

Abstract
Oral squamous cell carcinoma (OSCC) is currently a highly prevalent disease worldwide. Cisplatin (CDDP) is widely used for the chemotherapy of OSCC. Yet, the molecular mechanisms responsible for cisplatin resistance have not been fully elucidated. In this study, we showed that overexpression of p21 (RAC1) activated kinase 1 (PAK1) induced epithelial to mesenchymal transition (EMT) and significantly promoted the invasion and migration of oral squamous cell carcinoma SCC25 cells. Emerging evidence indicates a strong link between resistance to therapy and the induction of EMT in cancer. We showed that overexpression of PAK1 induced cisplatin resistance in SCC25 cells. ERCC1 and YAP can promote cisplatin resistance in human OSCC. We showed that ERCC1 and YAP protein were upregulated by PAK1 in SCC25 cells. We found that miR-485-5p inhibited PAK1 protein expression in the SCC25 cells. Contrary to PAK1, we demonstrated that overexpression of miR-485-5p reversed EMT and significantly inhibited invasion and migration. Moreover, its overexpression sensitized SCC25-CR cells (cisplatin-resistant cells) to cisplatin. Thus, we conclude that miR-485-5p reverses EMT and promotes cisplatin-induced cell death by targeting PAK1 in oral tongue squamous cell carcinoma. This study suggests that PAK1 plays an essential role in the progression of OSCC and it is a potential therapeutic target for OSCC.
Keywords: oral squamous cell carcinoma, cisplatin resistance, miR-485-5p, p21 (RAC1) activated kinase 1
Introduction
Oral squamous cell carcinoma (OSCC) is currently a highly prevalent disease worldwide (1). More than half of patients die of this disease or the associated complications within 5 years even under available therapies (2). The prognosis of OSCC remains dismal (2). The low median survival rate is associated with chemotherapeutic resistance (3,4). Presently, there is limited information regarding the regulatory mechanisms of chemoresistance in oral cancer.
Epithelial to mesenchymal transition (EMT) is an essential process for driving plasticity during development and in the context of different morphogenetic events; however it is also an unintentional behavior of cells during malignant transformation (5–6). During this process, the cells lose their epithelial characteristics, including their polarity and specialized cell-cell contacts, and acquire a migratory behavior, allowing them to move away from their epithelial cell community and to integrate into surrounding tissue, even at remote locations. EMT illustrates the differentiation plasticity during development and is complemented by another process, called mesenchymal to epithelial transition (MET) (8). Emerging evidence suggests that there is a strong link between therapeutic resistance and the induction of EMT in cancer (9). Identifying the mechanisms that promote EMT and the development of drug resistance could be a key approach for the development of novel therapeutic targets.
p21 (RAC1) activated kinase 1 (PAK1) lies within the 11q13 region. Aberrant expression/activation of PAK1 has been described in OSCC as well as in several other types of cancers including breast, brain, pancreatic, colon, bladder, ovarian, hepatocellular, urinary tract, renal cell carcinoma and thyroid cancers (10). Stimulating OSCC cells with serum growth factors was found to lead to PAK1 re-localization and caused profound cytoskeletal remodeling (11). PAK1 was also found to be involved in the invasion, migration and cytoskeletal remodelling for OSCC cells (11). In this study, we showed that PAK1 could be a potential therapeutic target for OSCC.
Materials and methods
Human OSCC cell lines, SCC25 and SCC25-res (cisplatin-resistant cells)
SCC25 cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). To obtain cisplatin-resistant tongue cancer cells, we treated SCC25 cells with escalating concentrations of cisplatin from 107 to 105 M. The established SCC25-res (cisplatin-resistant SCC25) cells grew at a similar rate in the presence or absence of 105 M cisplatin for 3 days (data not shown). The IC50 is the cisplatin concentration that reduces proliferating cells by 50%. The IC50 of SCC25-res cells increased by 12-fold, respectively, as compared with the SCC25 cells (data not shown). All cancerous cell lines were grown in RPMI-1640 medium (Thermo Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS) (HyClone, Rockford, IL, USA) and 100 U/ml penicillin and streptomycin.
