FAM and PET label, no LIZ-labelled size standard was added to remove the possibility of bleed-over of the LIZ emission into the PET channel. A separate capillary electrophoresis run, containing the LIZ500 size standard, was made to allow for alignment with the G+A ladder for peak identification. The identity of the peaks from the no-LIZ500 run was then made by profile comparison with the LIZ500 run.
The capillary electrophoresis data were analysed in GeneMapper v3.7 (Applied Biosystem) as previously described [33, 35, 36]. The cleavage sites were quantified by peak area as determined by GeneMapper after subtraction of the no BLM blank control. The intensity of the cleavage sites were expressed as percentages of the sum of all the cleavage including the full length (intact, uncleaved PCR products) peak areas. To remove end-label bias, an algorithmic correction was made [34, 35, 47]. For the comparison of the cleavage profiles between the BLM analogues, the corrected data was then normalised. The percentage cleavage of each site was determined over the sum of the corrected cleavage intensity of all the sites analysed in each profile.
3. RESULTS
The novel RTGTAY plasmid contained a number of DNA sequence motifs that were expected to be sites of BLM cleavage (Fig. 2). PCR products were generated from the RTGTAY plasmid and were fluorescently labelled with either PET at the 5′-end or FAM at the 3′-end. These PCR products were combined and treated with BLM or one of the analogues at concentrations that ranged from 0.08–0.24 mM. Both labelled PCR products were present in each assay reaction. Labelled fragments resulting from the assay were analysed by CE-LIF and displayed as electropherograms using GeneMapper software (Supplementary Figs. 1 and 2).
3.1 DNA cleavage profiles for BLM and analogues
The no BLM blank control electropherograms (Supplementary Figs. 1A and 2A) were used to evaluate the background noise and also the presence of any artefact peaks that might interfere with quantification. As expected, only the intact full-length PCR product was observed in the no BLM blank control and can be seen as the large peak at the right of the electropherogram.
The electropherograms in Supplementary Figs. 1B–E and 2B–E show the cleavage profiles of BLM and analogues, after treatment of the fluorescently end-labelled PCR products at 0.20 mM. The electropherograms in Supplementary Figs. 1 and 2 were from the same set of samples, with the 3′-end labelled fragments shown in Supplementary Fig. 1 (blue channel, FAM) and the corresponding 5′-end fragments in Supplementary Fig. 2 (red channel, PET).
3.2 DNA sequence specificity analysis
As expected, BLM cleaved at sites that contained 5′-GT-3′ and 5′-GC-3′ dinucleotides (Fig. 2). Cleavage at 5′-GA-3′ was also observed, albeit at a very low frequency (e.g. position 356). In the sequence analysed, all 63 5′-GT sites (100%), 6 out of 8 GC sites (75%) and only 1 out of 7 GA sites (14%) were cleaved. All sites cleaved by BLM were also cleaved by the three analogues. Comparing the PET and FAM electropherograms in Supplementary Figs. 1 and 2, the cleavage profiles appeared similar for the 3′- and 5′-labelled PCR products. A correlation coefficient was calculated for the 3′-PET and 5′-FAM labelled cleavage sites and the R2 values were found to range from 0.822 to 0.937 for BLM and the three analogues. This shows a high level of correlation as previously found for this double-label technique [37].
The electropherograms of the labelled PCR products treated with BLM and the analogues at 0.20 mM were analysed and quantified. The intensity of each cleavage site in the sequence was measured by the area under their respective peaks. The cleavage intensity of each site was corrected for end-label bias [35, 36]. The experiments were performed at a sufficiently low concentration that the end-label bias correcting algorithm altered the percentage damage to a very small degree. This indicated that the experiments were performed at close to single hit kinetics. The 3′-PET and 5′-FAM labelled data was amalgamated and depicted as a bar chart in Fig. 3.
Figure 3. The cleavage profiles of BLM and the three analogues.
