

Abstract
Catalytic promiscuity is a useful, but accidental, enzyme property, so finding catalytically promiscuous enzymes in nature is inefficient. Some ancestral enzymes were branch points in the evolution of new enzymes and are hypothesized to have been promiscuous. To test the hypothesis that ancestral enzymes were more promiscuous than their modern descendants, we reconstructed ancestral enzymes at four branch points in the divergence hydroxynitrile lyases (HNL’s) from esterases ~100 million years ago. Both enzyme types are α/β-hydrolase-fold enzymes and have the same catalytic triad, but differ in reaction type and mechanism. Esterases catalyze hydrolysis via an acyl enzyme intermediate, while lyases catalyze an elimination without an intermediate. Screening ancestral enzymes and their modern descendants with six esterase substrates and six lyase substrates found higher catalytic promiscuity among the ancestral enzymes (P <0.01). Ancestral esterases were more likely to catalyze a lyase reaction than modern esterases and the ancestral HNL was more likely to catalyze ester hydrolysis than modern HNL’s. One ancestral enzyme (HNL1) along the path from esterase to hydroxynitrile lyases was especially promiscuous and catalyzed both hydrolysis and lyase reactions with many substrates. A broader screen tested mechanistically related reactions that were not selected for by evolution: decarboxylation, Michael addition, γ-lactam hydrolysis and 1,5-diketone hydrolysis. The ancestral enzymes were more promiscuous than their modern descendants (P = 0.04). Thus, these reconstructed ancestral enzyme are catalytically promiscuous, but HNL1 is especially so.
Graphical abstract
Introduction
Catalytic promiscuity is the ability of enzymes to catalyze additional reactions beyond those beneficial for life.1, 2 Catalytic promiscuity differs from substrate promiscuity in that different transition states, rather than the same transition state, must be stabilized. For example, the natural function of carbonic anhydrase is to catalyze the reversible hydration of carbon dioxide, yet it also catalyzes the promiscuous hydrolysis of the ester p-nitrophenyl acetate.3
Catalytic promiscuity is useful because it expands the range of available reactions for industrial biocatalysis applications.4 For example, halohydrin dehalogenase normally catalyzes the hydrolysis of vicinal haloalcohols to vicinal diols and also catalyzes a promiscuous substitution of the halide with other nucleophiles.5 Fox and coworkers6 exploited this promiscuous reaction, substituting chloride with cyanide to make a key intermediate in the synthesis of a cholesterol-lowering drug.
Catalytic promiscuity is difficult to search for because it is an accidental property of enzymes. It is easier to search in places where the enzymatic property of interest is likely adaptive. For example, searching high temperature environments will likely yield thermostable enzymes. Searching chemical spill sites may yield enzymes that degrade environmental contaminants. Although there is no location where catalytic promiscuity is adaptive, there may a time when it was adaptive.
Several groups7–10 have suggested that ancestral enzymes must have been catalytically promiscuous. Some ancestral enzymes lie at key branch points where divergent evolution selected new catalytic functions as new metabolic opportunities arose. These ancestors might also have been promiscuous with regard to other mechanistically related reactions. If so, they would provide suitable places to search for catalytic promiscuity.
That ancestral enzymes no longer exist poses no problem – they can be resurrected. Analysis of modern sequences allows researchers to infer the sequences of ancestral enzymes.11, 12 These analyses use the topology of phylogenetic trees, the likelihood of different amino acid replacements and various assumptions about evolution such as whether or not different sites in a gene evolve at different rates. Chemical synthesis of the gene and expression of the protein in a suitable host yields the ancestral enzyme for characterization. In cases where analyses suggest several possible amino acids at a site, several ancestors can be made and characterized. In most cases such ancestors have similar properties.
Reconstructed ancestral enzymes show broader substrate ranges than their modern descendants. Voordeckers et al. showed that the reconstructed ancestors of modern α-glycosidases catalyzed hydrolysis of a wider range of substrates (maltose and isomaltose analogs).13 Likewise, reconstructed β-lactamases14 and carboxyl methyltransferases15 showed higher substrate promiscuity. An ancestral protein kinase bound an inhibitor with an affinity intermediate between the modern descendants.16 Similarly, in vitro divergent evolution of glucuronidase to new substrates specificity proceeded through non-specific intermediates. However, whether or not ancestral enzymes were more catalytically promiscuous than modern enzymes has not been explored. In this paper, we reconstruct ancestral hydroxynitrile lyases and esterases and assess their catalytic promiscuity. The transition states for these two reaction types differ significantly, so finding a single enzyme that catalyzes both reaction types efficiently would be remarkable.
Hydroxynitrile lyases (HNL’s) are plant enzymes that catalyze the elimination of hydrogen cyanide from cyanohydrins as a defense against herbivorous insects.17 HNL’s occur in at least five protein folds; our focus here is on HNL’s in the α/β-hydrolase-fold super-family. These HNL’s evolved from esterases ~100 million years ago when flowering plants and insects first diversified. Presumably, a weak promiscuous ability to cleave cyanohydrins first arose serendipitously in an ancient esterase. Providing some protection against herbivorous insects, selection for higher activity favored gene duplication, freeing the new copy to functionally diverge into the modern specialized hydroxynitrile lyases.
The reaction mechanism for the hydrolysis reaction catalyzed by esterases (Enzyme Classification (EC) group 3.1.1) differs in three key ways from the elimination reaction catalyzed by hydroxynitrile lyases (EC 4.1.2). Hydrolysis follows the canonical serine hydrolase mechanism with an acyl enzyme intermediate (Figure 1A). The elimination has no acyl enzyme intermediate and uses only binding and general acid-base chemistry (Figure 1B). Second, the two mechanisms require conflicting substrate orientations. Hydrolysis requires the carbonyl oxygen to bind in the oxyanion hole, while the lyase mechanism requires that it not bind in the oxyanion hole. Third, hydrolysis involves a hydrophobic leaving group, while the lyase reaction creates a polar leaving group. These clearly different mechanisms provide a good test of the hypothesis that ancestral enzymes were catalytically promiscuous. The prediction is that while modern enzymes catalyze only one of these reaction types, ancestral α/β-hydrolases may catalyze both hydrolysis and eliminations.
Figure 1.

Esterases catalyze hydrolysis of carboxylic acid esters while hydroxynitrile lyases catalyze an elimination of hydrogen cyanide from cyanohydrins. Both active sites contain a serine- histidine-aspartate catalytic triad. Aspartate not shown for clarity. A) Salicylic acid binding protein 2 (SABP2) catalyzes the hydrolysis of methyl salicylate. The substrate ester binds with the carbonyl oxygen in the oxyanion hole. The first step of the reaction is the nucleophilic attack by the serine Oγ on the carbonyl carbon. The mechanism involves an acyl enzyme intermediate (not shown). B) Hydroxynitrile lyase from Hevea brasiliensis (rubber tree) catalyzes the cleavage of acetone cyanohydrin. The substrate oxygen binds outside the oxyanion hole, which is blocked by Thr11. Elimination proceeds in one step without an acyl enzyme intermediate.
Experimental section
General
Enzymes and chemicals were bought from commercial suppliers and used directly without further purification. Racemic mandelonitrile (Sigma Aldrich, St. Louis, MO) was aliquoted in 10-mL portions and stored at −18 °C. 2-Hydroxy-6-oxo-6-phenylhexa-2, 4-dienoic acid (HOPDA) – was a gift from Lindsay Eltis’ research group at the University of British Columbia and 2-azabicyclo[2.2.1]hept-5-en-3-one (Vince lactam) was a gift from Robert Vince’s research group at the University of Minnesota. Protein concentrations were determined from the absorbance at 280 nm using extinction coefficients obtained from the ExPASy Bioinformatics Resource Portal.18 Protein gels were run on sodium dodecyl sulfate polyacrylamide gradient gels (NuPage 4–12% Bis-Tris gel from Invitrogen) using the BenchMark protein ladder (5 μL/lane) as a standard. DNA gels were run using 0.7% ultrapure agarose with 1×TAE buffer and 1 kb DNA ladder as a standard. Micro titer plate assays were performed in triplicate and the mean of the measurements is reported. One unit of enzyme activity corresponds to the amount of protein required to release of 1 μmol of product per minute. Steady state kinetic data were fitted to the Michaelis-Menten equation using the nonlinear fit routine in the Solver function of Microsoft Excel. NiNTA resin was regenerated according to Qiagen protocol. 1H-NMR spectra were run at 400 MHz in deuteriochloroform. Thin layer chromatography on silica gel was eluted with hexanes: ethylacetate (8:2). Lysogeny broth (LB) and terrific broth (TB) were prepared according to Sambrook et al.19
Ancestral enzyme reconstruction
Phylogenetics
Five thousand protein sequences, 150–600 amino acids long and sharing a minimum 30% sequence identity with Hevea brasiliensis acetone cyanohydrin lyase (GI: 1223884), were obtained from the NCBI protein sequence database. Identical copies, mutant peptides and all patents were removed and the remaining sequences aligned using Muscle20 in SeaView.21 A preliminary neighbor joining tree22 was used to identify a cluster of 1285 sequences between 30% and 99% identical, and that included the ACLs and salicylic acid binding protein 2 from Nicotiana tabacum, for further analysis.
The 1285 amino acid sequence alignment was adjusted manually guided by super-positioned protein structures (H. brasiliensis 1QJ4, Manihot esculenta 1E8D, Arabidopsis thaliana 3DQZ, Nicotiana tabacum 1XKL) obtained with DeepView/Swiss PDB-Viewer23 to adjust insertions and deletions into surface loops. The final alignment of 1285 sequences is available upon request from the authors.