MTT assay
Cell proliferation was assessed by 3-(4,5–dimethylthiazol-2–yl)-2,5-diphenyltetrazolium (MTT) assay (Sigma, St. Louis, MO, USA). MTT assay was performed as previously described (12–14). In brief, the cells were plated in 96-well plates in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum at a density of 8×103 cells per well at 37°C in a 5% CO2 incubator for 12 h. The cells were treated as indicated in each figure for 12 h. MTT solution (5 mg/ml) was then added to the wells (20 µl per well). The plates were incubated in a cell incubator for 4 h, and the supernatant was then removed and 150 µl of dimethyl sulfoxide were added to each well. Following incubation for 10 min, the absorbance of each well was measured using a Synergy™ 4 (BioTek Instruments, Winooski, VT, USA) at a wavelength of 570 nm, with the reference wavelength set at 630 nm. Absorbance was directly proportional to the number of surviving cells. The viability of the control group (cells transfected with pcDNA3.1) was considered to be 100%.
BrdU incorporation assay
Cell proliferation was also assessed using a colorimetric BrdU proliferation kit by following the manufacturer's instructions (Cat. no. 11647229001; Roche; Basel, Switzerland). Briefly, the cells treated with the peptides were labeled with BrdU for 3 to 4 h. The genomic DNA was then fixed and denatured, followed by incubation with peroxidase-conjugated anti-BrdU antibody for 90 min. The substrate of the conjugated peroxidase was then added and the reaction product was measured by determining the absorbance (A370–A492 nm). The results were then normalized by the number of total viable cells, which was determined by a side-by-side cell viability assay as described above.
PAK1-expressing plasmids/empty vectors and transfection experiments
PAK1-expressing plasmids and empty vectors (pcDNA3.1) were obtained from Tiangen Biotech (Beijing, China). The cells were seeded at a density of 1.5×105 per well in 6-well plates or 60-mm dishes in 2 ml of complete medium containing 10% FCS for 24 h. Transfections were performed using Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer's instructions.
Western blot analysis
The total proteins in cells were extracted with protein lysis solution (Tiangen Biotech). The protein concentration was measured with a bicinchoninic acid kit (Tiangen Biotech). Protein extracts were resolved through 8% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to polyvinylidene difluoride membranes (Bio-Rad, Berkeley, CA, USA), probed with anti-bodies against human ERCC1 (1:500; ab129267), YAP (1:500; ab52771), PAK1 (1:500; ab40795), vimentin (1:500; ab92547), E-cadherin (1:500; ab40772) or β-actin (1:500; ab8227) (all from Abcam, Cambridge, MA, USA) and then with IRDye™-800 conjugated anti-rabbit secondary antibodies (ab218695; Abcam) for 30 min at room temperature. The specific proteins were visualized by Odyssey™ Infrared Imaging System (Gene Company, Lincoln, NE, USA).
Real-time PCR for microRNAs
Total RNA from the cultured cells, with efficient recovery of small RNAs, was isolated using the mirVana miRNA isolation kit (Ambion, Austin, TX, USA). Detection of the mature form of miRNAs was performed using the mirVana qRT-PCR miRNA detection kit and qRT-PCR primer sets, according to the manufacturer's instructions (Ambion). The sequences of the primers were as follows: miR-485-5p forward, 5′-CCAAGCTTCACCCATTCCTAACAGGAC-3′ and reverse, 5′-CGGGATCCGTAGGTCAGTTACATGCATC-3′. The U6 small nuclear RNA was used as an internal control.
Immunofluorescence staining
Cells were stained for immuno-fluorescence on coverslips. After fixation and permeabilization, the cells were incubated with primary antibodies against PAK1, E-cadherin or vimentin (Abcam) and then incubated with FITC-conjugated secondary antibodies (Invitrogen). The coverslips were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) and imaged under a confocal microscope TCS SP5 (Lecia, Solms, Germany).
Reverse transcription-PCR and real-time PCR for mRNA
PCR was performed as previously described (12,15). The PCR primer sequences are as follows, glyceraldehyde 3-phosphate dehydrogenase (GAPDH) forward, 5′-ATTCAACGGCACAGTCAAGG-3′ and reverse, 5′-GCAGAAGGGGCGGAGATGA-3′; E-cadherin forward, 5′-TCAACGATCCTGACCAGCAGTTCG-3′ and reverse, 5′-GGTGAACCATCATCTGTGG CGATG-3′; vimentin forward, 5′-GACAATGCGTCTCTGGCACGTCTT-3′ and reverse, 5′-TCCTCCGCCTCCTGCAGGTTCTT-3′.