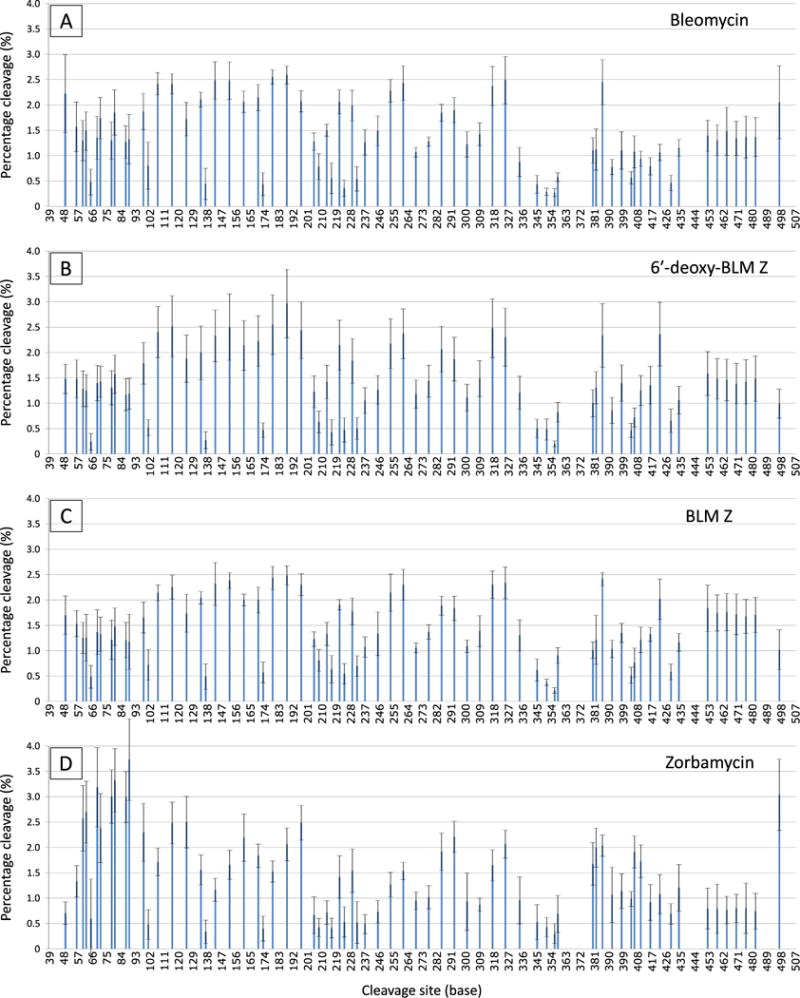
The cleavage profiles of BLM and the three analogues at 0.20 mM were corrected for end-label bias and the 3′- and 5′-end labelled data was amalgamated. Each bar represents a cleavage site on the RTGTAY sequence. The intensity of cleavage (percentage cleavage) was normalised to enable profile comparison between the analogues. Error bars indicate the standard deviation of the mean.
For the DNA sequence analysed, 63 (out of 70) of the cleavage sites contained the sequence 5′-GT-3′. The cleavage proportion of these sites ranged from 0.24–2.96%. Of these sites, 36 contained the sequence 5′-GTA-3′; one 5′-GTC-3′; five 5′-GTG-3′; and 21 sites with 5′-GTT-3′. Cleavage sites containing the sequence 5′-GTA-3′ appeared to be preferentially cleaved by BLM and the analogues since the 5′-GTA-3′ sites were cleaved at intensities in the upper half of the observed range. In contrast, 5′-GTT-3′ were cleaved at proportions in the lower half of the range. Interestingly, sites that contained the sequence 5′-GTG-3′ were highly cleaved by ZBM. These sites were only moderately cleaved by BLM, 6′-deoxy-BLM Z and BLM Z.
3.3 Zorbamycin has an altered DNA sequence specificity compared with BLM
The cleavage profiles in Fig. 3 were similar for BLM, 6-deoxy-BLM Z and BLM Z; however, the profile for ZBM appeared to be different to BLM and the other analogues. For example, the proportion of cleavage at the telomeric region (T7), at position 453–480, was lower for ZBM. ZBM cleaved the sites at this region at proportions that ranged from 0.77–0.96%. In contrast, BLM cleaved the sites at this region at 1.31–1.48%; 6′-deoxy-BLM Z at 1.39–1.59%; and BLM Z at 1.67–1.84%. In addition, ZBM exhibited a high proportion of cleavage at the region located at position 57–93. This region contained four pairs of 5′-GT-3′ sites that were directly adjacent to each other (i.e. 5′-GTGT-3′). ZBM cleaved at these sites at proportions that ranged from 2.38–3.73%. In fact, these sites were among the most cleaved sites on the construct by ZBM. In contrast, BLM cleaved these sites at 1.27–1.85%; 6′-deoxy-BLM Z at 1.17–1.57%; and BLM Z at 1.18–1.47%.
For each of the cleavage sites, the cleavage intensity for BLM and the three analogues were plotted (Fig. 4). A correlation coefficient between each of the profiles was then calculated to examine their similarity. The cleavage profiles of 6′-deoxy-BLM Z with BLM Z (R2 - 0.9552), 6′-deoxy-BLM Z with BLM (R2 - 0.8306) and BLM Z with BLM (R2 - 0.8357) were similar with high R2 values. However, the cleavage profile of ZBM was very different, when compared to BLM (R2 - 0.2618), 6′-deoxy-BLM Z (R2 - 0.1947) and BLM Z (R2 - 0.1443), with low R2 values.
Figure 4. Comparison of the cleavage profile between BLM and the three analogues.