Bootstrapped (1000 replicates) maximum likelihood trees were obtained using RAxML24 using the ML + Bootstrap + GAMMAPROT + LG settings. Maximum likelihood uses a probabilistic model of sequence evolution to construct a tree from a given alignment. The GAMMAPROT setting to allow some sites to evolve faster than others and the LG25 setting provides an empirical amino acid exchange matrix to accommodate differences in the rates of exchange between different amino acids. The most likely tree is one with topology, branch lengths and other parameters that maximize the likelihood of the observed alignment. An advantage of the maximum likelihood approach is that it provides a natural means to test alternative hypotheses - branch lengths, topologies and ancestral states. Similar trees were obtained using FastML with GAMMAPROT and a JTT amino acid exchange matrix26 in RAxML with SH support27 and bootstrapped neighbor joining with a Poisson correction in Seaview.
Ancestral sequences (Table S1) were inferred using maximum likelihood as implemented in RAxML using the ML + Bootstrap + GAMMAPROT + LG tree obtained above. Maximum like-lihood calculates the most likely amino acid at each site at each node using an empirical Bayes approach.28 The likelihood of observing a particular amino acid, x, at a particular site at a particular node is given as
where a is one of twenty amino acids at the focal site, t is the topology of the phylogenetic tree, m is an evolutionary model, q represents the various model parameters (rates of amino acid exchange, variable rates across sites etc.) and px is the prior probability of observing x. Maximum likelihood resolves ambiguities in favour of the most likely model. For example, in the case of tree (((Leu,Leu),(Met,Met)),Arg) the most likely ancestor of sequences had an Met because exchanges between Met and Leu and between Met and Arg are commonplace while those between Leu and Arg are rare. At branch points EST3 and HNL1, ancestral sequences were also inferred for the neighbor joining tree using maximum likelihood as implemented in MEGA.29
Gene synthesis and cloning
Genes for the ancestral enzymes were synthesized by GenScript and subcloned into a pET21a(+) vector at NdeI and XhoI restriction sites resulting in an upstream T7 promoter and lac operator and an in-frame C-terminal six His-tag. Fidelity of cloning and of the gene synthesis was confirmed by DNA sequencing the entire gene (ACGT, Wheeling, IL).
Protein expression and purification
Lysogeny broth media containing 100 μg/mL ampicillin (LB-amp, 5 mL) was inoculated with a single bacterial colony from an agar plate and incubated in an orbital shaker at 37 °C and 200 rpm for 15 h. This culture was used to inoculate terrific broth (TB)-amp media (500 mL) in 2-L baffled flasks, which was incubated at 37 °C and 250 rpm until the absorbance at 600 nm reached 1.0 (approximately 3–4 h). This culture was transferred to 17 °C and 200 rpm for 1 h to cool, and then isopropyl β-D-1-thiogalactopyranoside (IPTG, 1 mM) was added to induce the protein expression. Cultivation was continued for 20 h. The cells were harvested by centrifugation (8000 rpm, 10 min at 4 °C) and resuspended in buffer A (20 mM imidazole, 50 mM NaH2PO4, 300 mM NaCl, pH 7.2, 20 mL). The cells were disrupted by sonication (400 W, 40% amplitude for 5 min) and centrifuged (4 °C, 12,000 rpm 45 min). The supernatant was loaded onto a column containing Ni-NTA resin (1 mL, Qiagen) pre-equilibrated with buffer A (10 mL). The column was washed buffer A (50 mL) followed by buffer B (50 mM imidazole, 50 mM NaH2PO4, 300 mM NaCl, pH 7.2, 50 mL). The His-tagged protein was eluted with elution buffer (250 mM imidazole, 50 mM NaH2PO4, 300 mM NaCl, pH 7.2, 15 mL) and concentrated to ~2 mL with an Amicon ultrafiltration centrifuge tube (10 kDa cutoff). The imidazole buffer was exchanged by four successive additions of BES buffer (5 mM, pH 7.0, 10 mL) to <15 μM imidazole remaining, followed by concentration to ~2 mL with the centrifuge tube. Typical yields were 20–100 mg protein from a 500-mL culture.
Racemic 2-nitro-1-phenylethanol was prepared according to a literature procedure.30 Purification by silica-gel column chromatography eluted with hexanes: ethyl acetate (85:15) yielded 1.22 g (70%) of a colorless oil. Rf 0.44, 1H-NMR: 2.86 (br, 1H), 4.45 (dd, J = 13.2, 2.9 Hz, 1H), 4.54 (dd, J = 13.2, 9.6 Hz, 1H), 5.43 ( dd, J = 9.8, 2.9 Hz, 1H), 7.33–7.39 (m, 5H).
Racemic 3-(2-nitro-1-phenylethyl)pentane-2,4-dione was prepared according to a literature procedure.31 Acidic alumina (1 g) was added to a stirred solution of acetylacetone (0.10 mL, 1.0 mmol) and trans-β-nitrostyrene (0.15 g, 1.0 mmol) in Et2O (1 mL). The suspension was stirred at 25 °C for 2 h, concentrated under vacuum, and purified by silica-gel column chromatography eluted with hexanes: ethyl acetate (80:20) yielding 0.106 g (43%) as pale yellow solid. mp 107–113 °C; lit.31 110–112 °C; Rf 0.22, 1H-NMR: 1.92 (s, 3H), 2.28 (s, 3H), 4.18 (m, 1H), 4.38 (d, J = 11.1 Hz, 1H), 4.61 (m, 2H) and 7.1–7.3 (m, 5H).
Benzoylacetic acid
Benzoylacetic acid was prepared according to the literature procedure.32 To a solution of acetophenone (12 g, 0.10 mol), dry tetrahydrofuran (50 mL) in 250 mL overnight-oven-dried round-bottomed flask, NaH (60%) in oil (10.5 g, 0.25 mol) and dimethylcarbonate (16.2 g, 0.18 mol) was added. The suspension was heated to reflux for 2 h and cooled to room temperature. The reaction was quenched with ice water (100 mL), acidified with 3 M HCl to pH 2–3 and extracted with ethyl acetate (3 × 25 mL). The organic layer was dried over sodium sulfate and concentrated under vacuum. Purification by silica-gel column chromatography eluted with hexanes: ethyl acetate (95:5) yielded 11.5 g (65% yield) of methyl 3-oxo-3-phenyl proponoate. Hydrolysis of methyl 3-oxo-3-phenyl propanoate (1.0 g) in sodium hydroxide solution (0.5 M) at room temperature. After 12 h the reaction was acidified with HCl to pH 2–3, extracted with ethyl acetate (3 × 10 mL), dried and concentrated under vacuum. Benzoyl acetic acid obtained in 55% (0.5 g) yield as a white solid. Rf 0.33; mp 85 °C; lit.33 99 °C; 1HNMR: 4.1 (s, 2H), 5.7 (s, vinyl 1H), 7.2–8.05 (m, 5H), 9.75 (br, 1H).
Activity and enantioselectivity of enzyme-catalyzed reactions
Hydroxynitrile lyase activity
Hydroxynitrile lyase activity was assayed as described previously with minor modifications.34 The assay monitors the release of benzaldehyde (ε280 = 1352 M−1cm−1) from mandelonitrile spectrophotometrically and was corrected for the spontaneous cleavage of mandelonitrile. The assay solution contained mandelonitrile (2.0 mM), citrate buffer (pH 5.0, 50 mM) and enzyme (0.05–8 μM from a stock in 5 mM BES buffer, pH 7) in a total volume of 200 μL (path length = 0.60 cm). Steady state kinetic constants were determined under identical conditions with mandelonitrile concentrations from 1–20 mM.
The enantioselectivity was measured using the reverse reaction: addition of HCN to benzaldehyde. The reaction (0.5 mL total volume) contained benzaldehyde (1 mM, from a stock solution of 50 mM of benzaldehyde in 50 mM in sodium citrate buffer pH 5.0) and HCN (50 mM, from a stock solution of 1.0 M of HCN in tert-butyl methyl ether) in sodium citrate buffer (50 mM, pH 5.0). Caution: HCN is toxic and requires careful handling.35 Enzyme (0.002 – 0.1 mg protein in 5 mM of BES buffer, 100 μL, pH 7.0) was added to start the reaction. After 2 h shaking at 600 rpm at 20 °C, the tert-butyl methyl ether layer was separated, evaporated by stream of nitrogen, and dissolved in isopropanol. The enantiomeric purity of the product mandelonitrile was determined by HPLC using a Chiralcel OD-H column (Diacel) eluted with hexane: isopropanol (98: 2) at flow rate of 1.25 mL/min: The S and R enantiomers elute at 31 and 34 min, respectively. Absolute configuration of mandelonitrile was established by comparison with a commercial sample of (R)-mandelonitrile (Alfa Aesar, Ward Hill, MA).
Cleavage of acetone cyanohydrin, lactonitrile, 2-hydroxypentanenitrile and 2-hydroxyhexanenitrile was assayed using a modified König reaction.36, 37 Substrate (1.28 mM to 10 mM in 0.1 M citric acid) was assayed with up to 8 μM enzyme in 5 mM citrate phosphate buffer, pH 5. After up to 20 min, the reaction was quenched by addition of aqueous N-chlorosuccinimide (2 mM, 62.5 μL, also containing 20 mM succinimide). After 2 min, barbituric acid (230 mM in 30% pyridine, 12.5 μL) was added and after 10 min, the absorbance at 580 nm was measured and compared to a calibration curve constructed using K2[Zn(CN)4] with concentrations of HCN ranging from 2.5–100 μM.