Methods of bioinformatics
The analysis of potential microRNA target sites was performed using the commonly used prediction algorithm, miRanda (http://www.microrna.org/).
Migration and invasion assays
Migration and invasion assays were performed as previously described (16). The migration and invasion assays were performed using 24 well Transwell chambers (8 µm; Corning Inc., Corning, NY, USA). For the migration assay, 1×105 cells suspended in 200 µl serum-free RPMI-1640 medium were seeded into the upper chamber of the Transwell invasion system, and 500 µl RPMI-1640 medium containing 10% FBS was added to the lower chamber. Following a 24-h incubation, cells on the upper surface of the membrane were scrubbed off, and the migrated cells were fixed with 95% ethanol and stained with 0.1% crystal violet for 10 min. The number of migrated cells was determined by counting 5 random fields on each membrane. The invasion assay protocol was similar to that of the migration assay, with the exception that the upper chambers were first covered with 1 mg/ml Matrigel, as previously described (12,14,15,17).
Statistical analysis
Data are presented as the means ± SEM. Student's t-test (two-tailed) was used to compare two groups (P<0.05 was considered significant).
Results
PAK1 does not affect the proliferation of oral squamous cell carcinoma SCC25 cells
To investigate whether PAK1 can affect the proliferation of SCC25 cells, firstly using western blot analysis, we tested whether PAK1-expressing plasmids could cause stable expression of PAK1 protein in the SCC25 cells. The results showed that PAK1 protein was significantly increased by PAK1-expressing plasmids in the cells (Fig. 1A). In addition, we performed MTT assay to detect the proliferation of SCC25 cells transfected with the PAK1-expressing plasmids. The results showed that PAK1 did not affect the proliferation of the SCC25 cells after 48 h of transfection (Fig. 1B). To further show the effects of PAK1 on proliferation, we performed BrdU incorporation assay to detect DNA synthesis in the cells. The results confirmed that PAK1 did not affect DNA synthesis in the cells (Fig. 1C).
Figure 1.
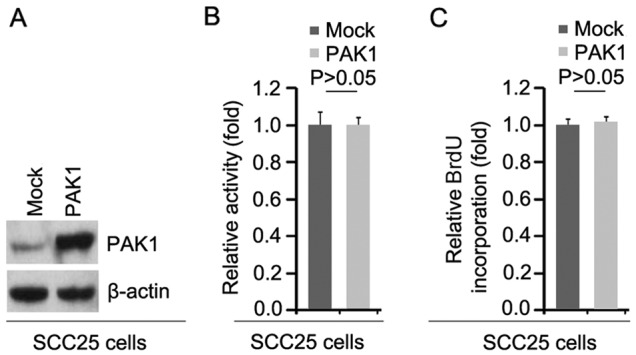
p21 (RAC1) activated kinase 1 (PAK1) does not affect the proliferation of oral squamous cell carcinoma SCC25 cells. (A) Western blotting for PAK1 protein in SCC25 cells transfected with PAK1-expressing plasmids and empty vectors (mock). β-actin was used as a loading control. n=3. (B) MTT assay of SCC25 cells. SCC25 cells were transfected with PAK1-expressing plasmids or empty vectors (mock) and then cell viability was measured. n=3. (C) BrdU incorporation assay of SCC25 cells. Cells were transfected with PAK1-expressing plasmids or empty vectors (mock). n=3.