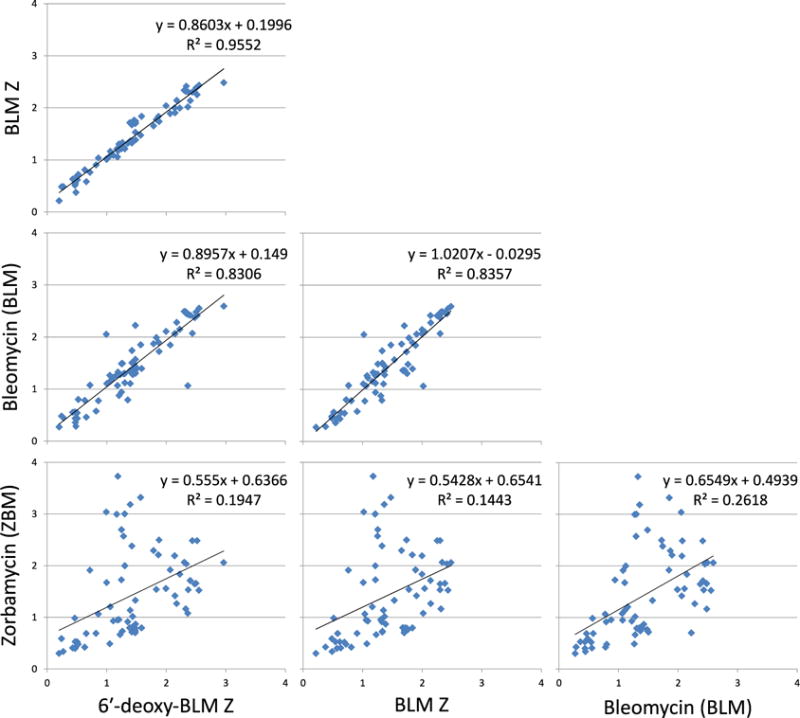
The cleavage site intensity data for BLM and each analogue were plotted against each other. At each cleavage site, the intensity was plotted for each of the compounds being compared as × and y values. A correlation coefficient between each of the profiles was then calculated to examine their similarity.
The 20 most highly cleaved sites by BLM and the three analogues are shown in Table 1. Table 2 is a frequency analysis of the nucleotides surrounding the 20 most highly cleaved sites by BLM and the analogues. With reference to the 20 most highly cleaved sites by BLM and the analogues shown in Tables 1 and 2, the following observations can be made that illustrate that ZBM has a different sequence specificity profile compared with BLM, 6′-deoxy-BLM Z and BLM Z. Seven of the eight most highly cleaved sites by ZBM (bp 89, 80, 69, 78, 87, 62, 60) do not appear in the top 20 for BLM, 6′-deoxy-BLM Z or BLM Z; and the eighth site (bp 497) only appears once for the other compounds at the 18th position for BLM (Table 1). At the -3 position, ZBM had a high number of G and low C, whereas BLM, 6′-deoxy-BLM Z and BLM Z had a low level of G and high C. For the -2 position, it was mainly T or C for all compounds. There was 100% G at the -1 position. At the 0 position that is destroyed during the cleavage reaction, there was 100% T for BLM and ZBM, and almost exclusively Ts with 6′-deoxy-BLM Z and BLM Z except for one C cleavage site. For the +1 position, ZBM had a high level of G while the other compounds had no G. At the +2 position, ZBM had a higher number of C compared with the other compounds that had a lower number of C.
Table 1.
The 20 most highly cleaved sites for BLM and the analogues with the RTGTAY plasmid.
|
|
||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BLM | 6′-deoxy-BLM Z | BLM Z | ZBM | |||||||||||||||||
|
| ||||||||||||||||||||
| Base | % Cleavage | Sequence | Base | % Cleavage | Sequence | Base | % Cleavage | Sequence | Base | % Cleavage | Sequence | |||||||||
| 1 | 188 | 2.59 | CT | GT | AT | 188 | 2.96 | CT | GT | AT | 188 | 2.48 | CT | GT | AT | 89 | 3.73 | GT | GT | AC |
| 2 | 179 | 2.55 | CT | GT | AA | 179 | 2.55 | CT | GT | AA | 179 | 2.43 | CT | GT | AA | 80 | 3.32 | GT | GT | AT |
| 3 | 325 | 2.49 | TC | GT | AT | 116 | 2.52 | AT | GT | AT | 386 | 2.41 | TT | GT | AT | 69 | 3.18 | TT | GT | GT |
| 4 | 143 | 2.48 | TT | GT | AA | 152 | 2.50 | TT | GT | AT | 152 | 2.38 | TT | GT | AT | 497 | 3.04 | GC | GT | AA |
| 5 | 152 | 2.48 | TT | GT | AT | 317 | 2.48 | TT | GT | AT | 325 | 2.34 | TC | GT | AT | 78 | 3.00 | TT | GT | GT |
| 6 | 386 | 2.45 | TT | GT | AT | 197 | 2.44 | CT | GT | AC | 143 | 2.32 | TT | GT | AA | 87 | 2.99 | TT | GT | GT |
| 7 | 261 | 2.43 | TC | GT | AA | 107 | 2.40 | AT | GT | AA | 197 | 2.30 | CT | GT | AC | 62 | 2.70 | GT | GT | AG |
| 8 | 107 | 2.42 | AT | GT | AA | 261 | 2.37 | TC | GT | AA | 317 | 2.30 | TT | GT | AT | 60 | 2.57 | TT | GT | GT |
| 9 | 116 | 2.41 | AT | GT | AT | 422 | 2.36 | TT | GC | CT | 261 | 2.30 | TC | GT | AA | 125 | 2.50 | AT | GT | AC |
| 10 | 317 | 2.37 | TT | GT | AT | 386 | 2.34 | TT | GT | AT | 116 | 2.25 | AT | GT | AT | 197 | 2.49 | CT | GT | AC |
| 11 | 253 | 2.28 | TT | GT | AA | 143 | 2.33 | TT | GT | AA | 253 | 2.14 | TT | GT | AA | 116 | 2.48 | AT | GT | AT |
| 12 | 49 | 2.22 | GG | GT | AA | 325 | 2.30 | TC | GT | AT | 107 | 2.14 | AT | GT | AA | 71 | 2.38 | GT | GT | AA |
| 13 | 170 | 2.15 | CT | GT | AG | 170 | 2.23 | CT | GT | AG | 134 | 2.04 | TT | GT | AG | 98 | 2.29 | AT | GT | AG |
| 14 | 134 | 2.11 | TT | GT | AG | 253 | 2.17 | TT | GT | AA | 422 | 2.02 | TT | GC | CT | 293 | 2.21 | TC | GT | AC |
| 15 | 197 | 2.07 | CT | GT | AC | 221 | 2.15 | TT | GT | AG | 161 | 2.00 | TT | GT | AC | 161 | 2.19 | TT | GT | AC |