Nitroaldolase (Henry reaction) activity
Nitroaldolase activity was assayed by monitoring the release of benzaldehyde from 2-nitro-1-phenylethanol as above for the hydroxynitrile lyase activity. The assay mixture (200 μL total volume, path length 0.60 cm) contained 2-nitro-1-phenylethanol (2 mM from stock of 50 mM 2-nitro-1-phenylethanol in 1 mM HCl), 50 mM citrate phosphate buffer, pH 5.5, and up to 5 μM enzyme in BES buffer (5 mM, pH 7.2). A blank reaction to monitor the spontaneous cleavage of 2-nitro-1-phenylethanol was identical, but the enzyme solution was replaced by BES buffer. Steady state kinetics used the same assay, but varied the concentration of 2-nitro-1-phenylethanol (0.5–6 mM).
The enantioselectivity was measured using the reverse reaction: addition of nitromethane to benzaldehyde. The reaction (0.5 mL total volume) contained benzaldehyde (1 mM from 50 mM of benzaldehyde in diisopropyl ether (DIPE) and nitromethane (1.0 M) in 50 mM in citrate: phosphate buffer (pH 6.0) and enzyme (0.1–0.7 μM) in 5 mM of BES buffer. After 2 h shaking at 500 rpm at room temperature, the organic layer was separated and evaporated by stream of nitrogen gas and dissolved in isopropanol. The enantiomeric purity of the product 2-nitro-1-phenylethanol was determined by HPLC using a Chiralcel OD-H column (Diacel) eluted with hexane: isopropanol (95: 5) at flow rate of 1.0 mL/min. The R and S enantiomers elute at 23 and 26 min respectively and it was reported to elute at 13.5 and 16.3 min with hexane: isopropanol (90: 10) at flow rate of 1.0 mL/min in Chiralcel OD-H column.38
Esterase activity
Esterase activity was measured by hydrolysis of p-nitrophenyl acetate (pNPAc)39 and was corrected for spontaneous hydrolysis. The assay mixture (100 μL total volume; path length 0.29 cm) contained 6 mM of pNPAc, 8 vol% acetonitrile, 5 mM BES, pH 7.0, and up to 5 μM enzyme in BES buffer. The increase in absorbance at 404 nm was monitored spectrophotometrically. The extinction coefficient used for calculations (ε404 = 11.4 × 103 M−1cm−1) takes into account the incomplete ionization of p-nitrophenol at this pH. Steady state kinetics used the same assay, but varied the concentration of pNPAc (0.5–6 mM). Methyl salicylate and methyl mandelate were assayed at 500 μM with up to 10 μM enzyme in 5 mM BES, pH 7. Methyl pentanoate, 1-naphthyl acetate, and 2-naphthyl acetate were assayed at 2 mM in 5 mM BES buffer pH 7 with up to 50 μM enzyme. Control reactions were performed by adding equal volumes buffer that was removed from the enzyme by centrifugal filtration. Reactions were shaken at room temperature for up to 18 hours before enzyme was removed by centrifugation using Amicon Ultra 0.5 mL regenerated cellulose 10 kDa cutoff centrifugal filters. Reactions with methyl salicylate, 1-naphthyl acetate and 2-naphthyl acetate were analyzed by HPLC on an Agilent Eclipse Plus C18 column eluted with methanol: water + 0.1% formic acid (80: 20) at 1.0 mL/min. Methyl mandelate reactions were analyzed on the same column but with a 60: 40 mix of solvents. The enantiomeric purity of unreacted methyl mandelate was measured using a Chiralpak AS-RH column (Diacel) eluted with acetonitrile: water + 0.1% trifluroacetic acid (30: 70) at 1.0 mL/min. The S and R enantiomers elute at 6.4 and 6.8 min, respectively. The absolute configuration was assigned by comparison to sample of R-methyl mandelate prepared from commercial R-mandelic acid. Methyl pentanoate hydrolysis was quantified p-nitrophenol as a pH-indicator as described previously.40
Lactonase activity
Lactonase activity corresponds to the hydrolysis of 4-phenyl-4-butryolactone. The reaction mixture (0.50 mL total volume) contained 4-phenyl-4-butyrolactone (0.1 mM), BES buffer (5 mM, pH 7.0), and enzyme (7–75 μM). Reaction was stirred at 500 rpm at 25 °C for 24 h. The enzyme was removed by filtration through a centrifugal filter (Amicon Ultra 0.5 mL regenerated cellulose 10 kDa cutoff spun at 4000 rpm) and the filtrate analyzed by HPLC. The conversion was measured using an Agilent Eclipse Plus C18 column eluted with acetonitrile: H2O + 0.1% formic acid (40: 60) where 4-hydroxy-4-phenylbutyric acid and 4-phenyl-4-butyrolactone eluted at 3.4 and 5.4 min respectively. A blank without enzyme showed ~5 % spontaneous hydrolysis. The amount was subtracted to determine the rate and conversion of the reaction. The enantiomeric purity of unreacted 4-phenyl-4-butyrolactone was determined by HPLC using a Chiralpak AS-RH column (Diacel) eluted with acetonitrile: water + 0.1% of formic acid (35: 65) at 1.0 mL/min. The R and S lactone enantiomers elute at 10.4 and 12.5 min, respectively. This configuration was established by comparison with an HPLC of a hydrolysis of the same lactone with pig liver esterase, which favors the (+)-enantiomer.41 The (+)-enantiomer was later identified as (R).42
Decarboxylase activity
Decarboxylase activity corresponds to the decarboxylation of benzoylacetic acid to acetophenone. The reaction mixture (0.50 mL total volume) contained benzoylacetic acid (0.25 mM), BES buffer (5 mM, pH 7.3) and enzyme (13–75 μM). The reaction mixture was stirred at 500 rpm at 25 °C for 6 h, then the enzyme was removed by filtration through an Amicon ultrafiltration centrifuge tube (10 kDa cutoff) at 4000 rpm and the filtrate analyzed by HPLC on an Agilent Eclipse Plus C18 column eluted with acetonitrile: H2O + 0.1% formic acid (50: 50) monitored at 254 nm where benzoylacetic acid and acetophenone elute at 4.3 and 7.4 min respectively. The molar absorbance of benzoylacetic acid is 3.03-fold larger than that for acetophenone and the peak areas were corrected by this factor. A blank without enzyme showed ~0.1% spontaneous cleavage.
Michael addition
A two-phase mixture of di-isopropyl ether (250 μL) and citrate: phosphate buffer (50 mM, pH 5.5, 250 μL) containing trans-β-nitrostyrene (2 mM), acetyl acetone (100 mM) and enzyme (80–140 μM) was stirred at 500 rpm at 25 °C for 8 h. The di-isopropyl ether layer was separated, flushed with stream of nitrogen and analyzed by HPLC using a Chiralpak OJ-R column (Diacel) eluted with acetonitrile: H2O (40: 60) at 1.0 mL/min. The S and R enantiomers of the product 3-(2-nitro-1-phenylethyl)pentane-2,4 dione elute at 7.1 and 9.1 min, respectively and the starting material trans-β-nitrostyrene elutes at 19.7 min. The absolute configuration was assigned with a Chiralcel OD-H column eluted with hexane: isopropanol (95: 5) where the S and R enantiomers were reported to elute at 25 and 27 min, respectively.43 A blank reaction without enzyme showed ~ 6% spontaneous formation of racemic product. The reported rates and enantiomeric purities were corrected for this spontaneous reaction.
Lactamase activity
Lactamase activity corresponds to the hydrolysis of 2-azabicyclo[2.2.1]hept-5-en-3-one. The reaction mixture (0.50 mL total volume) contained 2-azabicyclo[2.2.1]hept-5-en-3-one (0.50 mM), p-amino benzoic acid (0.1 mM, internal standard), Tris buffer (5 mM, pH 8.5), and enzyme (7–75 μM). The reaction was stirred at 500 rpm at 25 °C for 24 h. The enzyme was removed by filtration through an Amicon ultrafiltration centrifuge tube (10 kDa cutoff) at 4000 rpm and the filtrate analyzed by HPLC to determine conversion and enantioselectivity. The amount of substrate reacted was determined from the relative peaks area upon HPLC analysis on Agilent Eclipse Plus C18 column eluted with acetonitrile: H2O + 0.1% formic acid (40:60). p-Aminobenzoic acid and 2-azabicyclo [2.2.1] hept-en-3-one elute at 3.9 and 4.4 min respectively (relative absorbance = 4.37). A reaction with no enzyme showed ~0.1% conversion. The enantiomeric purity of unreacted 2-azabicyclo [2.2.1] hept-en-3-one as determined by HPLC using a Chiralpak AS-RH column (Diacel) eluted with acetonitrile: water + 0.1% formic acid (20: 80) at 1.0 mL/min. The (2S, 4R) and (2R, 4S) enantiomers eluted at 3.4 and 3.9 min, respectively. The absolute configuration was established by comparison of HPLC traces with a sample of the (2R, 4S) enantiomer (Sigma Aldrich, St. Louis, MO).
C-C Bond hydrolysis activity
Hydrolysis of C-C bonds was determined with 2-hydroxy-6-oxo-6-phenylhexa-2, 4-dienoic acid (HOPDA) as the substrate to yield benzoic acid and 2-hydroxypenta-2, 4-dienoic acid.44 This substrate was a gift from the Eltis group, who synthesized it enzymatically. The assays were performed at pH 7.5 in 100 mM phosphate buffer with 100 μM HOPDA and up to 50 μM enzyme. The decrease in HOPDA was monitored at 434 nm (ε = 25.7 mM−1cm−1).