PAK1 promotes EMT, migration and invasion in SCC25 cells
In order to assess the role of PAK1 in the EMT of SCC25 cells, we transfected SCC25 cells with PAK1-expressing plasmids and then we found that its overexpression caused significant changes in the cell morphology (EMT, phenotype from a cobblestone-like to a spindle-like morphology) (Fig. 2A). To further verify that the changes in cell morphology were caused by EMT, we performed immunofluorescence to detect expression of epithelial marker, E-cadherin, and mesenchymal marker, vimentin, in the SCC25 cells transfected with the PAK1-expressing plasmids and the cells transfected with the empty vectors. The results demonstrated that PAK1 promoted vimentin protein expression (Fig. 2B) and inhibited E-cadherin protein expression (Fig. 2C). To further verify the results of the immunofluorescence, we performed western blot analysis to detect the expression of vimentin and E-cadherin in the SCC25 cells transfected with the PAK1-expressing plasmids and the cells transfected with the empty vectors. The results revealed that vimentin protein was increased and E-cadherin protein was decreased by PAK1 (Fig. 2D). We also performed RT-PCR to detect vimentin and E-cadherin mRNA in the cells transfected with PAK1. Consistent with the western blot analysis, we found that vimentin mRNA was upregulated and E-cadherin mRNA was downregulated by PAK1 (Fig. 2E). We next sought to determine whether PAK1 has any impact on migration and invasion in the cells. The migration and invasion assays showed that overexpression of PAK1 promoted the migration and invasion of the PAK1-overexpressing SCC25 cells (Fig. 2F).
Figure 2.

p21 (RAC1) activated kinase 1 (PAK1) promotes epithelial to mesenchymal transition (EMT), migration and invasion in SCC25 cells. (A) SCC25 cells were transfected with PAK1-expressing plasmids or empty vectors (mock). Cells were then photographed after 72 h of transfection. n=3. (B) Representative images of vimentin immunofluorescence staining of SCC25 cells transfected as indicated. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Red indicates vimentin fluorescence, and blue indicates DAPI. (C) Representative images of E-cadherin immunofluorescence staining of SCC25 cells transfected as indicated. Nuclei were counterstained with DAPI. Red indicates E-cadherin fluorescence, and blue indicates DAPI. (D) Western blotting for E-cadherin and vimentin in SCC25 cells transfected with the PAK1-expressing plasmids or empty vectors (mock). β-actin was a loading control. n=3. (E) RT-PCR for E-cadherin and vimentin in SCC25 cells transfected with PAK1 expressing plasmids or empty vectors (mock). GAPDH was used as a loading control. n=3. (F) Matrigel invasion and Transwell migration assays for SCC25 cells transfected with the PAK1-expressing plasmids or empty vector (mock). n=3.
Overexpression of the PAK1 promotes cisplatin resistance in SCC25 cells
To further identify whether PAK1 affects cisplatin efficacy in SCC25 cells, we transfected the cells with PAK1-expressing plasmids. We then performed MTT assay in the SCC25 cells treated as indicated. The results showed that overexpression of PAK1 could transform SCC25 cells to SCC25-CR cells (Fig. 3A), suggesting that its overexpression promoted cisplatin resistance. We also performed western blot analysis to detect ERCC1 and YAP protein expression in SCC25 cells transfected with the PAK1-expressing plasmids and empty vectors. The results demonstrated that the levels of ERCC1 and YAP protein were increased by PAK1 (Fig. 3B).
Figure 3.
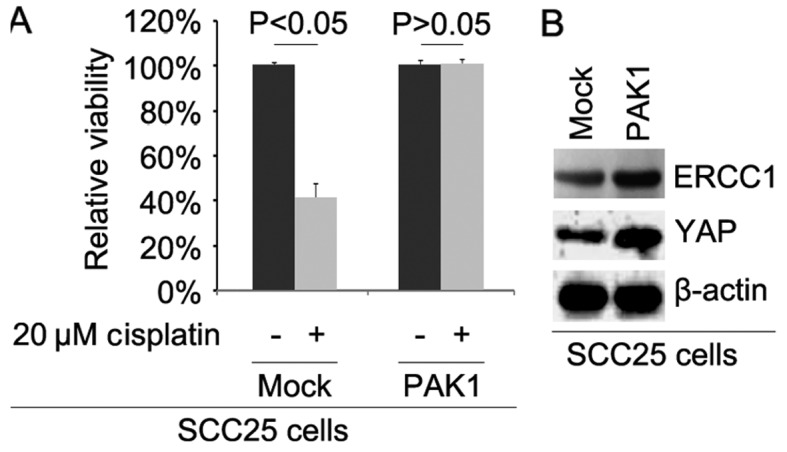
Overexpression of p21 (RAC1) activated kinase 1 (PAK1) promotes cisplatin resistance in SCC25 cells. (A) MTT assay of the cell viability of the SCC25 cells. SCC25 cells transfected with the PAK1-expressing plasmids and empty vectors (mock) were untreated or treated with cisplatin. n=3. (B) Western blotting for ERCC1 and YAP in the SCC25 cells transfected with the PAK1-expressing plasmids and empty vectors (mock). β-actin was used as a loading control. n=3.