| 16 | 161 | 2.07 | TT | GT | AC | 161 | 2.14 | TT | GT | AC | 170 | 2.00 | CT | GT | AG | 325 | 2.06 | TC | GT | AT |
| 17 | 221 | 2.06 | TT | GT | AG | 285 | 2.07 | TT | GT | AC | 221 | 1.91 | TT | GT | AG | 188 | 2.06 | CT | GT | AT |
| 18 | 497 | 2.05 | GC | GT | AA | 134 | 2.00 | TT | GT | AG | 285 | 1.89 | TT | GT | AC | 386 | 2.03 | TT | GT | AT |
| 19 | 229 | 1.99 | TC | GT | AG | 125 | 1.88 | AT | GT | AC | 452 | 1.84 | GG | GT | TA | 382 | 2.00 | GT | GT | TT |
| 20 | 293 | 1.90 | TC | GT | AC | 293 | 1.87 | TC | GT | AC | 293 | 1.83 | TC | GT | AC | 285 | 1.92 | TT | GT | AC |
The 20 most highly cleaved sites for each compound were sorted based on their relative cleavage intensity in decreasing order. The values of the cleavage intensity (percentage cleavage) has been normalised to allow for profile comparison between the analogues. In addition, the sequence of the cleavage sites are shown - the primary site in bold and their flanking nucleotides in plain text.
Table 2.
Nucleotide frequency analysis of the 20 most highly cleaved sites formed by BLM and the analogues with the RTGTAY plasmid.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| −3 | −2 | −1 | 0 | +1 | +2 | ||
|
|
|||||||
| BLM | G | 10 | 5 | 100 | 0 | 0 | 20 |
| A | 10 | 0 | 0 | 0 | 100 | 35 | |
| T | 60 | 70 | 0 | 100 | 0 | 30 | |
| C | 20 | 25 | 0 | 0 | 0 | 15 | |
|
|
|||||||
| 6′-deoxy-BLM Z | G | 0 | 0 | 100 | 0 | 0 | 15 |
| A | 15 | 0 | 0 | 0 | 95 | 25 | |
| T | 65 | 85 | 0 | 95 | 0 | 35 | |
| C | 20 | 15 | 0 | 5 | 5 | 25 | |
|
|
|||||||
| BLM Z | G | 5 | 5 | 100 | 0 | 0 | 15 |
| A | 10 | 0 | 0 | 0 | 90 | 30 | |
| T | 65 | 80 | 0 | 95 | 5 | 35 | |
| C | 20 | 15 | 0 | 5 | 5 | 20 | |
|
|
|||||||
| ZBM | G | 30 | 0 | 100 | 0 | 20 | 10 |
| A | 15 | 0 | 0 | 0 | 75 | 10 | |
| T | 45 | 85 | 0 | 100 | 5 | 50 | |
| C | 10 | 15 | 0 | 0 | 0 | 30 | |
|
|
|||||||
The frequency of occurrence of each nucleotide was calculated at the −3, −2, −1, 0, +1 and +2 positions at the 20 most highly cleaved sites for BLM and the analogues with the RTGTAY plasmid and presented as a percentage for each compound. The values have been corrected for the frequency of occurrence of nucleotides in the analysed sequence.
A graphical representation of the nucleotide present at particular positions of the cleavage site are listed in decreasing order of relative intensity in Supplementary Fig. 3. This graphic enables a visual comparison of the important differences between the cleavage profiles of ZBM and BLM and the other two analogues. It confirms the differences at the −3 and +1 positions observed in Tables 1 and 2; and it also reveals more subtle differences at positions −2 and +2.
It is also interesting to note that the presence of a second cleavage site in the flanking DNA sequences, influenced the resulting cleavage intensity of the first cleavage site. In this regard, the most cleaved sites by ZBM were directly flanked (3′ or 5′) by a GT or GC dinucleotide. In the analysed DNA sequence, there were 12 cleavage sites that contained at least another flanking GC or GT. Of these, 8 were found to be among the 10 most cleaved site by ZBM. In contrast, only one such site was found in the 20 most cleaved sequences for BLM and none for 6′-deoxy-BLM Z and BLM Z.
4. DISCUSSION
In this study we compared the DNA sequence specificity of three BLM analogues, BLM Z, 6′-deoxy-BLM Z and ZBM, that were produced in S. flavoviridis, with BLM that was produced in S. verticillis. BLM Z and BLM have identical chemical structures except for the C terminal tail. A specially-designed plasmid was utilised to investigate the DNA sequence selectivity. This plasmid contained a number of DNA sequence motifs that were expected to be BLM cleavage sites with systematic variations in the flanking DNA sequences.