Epoxide hydrolase activity
Epoxide hydrolase activity corresponds to the hydrolysis 2-(4-nitrophenyl)oxirane to the corresponding 1-(4-nitrophenyl)ethane-1,2-diol. The reaction mixture (0.50 mL total volume) contained 2-(4-nitrophenyl)oxirane (0.1 mM), BES buffer (5 mM, pH 7.0), and enzyme (7–75 μM). Reaction was stirred at 500 rpm at 25 °C for 12 h. The enzyme was removed by filtration through a centrifugal filter (Amicon Ultra 0.5 mL regenerated cellulose 10 kDa cutoff spun at 4000 rpm) and the filtrate analyzed by HPLC on an Agilent Eclipse Plus C18 column eluted with acetonitrile: H2O + (40: 60). 1-(4-Nitrophenyl)ethane-1,2-diol and 2-(4-nitrophenyl)oxirane elute at 3.4 and 10.7 min, respectively. Reactions without enzyme showed ~6 % spontaneous hydrolysis. No additional hydrolysis was detected in the reactions containing enzyme.
Aldol addition activity
Aldol activity was measured by the formation of 4-hydroxy-4-(nitrophenyl)butan-2-one from p-nitrobenzaldehdye and acetone. The reaction mixture (1 mL total volume) contained p-nitrobenzaldehdye (0.5 mM), acetone (10 mM), BES buffer (5 mM, pH 7.0), and enzyme (3–37 μM). Reaction was stirred at 500 rpm at 25 °C for 24 h. The enzyme was removed by filtration through a centrifugal filter (Amicon Ultra 0.5 mL regenerated cellulose 10,000 NMWL spun at 4000 rpm) and the filtrate analyzed by HPLC on an Agilent Eclipse Plus C18 column eluted with acetonitrile: H2O + (50: 50). 4-Hydroxy-4-(nitrophenyl)butan-2-one and p-nitrobenzaldehyde elute at 4.5 and 6.1 min, respectively. A blank without enzyme showed ~ 0.1 % spontaneous addition. No additional product was detected in the reactions containing enzyme.
Baylis-Hillman reaction
Baylis-Hillman reaction activity was measured by the formation of 3-(hydroxy-4-(nitrophenyl)methyl)but-3-en-2-one from p-nitrobenzaldehdye and methyl vinyl ketone. The reaction mixture (1 mL total volume) contained p-nitrobenzaldehdye (0.5 mM), methyl vinyl ketone (10 mM), BES buffer (5 mM, pH 7.0), and enzyme (3–37 μM). Reaction was stirred at 500 rpm at 25 °C for 24 h. The enzyme was removed by filtration through a centrifugal filter (Amicon Ultra 0.5 mL regenerated cellulose 10 kDa cutoff spun at 4000 rpm) and the filtrate analyzed by HPLC on an Agilent Eclipse Plus C18 column eluted with acetonitrile: H2O + (50: 50). 3-(Hydroxy-4-(nitrophenyl)methyl)but-3-en-2-one and nitrobenzaldehyde elute at 4.1 and 6.1 min, respectively. A blank without enzyme showed ~ 0.1 % spontaneous addition. No additional product was detected in the reactions containing enzyme.
Pixel plots in Figures 3 and 4
Figure 3.

Aldehyde lyase and ester hydrolase activities of modern and ancestral hydroxynitrile lyases and esterases. Columns correspond to different enzymes, while rows correspond to the reaction with the substrate at the left. Modern esterases catalyze mainly hydrolysis of esters and modern lyases catalyze mainly elimination reactions, while ancestral enzymes often catalyze both reactions. Darker squares correspond to faster reaction. For enantioselective reactions, green indicates R-selective reactions and red S-selective reactions. Color intensity reflects enantioselectivity. Supporting information Table S2 contains the data from which this figure was derived.
Figure 4.

Other eliminations and hydrolyses catalyzed by modern and ancestral hydroxynitrile lyases and esterases. Columns correspond to different enzymes, while rows correspond to reactions with the substrate at the left. Only two of the six modern enzymes catalyze one of these reactions, while seven of the ten ancestral enzymes catalyze at least one of these reactions. Darker squares correspond to faster reaction. For enantioselective reactions, green indicates R-selective reactions and red S-selective reactions. Color intensity reflects enantioselectivity. Supporting information Table S3 contains the data from which this figure was derived. The substrates for these reaction were: decarboxylation, 3-oxo-3-phenylpropanoic acid; Michael addition, acetylacetone and 2-nitrovinylbenzene; C-C hydrolysis, 2-hydroxy-6-oxo-6-phenylhexa-2,4-dienoic acid; lactam hydrolysis, 2-azabicylo[2.2.1]hept-5-en-3-one (Vince lactam).
The data in Tables S2 and S3 were used to create Figures 3 and 4 using the approach similar to Wahler et al.45 To show the wide range of reaction rates, the pixel darkness is proportional to the log10 of the rates, but color is linearly proportional to the enantioselectivity. Green indicates R-selectivity, red indicates S-selectivity and gray indicates no enantioselectivity data or no enantioselectivity. The RGB color values for an achiral substrate or for chiral substrate where enantioselectivity was not measured were: R = G = B = 255 – RATE where RATE = log10(rate in min−1) scaled to a whole number between 255 (for the fastest rate 12,600 min−1 for the MeHNL-catalyzed cleavage of acetone cyanohydrin in Table S2) and 0 (for the slowest rate 0.00023 min−1 for the EST1-catalyzed hydrolysis of the lactone in Table S3). The RATE values are subtracted from 255 because color values closer to zero give darker colors. For R-selective enantioselective reactions, the color values were R = 255 − (E*RATE)/(E+1), G = 255 − RATE/(E+1), B = (R + G)/2 and for S-selective reactions R = 255 − RATE/(E+1), G = 255 − (E*RATE)/(E+1), B = 255 − (R + G)/2 where E is the enantioselectivity.
Statistical analysis
Fisher’s exact tests46 were used to compare the substrate promiscuity and catalytic promiscuity of ancestral and modern enzymes. First, all enzymes-substrate combinations were classified as active or not active. Tables in supporting information. Classifications for resurrected ancestral enzymes at nodes EST3 and HNL1 were each averaged and rounded (e.g. a mean of 2/3 active is classified as active and a mean of 1/3 active is classified as not active). Substrate promiscuity compared the ability of ancestral and modern enzyme to catalyze their natural reactions - ester hydrolysis for esterases and hydroxynitrile cleavage for the HNLs. Catalytic promiscuity compared the ability of the ancestral and modern enzyme to catalyze unnatural reactions - hydroxynitrile cleavage for esterases and ester hydrolysis for HNL’s. The reactions in Table 3 were classified as unnatural reactions for both esterase and HNL’s.
Results
The α/β-hydrolase-fold superfamily contains >60,000 proteins47, 48 with wide-ranging catalytic activities.49–51 Most are hydrolases (Enzyme Classification group 3), although the family also includes hydroxynitrile lyases (Enzyme Classification group 4) that catalyze an elimination reaction.
These hydroxynitrile lyases cluster within a larger group of plant esterases (Figure S1; selected sequence alignments in Figure S2, also see reference 52) suggesting that they diverged from esterases ~100 million years ago when flowering plants (angiosperms) diversified. Hydroxynitrile lyases are plant enzymes involved in defense from insects.53 The hydroxynitrile lyases include enzymes from the rubber tree, Hevea brasiliensis, (HbHNL), cassava, Manihot esculenta, (MeHNL)54–56 and wild castor, Baliospermum montanum.57 The esterases include salicylic acid binding protein 2 (SABP2) from tobacco (Nicotiana tabacum)58 and polyneuridine-aldehyde esterase from snakeroot (Rauvolfia serpentina) (RsEST).59 This group of plant esterases and hydroxynitrile lyases share >40% amino acid identity (Table S1 in the supporting information lists pairwise sequence identities).
Although esterase from Arabidopsis thaliana (AtEST) catalyses ester hydrolysis,60 it also catalyzes fast promiscuous cleavage of mandelonitrile.61 It does not catalyze cleavage of acetone cyanohydrin, the natural cyanohydrin substrate in most plants. Cruciferous plants, which include Arabidopsis, lack the metabolic pathway to make cyanogenic glucosides,62 so do not need hydroxynitrile lyases. However, Arabidopsis does contain other cyanogenic metabolites like an acyl cyanide,63 where a hydrolase may contribute to cyanogenesis and plant defense.
More distantly related α/β-hydrolase-fold enzymes include more esterases (e.g. esterase from R. communis (RcEST)64 as well as enzymes that catalyze even more diverse reactions. The decarboxylase methylketone synthase I from tomato has 35–50% aa identity to the HNL/EST group65, 66 in Figure S1. More divergent α/β-hydrolase-fold enzymes (only 10–20% aa identify) found in various bacteria or animals are C–C hydrolases (e.g., BphD from Burkholderia cepacia44), epoxide ring hydrolases (e.g., human soluble epoxide hydrolase67–69), and haloalkane hydrolases (e.g., LinB from Sphingobium sp.70). These are too distantly related to be included in the tree in Figure S1.
Ancestral enzyme reconstruction
Many of these plant esterases and hydroxynitrile lyase share >40% amino acid identity making it feasible to infer ancestral enzyme sequences. We predicted the sequences of the most recent common ancestors at four different divergence points (Figures 2 and S1; sequences in Figure S2). These sequences were inferred from the sequences of the modern descendant enzymes using either neighbor joining, maximum parsimony, maximum likelihood, or a combination of maximum parsimony & maximum likelihood (Table 1).11 Some sites, typically at the protein surface, evolve rapidly because they do not affect protein function and their ancestral composition is less certain. The different algorithms predict slightly different sequences. If all reconstructed ancestors at a node behave similarly, then we assume that these reconstructions are a close approximation to the actual historical ancestor.