miR-485-5p inhibits PAK1 protein expression in SCC25 cells
Having demonstrated that overexpression of PAK1 promoted EMT, migration and invasion in SCC25 cells, we next studied the mechanisms regulating PAK1 expression in SCC25 cells. MicroRNAs (miRs) are a class of small noncoding RNAs (~22 nucleotides) that negatively regulate protein-coding gene expression by targeting mRNA degradation or translation inhibition (18–20). To further confirm whether PAK1 is regulated by microRNAs, we used the commonly used prediction algorithm, miRanda (http://www.microrna.org/microrna/home.do), to analyze the 3′ untranslated region (3′UTR) of PAK1. A number of microRNAs were found by the algorithm. But we were interested in miR-485-5p, as it has been reported that miR-485-5p is a tumor-suppression gene that inhibits oncogene expression (21–24). Thus, we reasoned that miR-485-5p could downregulate PAK1 protein expression by targeting its 3′UTR in SCC25 cells. The target sites on the 3′UTR of PAK1 are shown in Fig. 4A.
Figure 4.

miR-485-5p inhibits p21 (RAC1) activated kinase 1 (PAK1) protein expression in SCC25 cells. (A) Schematic of the predicted miR-485-5p-binding sites in the 3′ untranslated region (3′UTR) of PAK1 mRNA by miRanda. (B) Real-time PCR of miR-485-5p in SCC25 cells transfected with pre-miR-485-5p and control miR (mock). U6 was used as a loading control. n=3. (C) Western blotting for PAK1 in SCC25 cells transfected with miR-485-5p and control miR (mock). β-actin was a loading control. n=3. (D) Immunofluorescence analyses for PAK1 in SCC25 cells transfected with pre-miR-485-5p and control miR (mock). n=3.
To identify the role of miR-485-5p in regulating PAK1 expression in SCC25 cells, we transfected SCC25 cells with pre-miR-485-5p and control miR. After transfection, miR-485-5p expression was detected by real-time PCR and the results showed that miR-485-5p was significantly increased by pre-miR-485-5p in the cells (Fig. 4B). To confirm that miR-485-5p can regulate PAK1 expression, we performed western blot analysis to detect PAK1 protein expression in the SCC25 cells transfected with pre-miR-485-5p and control miR. The results showed that PAK1 protein was significantly inhibited by miR-485-5p (Fig. 4C). We next performed immunofluorescence analyses in SCC25 cells transfected with pre-miR-485-5p and control miR. The results showed that PAK1 protein was evidently inhibited in the cells transfected with pre-miR-485-5p (Fig. 4D).
miR-485-5p inhibits the proliferation of SCC25 cells
We performed MTT assay to detect the proliferation rate of the SCC25 cells transfected with miR-485-5p and control miR. The results demonstrated that miR-485-5p inhibited the proliferation of the SCC25 cells after 48 h of transfection (Fig. 5A). To further show the effects of miR-485-5p on proliferation, we performed BrdU incorporation assay to detect DNA synthesis in the cells. The results confirmed that DNA synthesis of the cells was significantly inhibited (Fig. 5B).
Figure 5.