The DNA sequence specificity of BLM Z and 6′-deoxy-BLM Z were very similar to BLM. These analogues were produced by expressing the BLM gene cluster in S. flavoviridis [44]. The BLM gene cluster can be manipulated by recombinant DNA techniques prior to DNA transformation with S. flavoviridis. Because of the modular nature of the BLM gene cluster, modifications can lead to the production of BLM analogues with predicted structural changes [38–42]. This provides a procedure for manipulation of the BLM gene cluster that avoids the DNA transformation problems associated with S. verticillis. Hence the similar DNA sequence specificity results give confidence that the strategy of expressing the BLM gene cluster in S. flavoviridis is sound and produces BLM analogues with similar properties to BLM produced in S. verticillis.
4.1 Zorbamycin has an altered DNA sequence specificity compared with BLM, BLM Z and 6′-deoxy-BLM Z
The DNA sequence specificity of ZBM was, however, different to that for BLM, BLM Z and 6′-deoxy-BLM Z. While ZBM cleaved at the same sites as the other analogues, it produced a different cleavage profile. We used several methods of analysis to highlight these sequence specificity differences. The 20 most highly cleaved sequences (Table 1) revealed that seven of the eight most highly cleaved sites for ZBM do not appear in the top 20 sites for BLM, 6′-deoxy-BLM Z or BLM Z. BLM, BLM Z and 6′-deoxy-BLM Z preferred 5′-TGTA at the cleavage site, while ZBM preferred 5′-TGTG as well as 5′-TGTA. In addition ZBM had a higher level of G at the −3 position and a higher level of C at the +2 position compared with the other three compounds (Tables 1 and 2). The correlation of the cleavage site intensities for BLM, BLM Z and 6′-deoxy-BLM Z were high, but for ZBM the correlations were low when compared with the other compounds (Fig. 4). The graphical representation in Supplementary Fig. 3 enabled other more subtle differences in sequence specificity to be visualised.
The highest intensity ZBM cleavage sites conform to an alternating purine-pyrimidine sequence that has previously been shown to be a hotspot for BLM cleavage [15, 28]. This has been speculated to be due to the DNA conformation at these alternating purine-pyrimidine sequences [28, 48]. However, other explanations e.g. cooperativity in the interaction or even binding as a dimer, could also be possible.
4.2 Differences in chemical structure between ZBM, BLM and analogues
There are five main differences in chemical structure between ZBM and the three other compounds (Fig. 1 with differences highlighted in red). Each of these five main differences in chemical structure will be discussed in turn to examine why ZBM has a different sequence specificity compared with other three compounds.
First, the C terminal tail is the same for BLM Z, 6′-deoxy-BLM Z and ZBM but is different for BLM. This implies that the C terminal tail is not important for the ZBM sequence specificity difference.
Second, ZBM and 6′-deoxy-BLM Z have a methyl group in the disaccharide region whereas BLM Z and BLM have a hydroxymethyl at this position and hence this implies that this change is also not important for the ZBM sequence specificity difference . However, this difference does appear to be important for DNA cleavage efficiency when comparing BLM Z and 6′-deoxy-BLM Z [44].
Third, ZBM has a thiazolinylthiazole tail while the other analogues have a bithiazole tail. This could be important for the ZBM sequence specificity difference. Phleomycin also has a thiazolinylthiazole tail and it has been reported that phleomycin did not appear to intercalate into DNA due to its thiazolinylthiazole tail but rather binds in the minor groove [49]. With the thiazolinylthiazole tail, the penultimate thiazolium ring is partially reduced and this leads to a non-planar molecule that is expected to disrupt binding by intercalation. The bithiazole tail of BLM, BLM Z and 6′-deoxy-BLM Z has been demonstrated to intercalate into DNA [50]. An alteration in ZBM that results in minor groove binding rather than intercalation, would be expected to give a different sequence specificity for ZBM. However, other data has shown that the thiazolinylthiazole tail was able to partially intercalate into DNA [51]. The thiazolinylthiazole tail bound more weakly to DNA than the bithiazole tail but the mode of binding was similar - partial intercalation. Differences in the model compounds employed and methods of analysis could be responsible for the alternative interpretations of DNA binding for thiazolinylthiazole and bithiazole tails.
The DNA sequence specificity of phleomycin containing a thiazolinylthiazole tail, has been compared with BLM containing a bithiazole tail [11, 13]. Kross et al. [13] observed no differences in DNA sequence specificity while Takeshita et al. [11] found a small number of differences between phleomycin and BLM. These earlier studies used primitive DNA sequence specificity methodology and would not be able to determine differences in sequence specificity with the accuracy and sensitivity utilised in this paper.
Fourth, there is an extra methoxy group at the alanine residue in the linker region for ZBM that is not present for the other three compounds. This changes the BLM L-alanine residue into an L-homoserine in ZBM. This alteration could be important for the ZBM sequence specificity difference since this will alter the structure of the analogue and potentially cause steric hindrance on binding to DNA.