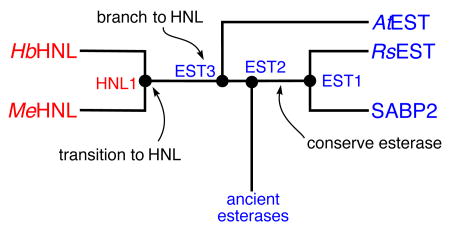
Figure 2.

Simplified phylogenetic tree of esterases and HNL’s identifies different types of ancestral enzymes. Ancestral enzymes EST1 and EST2 are conserved function enzymes. They lie along a path from ancient to modern esterases and are expected to be esterases. Ancestral enzyme EST3 is a functional branch point enzyme. It lies between ancient esterases and modern HNL’s and is expected to be an esterase with promiscuous HNL activity. Ancestral enzyme HNL1 is a transitional ancestral enzyme because it leads only to modern HNL’s. This is expected to be an HNL, but may show remaining esterase activity.
Table 1.
Extant and ancestral enzymes in this study and their amino acid sequence identity with hydroxynitrile lyase HbHNL and esterase SABP2.a
| Enzyme | Construct Originb | % ID to HbHNL | % ID to SABP2 |
|---|---|---|---|
| HbHNL | Hevea brasiliensis | 100 | 44 |
| MeHNL | Manihot esculenta | 76 | 41 |
| HNL1 | max parsimony & likelihood | 79 | 49 |
| HNL1-ML | maximum likelihood | 75 | 51 |
| HNL1-NJ | neighbor joining | 79 | 50 |
| EST3 | max parsimony & likelihood | 67 | 55 |
| EST3-ML | maximum likelihood | 58 | 60 |
| EST3-NJ | neighbor joining | 56 | 59 |
| EST2 | max parsimony & likelihood | 48 | 71 |
| EST1 | max parsimony & likelihood | 49 | 70 |
| AtEST | Arabidopsis thaliana | 47 | 50 |
| RcEST | Ricinus communis | 21 | 28 |
| RsEST | Rauvolfia serpentina | 41 | 56 |
| SABP2 | Nicotinia tabacum | 44 | 100 |
Supporting information Table S1 compares the sequence identity for each enzyme pair and Figure S1 shows the aligned amino acid sequences.
Plant species origin for extant enzymes and methods used to derive the ancestral sequences.
The sequences of the ancestral enzymes differ substantially from the modern descendants, but these differences are similar to the differences between modern enzymes. For example, the amino acids in the modern enzyme MeHNL are 76% identical to those in HbHNL; similarly, the ancestral enzymes at node HNL1 are 72–79% identical to HbHNL. Modern esterases are 41–47% identical to HbHNL; ancestral enzymes at nodes EST1 and EST2 are 48–49% identical to HbHNL, while ancestral enzymes at node EST3 are 56–67% identical to HbHNL. These differences correspond to large numbers of amino acid substitutions; HNL1 and HbHNL differ by 54–72 amino acid substitutions. Such large numbers of substitutions can be expected to substantially change their properties. Nakano and Asano71 created hybrid HNL’s using a consensus sequence approach, but these cannot correspond to ancestral enzymes. No information about the catalytic promiscuity of these hybrids is available.
Ancestral enzymes are branch points in the evolution of modern enzymes. We classify these branch points into three types as related to divergent evolution. Some ancestral enzymes lie at points where all connecting branches lead to enzymes with the same catalytic function. These, which we call conserved function ancestors, are expected to have the ancestral function. Second, other ancestral enzymes are branch points to younger nodes with different functions. These, which we call function branch point ancestors, are also expected to have the ancestral function, since one of the branches retains the ancestral function. However these functional branch points may have already undergone selection toward both activities, and thus were likely to be bifunctional or promiscuous for the new function as well as active for the ancestral function. The third type of ancestral enzyme is the branch point from an older node with the ancestral activity to a clade of modern enzymes, all with the new function. These, which we call transitional ancestors, are expected to be specialized for the new function, but may retain vestigial activity toward the ancestral reaction.
In this classification, EST1 and EST2 are conserved function ancestors, predicted to be esterases since all descendant branches from theses enzymes contain esterases. EST3 is a branch point ancestor, also predicted to be an esterase, likely with promiscuous HNL activity, since some descendant enzymes are esterases (ancestral function) and some are hydroxynitrile lyases. Finally, HNL1 is a transitional ancestor, since all modern descendants are HNL’s, and the node immediately ancestral to HNL1, (i.e. EST3) is predicted to be an esterase. HNL1 is predicted to be an HNL like its descendants, yet to possibly retain some of the esterase activity of its ancestors.
Esterase & hydroxynitrile lyase activities of modern enzymes
All predicted ancestral enzyme sequences conserve the catalytic triad of Ser-His-Asp found in modern descendants. The ancestral enzymes also contain active site differences that match their modern descendants. Esterases contain a glycine on the oxyanion loop that allows access to the oxyanion hole. Ancestral enzymes EST1 and EST2 also contain a glycine at this location. Modern hydroxynitrile lyases contain a threonine as this location; ancestral enzymes at HNL1 also contain a threonine. Esterases contain a hydrophobic site to bind the alcohol (often phenylalanine or histidine, which would be uncharged with a non polar region), while HNL’s contain a polar site – a lysine-glutamate pair. The ancestral enzymes EST1 and EST2 contain histidine, while enzymes at HNL1 contain the lysine-glutamate pair. EST3 is an exception to this generalization. Three of the four reconstructions contain an asparagine on the oxyanion loop, suggesting an HNL-like block of the oxyanion hole, but non-polar residues in the alcohol binding site suggest an EST-like active site. The modern enzyme AtEST has a similar mix of residues in the active site. Based upon these sequence differences, we expect HNL1 to be a hydroxynitrile lyase, EST1 and EST2 to be esterases, but the function of EST3 is not easily predicted from its sequence. Since it is most similar to AtEST, it may be, like AtEST, an esterase with a promiscuous hydroxynitrile lyase activity.
Since the substrate specificities of the various enzymes are unknown, we used six esters with varying structure to measure the carboxylic acid esterase activities (E.C. group 3.1.1): methyl salicylate, methyl mandelate, 1- and 2-naphthyl acetate, methyl pentanoate and 4-phenyl-4-butyrolactone. We omitted the often-used p-nitrophenyl acetate because its high reactivity makes it different from typical esters. Similarly, we used six substrates of varying structure to measure aldehyde lyase activity (E.C. group 4.1.2): acetone cyanohydrin (natural substrate for hydroxynitrile lyases), mandelonitrile, lactonitrile, 2-hydroypentanenitrile, 2-hydroxyhexanenitrile and 2-nitro-1-phenylethanol. This last substrate involves a nitro-aldol cleavage, whose mechanism appears similar to that for cyanohydrin cleavage.72 The modern esterases catalyzed hydrolysis of almost all of the ester substrates and similarly, the modern hydroxynitrile lyases catalyzed cleavage of almost all the hydroxynitrile lyase substrates. Thus, this range of substrate cannot test for increased substrate promiscuity since the ancestral enzymes cannot do more than the modern enzyme with this set of substrates.
The modern enzymes showed little catalytic promiscuity; that is, the hydroxynitrile lyases catalyzed mainly hydroxynitrile cleavage and the modern esterases catalyzed mainly ester hydrolysis, Figure 3, Table S2. The modern hydroxynitrile lyases, HbHNL and MeHNL, catalyzed cleavage of all five cyanohydrins and the nitroaldol compound. The best substrate is the natural one, acetone cyanohydrin, with kcat values of 2,400 and 12,600 min−1, respectively. They also efficiently cleaved their unnatural substrate mandelonitrile (kcat = 1530 and 1340 min−1, respectively) with high enantioselectivity for the (S)-enantiomer, as previously reported.73, 74 These two HNL’s catalyzed the slower cleavage of three other cyanohydrins and the nitroaldol compound72, 75–77 with kcat values ranging from 0.3 to 50 min−1. The enantioselectivity of the nitroaldol reaction was measured in the synthesis direction. Both MeHNL and HbHNL favored the (S)-enantiomer, as was reported previously.
The modern hydroxynitrile lyases showed little promiscuous esterase activity. HbHNL catalyzed the slow hydrolysis of methyl salicylate, 0.07 min−1 while MeHNL showed no detectable esterase activity.
The modern esterases all catalyzed ester hydrolysis. SABP2 catalyzed hydrolysis of all six esters tested. The natural function of SABP2 is to hydrolyze methyl salicylate to salicylic acid with a reported kcat value of 27 min−1 and KM of 8.6 μM.78 We measured a lower value of 0.5 min−1 for this substrate likely due to strong inhibition by the product salicylic acid under our conditions. Among the other five esters, the best substrate was methyl pentanoate, with a rate of 18,000 min−1. RcEST is a predicted, but not experimentally confirmed, polyneuridine aldehyde esterase from sequence similarities. RsEST is experimentally confirmed as a polyneuridine aldehyde esterase, but closely related analogs of polyneuridine aldehyde did not react.79 Both esterases catalyzed hydrolysis of all five esters tested, but some rates were as low as 0.01 min−1. The good ester substrates for RcEST were methyl salicylate (7.5 min−1) and methyl pentanoate (260 min−1), while the good esterase substrate for RsEST was only methyl pentanoate (650 min−1). RsEST also catalyzed very slow hydrolysis of the lactone (0.00077 min−1 or 1 d−1). AtEST catalyzed hydrolysis of three of the five esters with rates ranging from 0.04 to 1.7 min−1 and the very slow hydrolysis of the lactone (0.0005 min−1 or 0.7 d−1) confirming its assignment as an esterase, albeit not an efficient one. Koo et al.60 also reported the AtEST catalyzes hydrolysis of esters. Neither SABP2, RcEST, or RsEST catalyzed cleavage of any of the hydroxynitriles or of the nitrolaldol substrate. As reported previously, AtEST is unusual in that it showed efficient cleavage of mandelonitrile (2,530 min−1) and the slow cleavage of the similar nitroaldol compound (5 min−1). In both cases, the (R)-enantiomer was cleaved in contrast to the (S)-enantiomer favored by HbHNL and MeHNL.