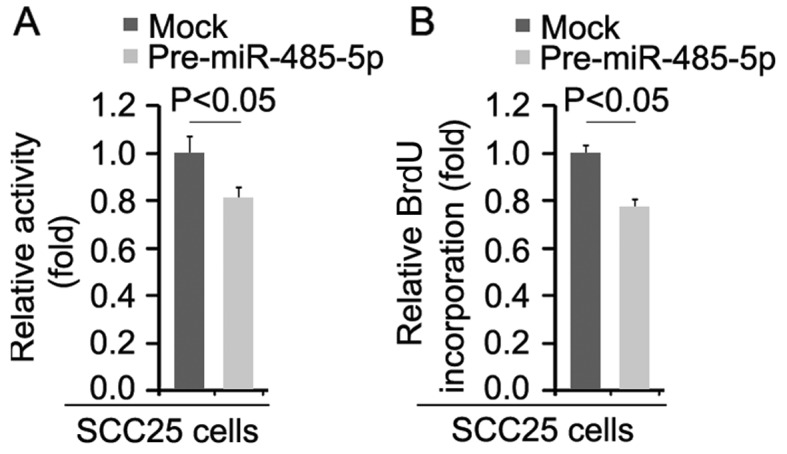
miR-485-5p inhibits the proliferation of SCC25 cells. (A) MTT assay of SCC25 cells. SCC25 cells were transfected with pre-miR-485-5p or control miR (mock) and then cell viability was assessed. n=3. (B) BrdU incorporation assay of SCC25 cells. Cells were transfected with pre-miR-485-5p or control miR (mock). n=3.
miR-485-5p reverses EMT and inhibits the migration and invasion of SCC25 cells
In order to assess the role of miR-485-5p in EMT of SCC25 cells, we transfected SCC25 cells with pre-miR-485-5p and then found that its overexpression caused significant changes in the cell morphology (MET, phenotype from a spindle-like morphology to a cobblestone-like) (Fig. 6A). To further verify that the changes in cell morphology were caused by MET, we performed western blot analysis to detect the expression of epithelial marker, E-cadherin, and mesenchymal marker, vimentin, in the SCC25 cells transfected with pre-miR-485-5p and the cells transfected with control miR. The results demonstrated that miR-485-5p inhibited vimentin protein expression and promoted E-cadherin protein expression (Fig. 6B). We next sought to determine whether miR-485-5p has any impact on migration and invasion in the cells. The migration and invasion assays showed that overexpression of miR-485-5p inhibited the migration and invasion of cells transfected with pre-miR-485-5p (Fig. 6C).
Figure 6.
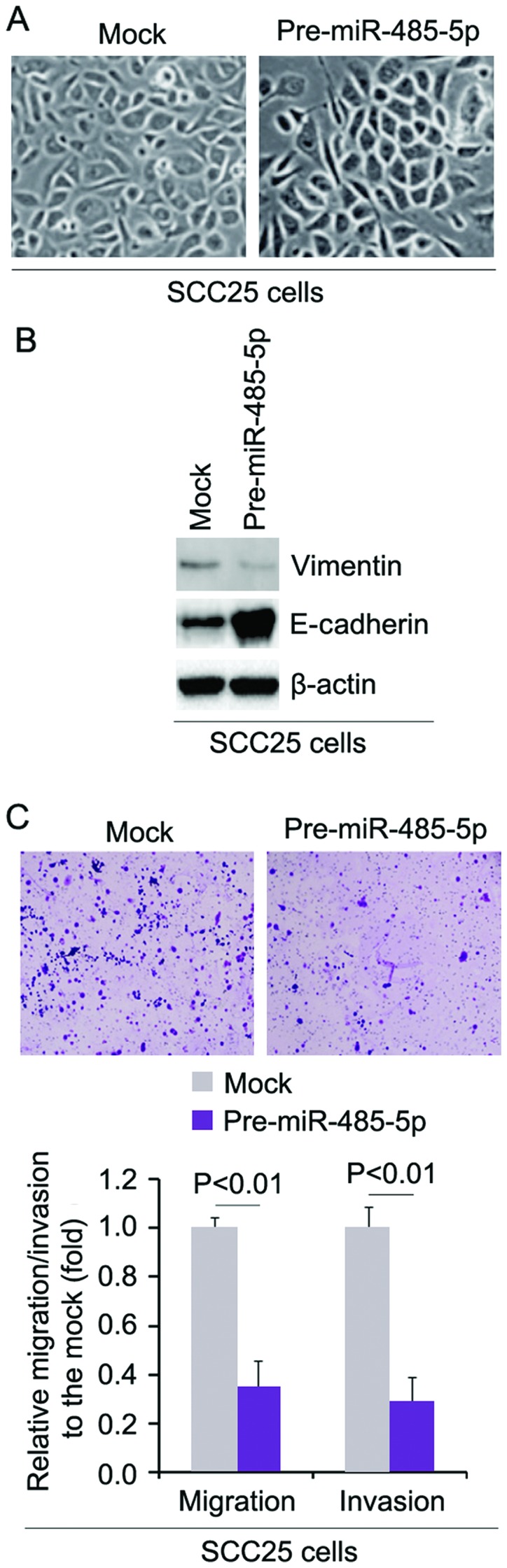
miR-485-5p reverses epithelial to mesenchymal transition (EMT) and inhibits the migration and invasion of SCC25 cells. (A) SCC25 cells were transfected with pre-miR-485-5p or control miR (mock). Cells were then photographed after 72 h of transfection. n=3. (B) Western blotting of E-cadherin and vimentin in the SCC25 cells transfected with pre-miR-485-5p or con-trol miR (mock). β-actin was used as a loading control. n=3. (C) Matrigel invasion and Transwell migration assays of SCC25 cells transfected with pre-miR-485-5p or control miR (mock). n=3.