Fifth, there is an extra methyl group at the threonine residue in the linker region for ZBM that is not present for the other three compounds. This modifies the BLM L-threonine residue to an L-OH-valine. This could be important for the ZBM sequence specificity difference and again cause steric hindrance on binding to DNA.
Hence these latter three alterations could be crucial for the ZBM sequence specificity difference. The change from a bithiazole tail to a thiazolinylthiazole tail and the subsequent possible alteration from intercalation to minor groove binding would be the most likely explanation for the ZBM sequence specificity difference [19]. However, if there is no change from intercalation to minor groove binding for ZBM, then the modifications in the linker region could be responsible for the ZBM sequence specificity difference. In order to clarify these differences, further analogues would need to be created that only differed by a single structural alteration so that definitive structure-activity relationships can be determined.
Supplementary Material
Acknowledgments
This work is supported in part by the University of New South Wales, Science Faculty Research Grant Scheme (to B.A.N and V.M.) and the US NIH grant CA094426 (to B.S.) and the Natural Products Library Initiative at The Scripps Research Institute (to B.S.).
Abbreviations
- CE-LIF
capillary electrophoresis with laser-induced fluorescence detection
- BLM
bleomycin
- PLM
phleomycin D1
- ZBM
zorbamycin
References
- 1.Umezawa H, Maeda K, Takeuchi T, Okami Y. New antibiotics, bleomycin A and B. J Antibiot. 1966;19:200–209. [PubMed] [Google Scholar]
- 2.Williams SD, Birch R, Einhorn LH, Irwin L, Greco FA, Loehrer PJ. Treatment of disseminated germ-cell tumors with cisplatin, bleomycin, and either vinblastine or etoposide. N Engl J Med. 1987;316:1435–1440. doi: 10.1056/NEJM198706043162302. [DOI] [PubMed] [Google Scholar]
- 3.Einhorn LH. Curing metastatic testicular cancer, Proc Natl Acad Sci USA. 2002;99:4592–4595. doi: 10.1073/pnas.072067999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duggan DB, Petroni GR, Johnson JL, Glick JH, Fisher RI, Connors JM, Canellos GP, Peterson BA. Randomized comparison of ABVD and MOPP/ABV hybrid for the treatment of advanced Hodgkin’s disease: report of an intergroup trial. J Clin Oncol. 2003;21:607–614. doi: 10.1200/JCO.2003.12.086. [DOI] [PubMed] [Google Scholar]
- 5.Burger RM. Cleavage of Nucleic Acids by Bleomycin. Chem Rev. 1998;98:1153–1170. doi: 10.1021/cr960438a. [DOI] [PubMed] [Google Scholar]
- 6.Chen J, Stubbe J. Bleomycins: towards better therapeutics. Nat Rev Cancer. 2005;5:102–112. doi: 10.1038/nrc1547. [DOI] [PubMed] [Google Scholar]
- 7.Tang C, Paul A, Alam MP, Roy B, Wilson WD, Hecht SM. A short DNA sequence confers strong bleomycin binding to hairpin DNAs. J Am Chem Soc. 2014;136:13715–13726. doi: 10.1021/ja505733u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roy B, Hecht SM. Hairpin DNA sequences bound strongly by bleomycin exhibit enhanced double-strand cleavage. J Am Chem Soc. 2014;136:4382–4393. doi: 10.1021/ja500414a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rabow LE, Stubbe J, Kozarich JW. Identification and quantitation of the lesion accompanying base release in bleomycin-mediated DNA-degradation. J Am Chem Soc. 1990;112:3196–3203. [Google Scholar]
- 10.Takeshita M, Grollman AP, Ohtsubo E, Ohtsubo H. Interaction of bleomycin with DNA. Proc Natl Acad Sci USA. 1978;75:5983–5987. doi: 10.1073/pnas.75.12.5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takeshita M, Kappen LS, Grollman AP, Eisenberg M, Goldberg IH. Strand scission of deoxyribonucleic acid by neocarzinostatin, auromomycin, and bleomycin: studies on base release and nucleotide sequence specificity. Biochemistry. 1981;20:7599–7606. doi: 10.1021/bi00529a039. [DOI] [PubMed] [Google Scholar]
- 12.D’Andrea AD, Haseltine WA. Sequence specific cleavage of DNA by the antitumor antibiotics neocarzinostatin and bleomycin. Proc Natl Acad Sci USA. 1978;75:3608–3612. doi: 10.1073/pnas.75.8.3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kross J, Henner WD, Hecht SM, Haseltine WA. Specificity of deoxyribonucleic acid cleavage by bleomycin, phleomycin, and tallysomycin. Biochemistry. 1982;21:4310–4318. doi: 10.1021/bi00261a021. [DOI] [PubMed] [Google Scholar]
- 14.Mirabelli CK, Ting A, Huang CH, Mong S, Crooke ST. Bleomycin and talisomycin sequence-specific strand scission of DNA: a mechanism of double-strand cleavage. Cancer Res. 1982;42:2779–2785. [PubMed] [Google Scholar]