Esterase & hydroxynitrile lyase activities of ancestral enzymes
In contrast, almost all of the ancestral enzymes catalyzed both an ester hydrolysis and a cyanohydrin cleavage, Figure 3, Table S2. At the HNL1 node, all three reconstructions catalyzed both cyanohydrin cleavage and ester hydrolysis. Cyanohydrins were better substrates than esters and acetone cyanohydrin is the best substrate. HNL1 has detectable activity with nine of the twelve substrates; HNL1-ML with ten substrates and HNL1-NJ with eleven substrates. HNL1 showed the highest cyanohydrin cleavage activity (880 min−1 with acetone cyanohydrin) and HNL1-NJ showed the highest ester hydrolysis activity (0.9 min−1 with methyl pentanoate).
Ancestral esterases, EST1, EST2, and one of the three reconstructions at node EST3 (EST3-ML) also catalyzed both cyanohydrin cleavage and ester hydrolysis. EST1 and EST2 show high activity (>1 min−1) toward the majority of ester substrates, whereas the modern esterases have high activity with only one or two of the substrates. Both EST1 and EST2 also have activity with mandelonitrile and, other than AtEST, the modern esterases had no detectable hydroxynitrile cleavage activity. The best substrate for EST3-ML was mandelonitrile, not an ester. Ester hydrolysis was very slow: 0.008 min−1 for EST3-ML. The other two ancestral enzyme reconstructions at EST3 catalyzed only ester hydrolysis. Methyl pentanoate was the best substrate: 42 min−1 for EST3-NJ; only 0.6 min−1 for EST3. Chemical steps are rate-limiting for both esterases80 and hydroxynitrile lyases.55 We assume that mechanisms of the ancestral enzymes are similar to those of modern descendants and that chemical steps are also rate-limiting.
Statistical comparison with Fisher’s exact test indicates that these ancestral and modern enzymes do not differ significantly in their substrate promiscuity, but do differ in their catalytic promiscuity. The 2×2 Fisher exact test with the natural reactions (ester hydrolysis catalyzed by esterases and hydroxynitrile cleavage by HNL’s) is not significant (P = 0.67) indicating that ancestral enzymes are no more substrate promiscuous than their modern descendants. While the ancestral enzymes accepted almost all of the substrates, so did the modern enzymes. The ancestral enzymes were not given the opportunity to demonstrate substrate promiscuity with this selection of substrates. Further testing with a wider range of substrate might show increased substrate promiscuity of these enzymes as has been observed for other ancestral enzymes. The 2×2 Fisher exact test with the unnatural reactions (ester hydrolysis catalyzed by HNL’s and hydroxynitrile cleavage catalyzed by esterases) is highly significant (P < 0.01) indicating that ancestral enzymes are more likely to be catalytically promiscuous than modern enzymes.
Among the ancestral enzymes, HNL1 is a transitional enzyme between true esterases and true HNLs. It shows good HNL activity while retaining some esterase activity. It can best catalyze both hydrolysis and elimination reactions. The other ancestors (EST1, EST2, EST3) were, in their time, modern specialist esterases. A statistical comparison of HNL1 to all other enzymes shows that HNL1 is more catalytically promiscuous than the group of modern and other ancestral enzymes (P = 0.012). The enhanced catalytic promiscuity of HNL1 does not come at the cost of stability. Its unfolding temperature (~80 °C) is higher than that for the modern descendants HbHNL and MeHNL (54–70 °C).81, 82 Other researchers also found increased stability for many ancestral enzymes.83
Other hydrolase and lyase activities of modern and ancestral enzymes
To test the ability of these enzymes to catalyze an even broader range of reactions, we tested two other hydrolyses (lactam hydrolysis, EC 3.5.2, and carbon-carbon bond hydrolysis, EC 3.7.1) and three other lyase reactions (decarboxylation, EC 4.1.1, Michael addition, EC 4.1.99) Figure 4, Table S3. All four are reactions catalyzed by enzymes in the α/β-hydrolase-fold family, but the branch points for these enzymes occur outside the region of the reconstructed ancestral enzymes. We refer to these as outside reactions. Two of the modern esterases catalyzed a slow hydrolysis reaction (SABP2, 0.003 min−1 for lactam hydrolysis; RsEST, 0.0004 min−1 or 0.5 d−1, for carbon-carbon bond hydrolysis), but none of the remaining four modern enzyme catalyzed any of these four reactions. In contrast, seven of the ten ancestral enzymes catalyzed at least one of the reactions. EST2 catalyzed two reactions, EST3-NJ and HNL1-NJ catalyzed three reactions. The reaction rates were all slow; the fastest reaction was the EST3-catalyzed hydrolysis of the carbon-carbon bond (0.03 min−1 or ~40 d−1). Both modern and ancestral enzymes were enantioselective.
The 2×2 Fisher’s exact test indicates that the ancestral enzymes are more likely (P = 0.04) to catalyze one of these mechanistically related reactions than is a modern descendant even though they were never subject to selection for this ability. Ancestral enzymes HNL1 and EST3 are transitional and functional branch point enzymes for HNL activity, but not for these novel reactions, so this catalytic promiscuity is accidental. Nevertheless, ancestral enzymes are more likely than modern enzymes to possess these accidental abilities.
None of the ancestral enzymes or modern enzymes catalyzed epoxide hydrolysis (hydrolysis of 2-(4-nitrophenyl)oxirane; <0.0001 min−1). This result is not surprising since the expoxide hydrolase mechanism requires an aspartate nucleophile, while all the ancestral and modern enzymes contain a serine as the nucleophile in the active site. None of the ancestral enzymes or modern enzymes catalyzed the aldol addition of acetone to p-nitrobenzaldehyde or the Baylis-Hillman reaction, addition of methyl vinyl ketone to p-nitrobenzaldehdye (<0.0001 min−1). We tested the aldol and Baylis-Hillman reactions because catalysis involves an amine and HNL’s contain a lysine residue near the active site. No α/β-hydrolase-fold enzyme naturally catalyzes these additions, but two groups reported weak promiscuous activity of α/β hydrolase-fold enzymes toward these reactions.84, 85
Discussion
Ancestral enzymes at branch points for new catalytic activities are good places to search for catalytic promiscuity. Random variation can create promiscuity both in modern and ancestral enzymes, but along branches for new catalytic function, there is an expectation of catalytic promiscuity and our results support this expectation. Modern catalytically promiscuous enzymes also exist, but there is no systematic method to find them. In contrast, examination of phylogenetic trees can identify which ancestral enzymes are most like to be promiscuous.
The major catalytic behavior of ancestral enzymes is consistent with our understanding of the hydrolysis versus lyase reaction mechanisms. The ancestral esterases, EST1, EST2, and EST3 have esterase-like active sites and are efficient hydrolases. The ancestral hydroxynitrile lyases at HNL1 have a hydroxynitrile-lyase-like active site and are efficient lyases. They also show significant esterase activity. We did not find such catalytic promiscuity when we engineered esterase variants for HNL activity.86 Site directed mutagenesis added three catalytic HNL residues to the esterases PFE and SABP2. In both cases, esterase activity was abolished. In the case of PFE, no HNL activity was detected, while for SABP2 the HNL activity was weak (0.09 min−1 vs. 60–170 min−1 for HNL1). Other amino acid must be responsible for the higher HNL activity of HNL1 and its ability to catalyze both reaction types.
Substrate promiscuity has been correlated with protein flexibility.87 Ubiquitin is a small protein (76 amino acids) that binds to many different proteins as part of its regulatory function. In solution and in the absence of protein ligands, ubiquitin is flexible and the range of observed conformations matches those seen in the crystal structures.88 Molecular dynamics simulations of glutathione S-transferases89 and cytochrome P450 monooxygenases80 correlated higher flexibility with broader substrate specificity. As substrates bind, they stabilize the conformation that creates the best contacts. For example, x-ray structures of an enzyme that catalyzes an isomerization in both histidine and tryptophan biosynthesis shows different active site conformations when bound to the two different substrates.91 In another example, x-ray structures of two amino glycoside antibiotics bound to a promiscuous kinase show different loop conformation for the two antibiotics.92
Conformational flexibility may also contribute to catalytic promiscuity. Chemically different reactions likely proceed via different protein conformations.93 The increase in the flexibility of a loop in a lactonase variant correlated with increased catalytic promiscuity.94 As the substrate orientations for the hydrolysis and lyase reactions differ, so an enzyme that catalyzes both reactions must bind the two substrates in at least two different orientations. Although the details are unknown, conformational flexibility of the ancestral enzymes may also account for their catalytic promiscuity.
Whatever the mechanism generating catalytic promiscuity it likely involves residues outside the active sites. The modern and ancestral enzymes differ in approximately fifty residues, only a few of which are located in the active site. Distant residues can change the binding affinity of proteins. For example, the protein kinases Abl and Src differ in their affinity for the inhibitor Gleevec. The difference stems from the shift of a loop in Abl to wrap around Gleevec.16 In contrast a hydrogen bond network involving distant residues prevents a similar shift in Src. Distant residues might similarly create several possible catalytic conformations.
Supplementary Material
Acknowledgments
This work was supported by NIH grant 5R01GM102205 and NSF grant 1152804 to AMD and RJK.
Footnotes
Author Contributions
T. D. and A. M. R. contributed equally. All authors approved the final version of the manuscript.
The authors declare no competing financial interest.
Cladogram showing the hydroxynitrile lyases within a larger group of plant esterases in the α/β-hydrolase fold superfamily. Amino acid sequence alignment of selected modern and reconstructed ancestral α/β-hydrolases. Pairwise sequence identities of modern and ancestral enzymes. Cyanohydrin cleavage and ester hydrolysis catalyzed by modern and ancestral enzymes. Rates and enantioselectivity of other nucleophilic additions and other hydrolyses catalyze by modern and ancestral hydroxynitrile lyases and esterases. Data (conversion and enantiomeric excess) to measure the enantioselectivity of modern and ancestral enzymes. Representative HPLC chromatograms to measure enantioselectivity of modern and ancestral enzymes. Categorial grouping of data from Tables S2 and S3 for Fisher’s exact test. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.O’Brien PJ, Herschlag D. Chem Biol. 1999;6:R91. doi: 10.1016/S1074-5521(99)80033-7. [DOI] [PubMed] [Google Scholar]
- 2.Humble MS, Berglund P. Eur J Org Chem. 2011:3391. [Google Scholar]
- 3.Pocker Y, Stone JT. Biochemistry. 1967;6:668. doi: 10.1021/bi00855a005. [DOI] [PubMed] [Google Scholar]
- 4.Bornscheuer UT, Kazlauskas RJ. Angew Chem Intl Ed. 2004;43:6032. doi: 10.1002/anie.200460416. [DOI] [PubMed] [Google Scholar]
- 5.Nakamura T, Nagasawa T, Yu F, Watanabe I, Yamada H. Biochem Biophys Res Commun. 1991;180:124. doi: 10.1016/s0006-291x(05)81264-1. [DOI] [PubMed] [Google Scholar]
- 6.Fox RJ, Davis SC, Mundorff EC, Newman LM, Gavrilovic V, Ma SK, Chung LM, Ching C, Tam S, Muley S, Grate J, Gruber J, Whitman JC, Sheldon RA, Huisman GW. Nat Biotechnol. 2007;25:338. doi: 10.1038/nbt1286. [DOI] [PubMed] [Google Scholar]
- 7.Waley SG. Comput Biochem Physiol. 1969;30:1. doi: 10.1016/0010-406x(69)91293-6. [DOI] [PubMed] [Google Scholar]
- 8.Ycas M. J Theor Biol. 1974;44:145. doi: 10.1016/s0022-5193(74)80035-4. [DOI] [PubMed] [Google Scholar]
- 9.Jensen RA. Annu Rev Microbiol. 1976;30:409. doi: 10.1146/annurev.mi.30.100176.002205. [DOI] [PubMed] [Google Scholar]
- 10.Weng JK, Philippe RN, Noel JP. Science. 2012;336:1667. doi: 10.1126/science.1217411. [DOI] [PubMed] [Google Scholar]
- 11.Thornton JW. Nat Rev Genet. 2004;5:366. doi: 10.1038/nrg1324. [DOI] [PubMed] [Google Scholar]
- 12.Harms MJ, Thornton JW. Nat Rev Genet. 2013;14:559. doi: 10.1038/nrg3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Voordeckers K, Vanneste K, van der Zande E, Voet A, Maere S, Verstrepen KJ. PLoS Biol. 2012;10:e1001446. doi: 10.1371/journal.pbio.1001446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Risso V, Gavira J, Mejia-Carmona DF, Gaucher E, Sanchez-Ruiz JM. J Am Chem Soc. 2013;135:2899. doi: 10.1021/ja311630a. [DOI] [PubMed] [Google Scholar]
- 15.Huang R, Hippauf F, Rohrbeck D, Haustein M, Wenke K, Feike J, Sorrelle N, Piechulla B, Barkman TJ. Proc Natl Acad Sci U S A. 2012;109:2966. doi: 10.1073/pnas.1019605109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilson C, Agafonov RV, Hoemberger M, Kutter S, Zorba A, Halpin J, Buosi V, Otten R, Waterman D, Theobald DL, Kern D. Science. 2015;347:882. doi: 10.1126/science.aaa1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poultan JE. Plant Physiol. 1990;94:401. doi: 10.1104/pp.94.2.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. In: The Proteomics Protocols Handbook. Walker JG, editor. Humana Press; Totowa, NJ: 2005. p. 571. [Google Scholar]
- 19.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. Cold Spring Harbor Laboratory; Cold Spring Harbor, New York: 1989. [Google Scholar]
- 20.Edgar RC. Nucl Acids Res. 2004;32:1792. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gouy M, Guindon S, Gascuel O. Mol Biol Evol. 2010;27:221. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- 22.Saitou N, Nei M. Mol Biol Evol. 1987;4:406. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 23.Guex N, Peitsch MC. Electrophoresis. 1997;18:2714. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 24.Stamatakis A. Bioinformatics. 2006;22:2688. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 25.Le SQ, Gascuel O. Mol Biol Evol. 2008;25:1307. doi: 10.1093/molbev/msn067. [DOI] [PubMed] [Google Scholar]
- 26.Jones DT, Taylor WR, Thornton JM. Comput Applic Biosci. 1992;8:275. doi: 10.1093/bioinformatics/8.3.275. [DOI] [PubMed] [Google Scholar]
- 27.Shimodaira H, Hasegawa M. Mol Biol Evol. 1999;16:1114. [Google Scholar]
- 28.Yang Z, Kumar S, Nei M. Genetics. 1995;141:1641. doi: 10.1093/genetics/141.4.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. Mol Biol Evol. 2013;30:2725. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yuryev R, Briechle S, Gruber-Khadjawi M, Griengl H, Liese A. ChemCatChem. 2010;2:981. [Google Scholar]
- 31.Ballini R, Maggi R, Palmieri A, Sartori G. Synthesis. 2007;19:3017. [Google Scholar]
- 32.Liu L, Zou Y. J Appl Polym Sci. 2012;123:554. [Google Scholar]
- 33.Martin S. J Am Chem Soc. 1959;89:2598. [Google Scholar]
- 34.von Langermann J, Nedrud DM, Kazlauskas RJ. ChemBioChem. 2014;15:1931. doi: 10.1002/cbic.201402081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wolfsie JH, Sheffer CB. J Occup Med. 1959:281. [Google Scholar]
- 36.Selmar D, Carvalho FJP, Conn EE. Anal Biochem. 1987;166:208. doi: 10.1016/0003-2697(87)90565-3. [DOI] [PubMed] [Google Scholar]
- 37.Andexer J, Guterl JK, Pohl M, Eggert T. Chem Commun. 2006;40:4201. doi: 10.1039/b607863j. [DOI] [PubMed] [Google Scholar]
- 38.Fuhshuku K, Asano Y. J Biotech. 2011;153:153. doi: 10.1016/j.jbiotec.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 39.Bernhardt P, Hult K, Kazlauskas RJ. Angew Chem Int Ed Engl. 2005;44:2742. doi: 10.1002/anie.200463006. [DOI] [PubMed] [Google Scholar]
- 40.Janes LE, Löwendahl AC, Kazlauskas RJ. Chem Eur J. 1998;4:2324. [Google Scholar]
- 41.Barton P, Page MI. J Chem Soc Perkin Trans. 1993;2:2317. [Google Scholar]
- 42.Corric I, Muller S, List B. J Am Chem Soc. 2010;132:17370. doi: 10.1021/ja108642s. [DOI] [PubMed] [Google Scholar]
- 43.Kasaplar P, Riente P, Hartmann C, Periquas MA. Adv Synth Catal. 2012;354:2905. [Google Scholar]
- 44.Seah SYK, Labbé G, Nerdinger S, Johnson MR, Snieckus V, Eltis LD. J Biol Chem. 2000;275:15701. doi: 10.1074/jbc.275.21.15701. [DOI] [PubMed] [Google Scholar]
- 45.Wahler D, Badalassi F, Crotti P, Reymond JL. Chem Eur J. 2002;8:3211. doi: 10.1002/1521-3765(20020715)8:14<3211::AID-CHEM3211>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 46.Agresti AA. Stat Sci. 1992;7:131. [Google Scholar]