- 15.Murray V, Martin RF. Comparison of the sequence specificity of bleomycin cleavage in two slightly different DNA sequences. Nucleic Acids Res. 1985;13:1467–1481. doi: 10.1093/nar/13.5.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murray V, Tan L, Matthews J, Martin RF. The sequence specificity of bleomycin damage in three cloned DNA sequences that differ by a small number of base substitutions. J Biol Chem. 1988;263:12854–12859. [PubMed] [Google Scholar]
- 17.Nightingale KP, Fox KR. DNA structure influences sequence specific cleavage by bleomycin. Nucleic Acids Res. 1993;21:2549–2555. doi: 10.1093/nar/21.11.2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murray V. A survey of the sequence-specific interaction of damaging agents with DNA: emphasis on anti-tumour agents. (Review) Prog Nucl Acid Res Mol Biol. 2000;63:367–415. doi: 10.1016/s0079-6603(08)60727-8. [DOI] [PubMed] [Google Scholar]
- 19.Roy B, Tang C, Alam MP, Hecht SM. DNA methylation reduces binding and cleavage by bleomycin. Biochemistry. 2014;53:6103–6112. doi: 10.1021/bi5010848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Segerman ZJ, Roy B, Hecht SM. Characterization of bleomycin-mediated cleavage of a hairpin DNA library. Biochemistry. 2013;52:5315–5327. doi: 10.1021/bi400779r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murray V, Martin RF. The sequence specificity of bleomycin-induced DNA damage in intact cells. J Biol Chem. 1985;260:10389–10391. [PubMed] [Google Scholar]
- 22.Cairns MJ, Murray V. Influence of chromatin structure on bleomycin-DNA interactions at base pair resolution in the human beta-globin gene cluster. Biochemistry. 1996;35:8753–8760. doi: 10.1021/bi9600207. [DOI] [PubMed] [Google Scholar]
- 23.Kim A, Murray V. A large “footprint” at the boundary of the human beta-globin locus control region hypersensitive site-2. Int J Biochem Cell Biol. 2000;32:695–702. doi: 10.1016/s1357-2725(00)00023-6. [DOI] [PubMed] [Google Scholar]
- 24.Kim A, Murray V. Chromatin structure at the 3′-boundary of the human beta-globin locus control region hypersensitive site-2. Int J Biochem Cell Biol. 2001;33:1183–1192. doi: 10.1016/s1357-2725(01)00087-5. [DOI] [PubMed] [Google Scholar]
- 25.Temple MD, Freebody J, Murray V. Genomic and phylogenetic footprinting at the epsilon-globin silencer region in intact human cells. Biochim Biophys Acta. 2004;1678:126–134. doi: 10.1016/j.bbaexp.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 26.Temple MD, Murray V. Footprinting the ‘essential regulatory region’ of the retinoblatoma gene promoter in intact human cells. Int J Biochem Cell Biol. 2005;37:665–678. doi: 10.1016/j.biocel.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 27.Nguyen HTQ, Murray V. The DNA sequence specificity of bleomycin cleavage in telomeric sequences in human cells. J Biol Inorgan Chem. 2012;17:1209–1215. doi: 10.1007/s00775-012-0934-8. [DOI] [PubMed] [Google Scholar]
- 28.Murray V, Chen JK, Tanaka MM. The genome-wide DNA sequence specificity of the anti-tumour drug bleomycin in human cells. Mol Biol Rep. 2016;43:639–651. doi: 10.1007/s11033-016-3998-7. [DOI] [PubMed] [Google Scholar]
- 29.Kuo MT, Hsu TC. Bleomycin causes release of nucleosomes from chromatin and chromosomes. Nature. 1978;271:83–84. doi: 10.1038/271083a0. [DOI] [PubMed] [Google Scholar]
- 30.Galea AM, Murray V. The influence of chromatin structure on DNA damage induced by nitrogen mustards and cisplatin analogues. Chem Biol Drug Design. 2010;75:578–589. doi: 10.1111/j.1747-0285.2010.00969.x. [DOI] [PubMed] [Google Scholar]
- 31.Murray V, Chen JK, Galea AM. The anti-tumour drug, bleomycin, preferentially cleaves at the transcription start sites of actively transcribed genes in human cells. Cell Mol Life Sci. 2014;71:1505–1512. doi: 10.1007/s00018-013-1456-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murray V, Chen JK, Galea AM. Enhanced repair of bleomycin DNA damage at the transcription start sites of actively transcribed genes in human cells. Mutat Res Fund Mol Mech Mutagen. 2014;769:93–99. doi: 10.1016/j.mrfmmm.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 33.Paul M, Murray V. Use of an Automated Capillary DNA Sequencer to Investigate the Interaction of Cisplatin with Telomeric DNA Sequences. Biomed Chromatog. 2012;26:350–354. doi: 10.1002/bmc.1664. [DOI] [PubMed] [Google Scholar]
- 34.Nguyen TV, Murray V. Human telomeric DNA sequences are a major target for the anti-tumour drug, bleomycin. J Biol Inorgan Chem. 2012;17:1–9. doi: 10.1007/s00775-011-0818-3. [DOI] [PubMed] [Google Scholar]