- 47.Lenfant N, Hotelier T, Velluet E, Bourne Y, Marchot P, Chatonnet A. Nucl Acid Res. 2013;41:D423. doi: 10.1093/nar/gks1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kourist R, Jochens H, Bartsch S, Kuipers R, Padhi SK, Gall MG, Böttcher D, Joosten HJ, Bornscheuer UT. ChemBioChem. 2010;11:1635. doi: 10.1002/cbic.201000213. [DOI] [PubMed] [Google Scholar]
- 49.Ollis D, Cheah E, Cygler M, Dijkstra BW, Frolow F, Franken SM, Harel M, Remington S, Silman I, Schrag JD. Protein Eng Des Sel. 1992;5:197. doi: 10.1093/protein/5.3.197. [DOI] [PubMed] [Google Scholar]
- 50.Nardini M, Dijkstra BW. Curr Opin Struct Biol. 1999;9:732. doi: 10.1016/s0959-440x(99)00037-8. [DOI] [PubMed] [Google Scholar]
- 51.Holmquist M. Curr Prot Pept Sci. 2000;1:209. doi: 10.2174/1389203003381405. [DOI] [PubMed] [Google Scholar]
- 52.Rauwerdink AM, Lunzer M, Devamani T, Jones B, Mooney J, Zhang Z-J, Xu J-H, Kazlauskas RJ, Dean AM. Mol Biol Evol. 2016;33 doi: 10.1093/molbev/msv338. http://doi.org/10.1093/molbev/msv338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hickel A, Hasslacher M, Griengl H. Physiol Plant. 1996;98:891. [Google Scholar]
- 54.Selmar D, Lieberei R, Biehl B, Conn EE. Physiol Plant. 1989;75:97. [Google Scholar]
- 55.Gruber K, Gartler G, Krammer B, Schwab H, Kratky C. J Biol Chem. 2004;279:20501. doi: 10.1074/jbc.M401575200. [DOI] [PubMed] [Google Scholar]
- 56.Hughes J, Carvalho FJP, Hughes MA. Arch Biochem Biophys. 1994;311:469. doi: 10.1006/abbi.1994.1267. [DOI] [PubMed] [Google Scholar]
- 57.Dadashipour M, Yamazaki M, Momonoi K, Tamura K, Fuhshuku K, Kanase Y, Uchimura E, Kaiyun G, Asano Y. J Biotechnol. 2011;153:100. doi: 10.1016/j.jbiotec.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 58.Forouhar F, Yang Y, Kumar D, Chen Y, Fridman E, Park SW, Chiang Y, Acton TB, Montelione GT, Pichersky E, Klessig DF, Tong L. Proc Natl Acad Sci U S A. 2005;102:1773. doi: 10.1073/pnas.0409227102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mattern-Dogru E, Ma X, Hartmann J, Decker H, Stöckigt J. Eur J Biochem. 2002;269:2889. doi: 10.1046/j.1432-1033.2002.02956.x. [DOI] [PubMed] [Google Scholar]
- 60.Koo YJ, Yoon ES, Seo JK, Kim JK, Choi YD. J Korean Soc Appl Biol Chem. 2013;56:27. [Google Scholar]
- 61.Andexer JN, von Langermann J, Mell A, Bocola M, Kragl U, Eggert T, Pohl M. Angew Chem Int Ed Engl. 2007;46:8679. doi: 10.1002/anie.200701455. [DOI] [PubMed] [Google Scholar]
- 62.Tattersall DB, Bak S, Jones PR, Olsen CE, Nielsen JK, Hansen ML, Høj PB, Møller BL. Science. 2001;293:1826. doi: 10.1126/science.1062249. [DOI] [PubMed] [Google Scholar]
- 63.Rajniak J, Barco B, Clay NK, Sattely ES. Nature. 2015;525:376. doi: 10.1038/nature14907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chan AP, Crabtree J, Zhao Q, Lorenzi H, Orvis J, Puiu D, Melake-Berhan A, Jones KM, Redman J, Chen G, Cahoon EB, Gedil M, Stanke M, Haas BJ, Wortman JR, Fraser-Liggett CM, Ravel J, Rabinowicz PD. Nat Biotechnol. 2010;28:951. doi: 10.1038/nbt.1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yu G, Nguyen TT, Guo Y, Schauvinhold I, Auldridge ME, Bhuiyan N, Ben-Israel I, Iijima Y, Fridman E, Noel JP, Pichersky E. Plant Physiol. 2010;154:67–77. doi: 10.1104/pp.110.157073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Auldridge ME, Guo Y, Austin MB, Ramsey J, Fridman E, Pichersky E, Noel JP. Plant Cell. 2012;24:1596. doi: 10.1105/tpc.111.093997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, Parker C, Graham L, Engler MM, Hammock BD, Zeldin DC, Kroetz DL. Mol Med. 2013;87:992. doi: 10.1161/01.res.87.11.992. [DOI] [PubMed] [Google Scholar]
- 68.Thalji RK, McAtee JJ, Belyanskaya S, Brandt M, Brown GD, Costell MH, Ding Y, Dodson JW, Eisennagel SH, Fries RE, Gross JW, Harpel MR, Holt DA, Israel DI, Jolivette LJ, Krosky D, Li H, Lu Q, Mandichak T, Roethke T, Schnackenberg CG, Schwartz B, Shewchuk LM, Xie W, Behm DJ, Douglas SA, Shaw AL, Marino JP., Jr Bioorg Med Chem Lett. 2013;23:3584. doi: 10.1016/j.bmcl.2013.04.019. [DOI] [PubMed] [Google Scholar]
- 69.Beetham JK, Tian TG, Hammock BD. Arch Biochem Biophys. 1993;305:197. doi: 10.1006/abbi.1993.1411. [DOI] [PubMed] [Google Scholar]
- 70.Okai M, Ohtsuka J, Imai LF, Mase T, Moriuchi R, Tsuda M, Nagata K, Nagata Y, Tanokura M. J Bacteriol. 2013;195:2642. doi: 10.1128/JB.02020-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nakano S, Asano Y. Sci Rep. 2015;5:8193. doi: 10.1038/srep08193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Purkarthofer T, Gruber K, Gruber-Khadjawi M, Waich K, Skranc W, Mink D, Griengl H. Angew Chem Int Ed Engl. 2006;45:3454. doi: 10.1002/anie.200504230. [DOI] [PubMed] [Google Scholar]
- 73.Wajant H, Forster S. Plant Sci. 1996;115:25. [Google Scholar]
- 74.Foerster S, Roos J, Effenberger F, Wajant H, Sprauer A. Angew Chem Int Ed Engl. 1996;35:437. [Google Scholar]
- 75.Gruber-Khadjawi M, Purkarthofer T, Skranc W, Griengl H. Adv Synth Catal. 2007;349:1445. [Google Scholar]
- 76.Yuryev R, Briechle S, Gruber-Khadjawi M, Griengl H, Liese A. ChemCatChem. 2010;2:981. [Google Scholar]
- 77.Fuhshuku K, Asano Y. J Biotech. 2011;153:153. doi: 10.1016/j.jbiotec.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 78.Forouhar F, Yang Y, Kumar D, Chen Y, Fridman E, Park SW, Chiang Y, Acton TB, Montelione GT, Pichersky E, Klessig DF, Tong L. Proc Natl Acad Sci U S A. 2005;102:1773. doi: 10.1073/pnas.0409227102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dogru E, Warzecha H, Seibel F, Haebel S, Lottspeich F, Stöckigt J. Eur J Biochem. 2000;267:1397. doi: 10.1046/j.1432-1327.2000.01136.x. [DOI] [PubMed] [Google Scholar]
- 80.Hogg JL, Elrod JP, Schowen RL. J Am Chem Soc. 1980;102:2082. [Google Scholar]
- 81.Guterl J-K, Andexer J, Sehl T, von Langermann J, Frindi-Wosch I, Rosenkranz T, Fitter J, Gruber K, Kragl U, Eggert T. J Biotechnol. 2009;141:166. doi: 10.1016/j.jbiotec.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 82.Yan G, Cheng S, Zhao G, Wu S, Liu Y, Sun W. Biotechnol Lett. 2003;25:1041. doi: 10.1023/a:1024182228057. [DOI] [PubMed] [Google Scholar]
- 83.review: Cole MF, Gaucher EA. Curr Opin Chem Biol. 2011;15:399. doi: 10.1016/j.cbpa.2011.03.005.
- 84.Reetz MT, Mondière R, Carballeira JD. Tetrahedron Lett. 2007;48:1679. [Google Scholar]
- 85.Li C, Feng XW, Wang N, Zhou YJ, Yu XQ. Green Chem. 2008;10:616. [Google Scholar]
- 86.Padhi SK, Fujii R, Legatt GA, Fossum SL, Berchtold R, Kazlauskas RJ. Chem Biol. 2010;17:863. doi: 10.1016/j.chembiol.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 87.Khersonsky O, Tawfik DS. Annu Rev Biochem. 2010;79:471. doi: 10.1146/annurev-biochem-030409-143718. [DOI] [PubMed] [Google Scholar]
- 88.Lange OF, Lakomek NA, Fares C, Schroder GF, Walter KFA, Becker S, Meiler J, Grubmuller H, Griesinger C, de Groot BL. Science. 2008;320:1471. doi: 10.1126/science.1157092. [DOI] [PubMed] [Google Scholar]
- 89.Hou L, Honaker MT, Shireman LM, Balogh LM, Roberts AG, Ng KC, Nath A, Atkins WM. J Biol Chem. 2007;282:23264. doi: 10.1074/jbc.M700868200. [DOI] [PubMed] [Google Scholar]
- 90.Skopalik J, Anzenbacher P, Otyepka M. J Phys Chem B. 2008;112:8165. doi: 10.1021/jp800311c. [DOI] [PubMed] [Google Scholar]
- 91.Due AV, Kuper J, Geerlof A, von Kries JP, Wilmanns M. Proc Natl Acad Sci U S A. 2011;108:3554. doi: 10.1073/pnas.1015996108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fong DH, Berghuis AM. EMBO J. 2002;21:2323. doi: 10.1093/emboj/21.10.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Babtie AC, Tokuriki N, Hollfelder F. Curr Opin Chem Biol. 2010;14:200. doi: 10.1016/j.cbpa.2009.11.028. [DOI] [PubMed] [Google Scholar]
- 94.Hiblot J, Gotthard G, Elias M, Chabriere E. PLoS One. 2013;8:e75272. doi: 10.1371/journal.pone.0075272. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.