- 35.Murray V, Nguyen TV, Chen JK. The Use of Automated Sequencing Techniques to Investigate the Sequence Selectivity of DNA Damaging Agents. Chem Biol Drug Design. 2012;80:1–8. doi: 10.1111/j.1747-0285.2012.01379.x. [DOI] [PubMed] [Google Scholar]
- 36.Nguyen TV, Chen JK, Murray V. Bleomycin DNA damage: Anomalous mobility of 3′-phosphoglycolate termini in an automated capillary DNA sequencer. J Chromat B. 2013;913:113–122. doi: 10.1016/j.jchromb.2012.11.029. [DOI] [PubMed] [Google Scholar]
- 37.Chen JK, Murray V. The determination of the DNA sequence specificity of bleomycin-induced abasic sites. J Biol Inorgan Chem. 2016;21:395–406. doi: 10.1007/s00775-016-1349-8. [DOI] [PubMed] [Google Scholar]
- 38.Du L, Sanchez C, Chen M, Edwards DJ, Shen B. The biosynthetic gene cluster for the antitumor drug bleomycin from Streptomyces verticillus ATCC15003 supporting functional interactions between nonribosomal peptide synthetases and a polyketide synthase. Chem Biol. 2000;7:623–642. doi: 10.1016/s1074-5521(00)00011-9. [DOI] [PubMed] [Google Scholar]
- 39.Shen B, Du L, Sanchez C, Edwards DJ, Chen M, Murrell JM. Cloning and characterization of the bleomycin biosynthetic gene cluster from Streptomyces verticillus ATCC15003. J Nat Prod. 2002;65:422–431. doi: 10.1021/np010550q. [DOI] [PubMed] [Google Scholar]
- 40.Shen B, Du L, Sanchez C, Edwards DJ, Chen M, Murrell JM. The biosynthetic gene cluster for the anticancer drug bleomycin from Streptomyces verticillus ATCC15003 as a model for hybrid peptide-polyketide natural product biosynthesis. J Ind Microbiol Biotechnol. 2001;27:378–385. doi: 10.1038/sj.jim.7000194. [DOI] [PubMed] [Google Scholar]
- 41.Galm U, Wang L, Wendt-Pienkowski E, Yang R, Liu W, Tao M, Coughlin JM, Shen B. In vivo manipulation of the bleomycin biosynthetic gene cluster in Streptomyces verticillus ATCC15003 revealing new insights into its biosynthetic pathway. J Biol Chem. 2008;283:28236–28245. doi: 10.1074/jbc.M804971200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Galm U, Wendt-Pienkowski E, Wang L, Huang SX, Unsin C, Tao M, Coughlin JM, Shen B. Comparative analysis of the biosynthetic gene clusters and pathways for three structurally related antitumor antibiotics: bleomycin, tallysomycin, and zorbamycin. J Nat Prod. 2011;74:526–536. doi: 10.1021/np1008152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Galm U, Wendt-Pienkowski E, Wang L, George NP, Oh TJ, Yi F, Tao M, Coughlin JM, Shen B. The biosynthetic gene cluster of zorbamycin, a member of the bleomycin family of antitumor antibiotics, from Streptomyces flavoviridis ATCC 21892. Mol Biosyst. 2009;5:77–90. doi: 10.1039/b814075h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang SX, Feng Z, Wang L, Galm U, Wendt-Pienkowski E, Yang D, Tao M, Coughlin JM, Duan Y, Shen B. A designer bleomycin with significantly improved DNA cleavage activity. J Am Chem Soc. 2012;134:13501–13509. doi: 10.1021/ja3056535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Inazuka M, Tahira T, Hayashi K. One-tube post-PCR fluorescent labeling of DNA fragments. Genome Res. 1996;6:551–557. doi: 10.1101/gr.6.6.551. [DOI] [PubMed] [Google Scholar]
- 46.Belikov S, Wieslander L. Express protocol for generating G+A sequencing ladders. Nucleic Acids Res. 1995;23:310. doi: 10.1093/nar/23.2.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.White RJ, Phillips DR. Sequence-dependent termination of bacteriophage T7 transcription in vitro by DNA-binding drugs, Biochemistry. 1989;28:4277–4283. doi: 10.1021/bi00436a024. [DOI] [PubMed] [Google Scholar]
- 48.Calladine CR. Mechanics of sequence-dependent stacking of bases in B-DNA. J Mol Biol. 1982;161:343–352. doi: 10.1016/0022-2836(82)90157-7. [DOI] [PubMed] [Google Scholar]
- 49.Povirk LF, Hogan M, Dattagupta N, Buechner M. Copper(II).bleomycin, iron(III).bleomycin, and copper(II).phleomycin: comparative study of deoxyribonucleic acid binding. Biochemistry. 1981;20:665–671. doi: 10.1021/bi00506a034. [DOI] [PubMed] [Google Scholar]
- 50.Goodwin KD, Lewis MA, Long EC, Georgiadis MM. Crystal structure of DNA-bound Co(III) bleomycin B2: Insights on intercalation and minor groove binding. Proc Natl Acad Sci USA. 2008;105:5052–5056. doi: 10.1073/pnas.0708143105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu W, Vanderwall DE, Turner CJ, Hoehn S, Chen J, Kozarich JW, Stubbe J. Solution structure of the hydroperoxide of Co(III) phleomycin complexed with d(CCAGGCCTGG)2: evidence for binding by partial intercalation. Nucleic Acids Res. 2002;30:4881–4891. doi: 10.1093/nar/gkf608. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.