Overexpression of miR-485-5p reverses cisplatin resistance in SCC25-CR (cisplatin-resistant cells) cells
To further identify whether miR-485-5p affects cisplatin efficacy in SCC25-CR cells, we transfected the cells with pre-miR-485-5p. We then performed MTT assay in the SCC25-CR cells treated as indicated (Fig. 7A). The results showed that overexpression of miR-485-5p transformed SCC25-CR cells to SCC25 cells (Fig. 7A), suggesting that its overexpression reversed cisplatin resistance. We also performed western blot analysis to detect ERCC1 and YAP protein expression in the SCC25-CR cells transfected with pre-miR-485-5p and control miR. The results demonstrated that levels of ERCC1 and YAP protein were decreased by miR-485-5p (Fig. 7B).
Figure 7.
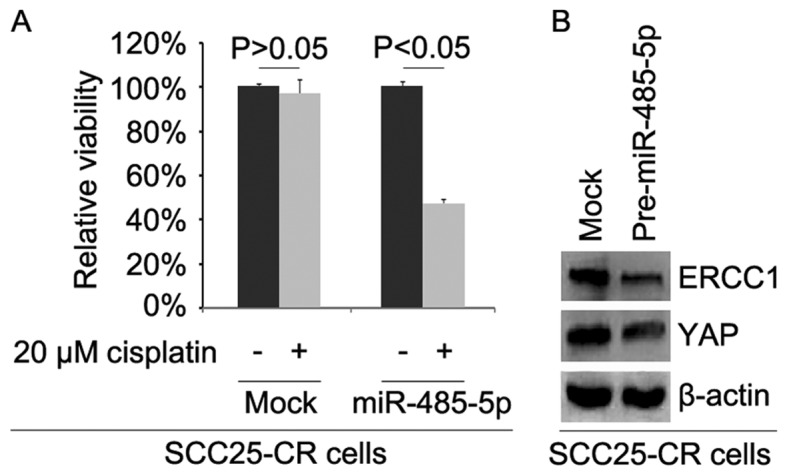
Overexpression of miR-485-5p reverses cisplatin resistance in SCC25-CR (cisplatin-resistant) cells. (A) MTT assay for cell viability of the SCC25-CR cells. SCC25-CR cells transfected with pre-miR-485-5p and empty vectors (mock) were untreated or treated with cisplatin. n=3. (B) Western blotting of ERCC1 and YAP in SCC25-CR cells transfected with pre-miR-485-5p and empty vectors (mock). β-actin was used as a loading control. n=3.
Discussion
Most OSCC-associated deaths are in part due to the spread of tumor cells resistant to conventional therapies (25). Aggressive cells acquire genetic and epigenetic changes that promote formation of their metastasis-associated abilities such as acquisition of increased motility and invasiveness (25). Increased abilities of motility and invasiveness are linked to enhanced local growth of tumor cells, decreased cell-cell adhesion as well as degradation of basement membranes and stroma, referred to as EMT (26,27), a frequently observed phenotypic change usually caused by oncogenes (28–30), and by degradation of the basement membranes initiated by increased matrix metalloproteinases and collagenases (31). In this study, we found that PAK1 induced EMT and promoted migration and invasion in SCC25 cells. Emerging evidence associates chemoresistance with acquisition of EMT in cancer (32). Consistent with the study, we showed that overexpression of PAK1 promoted cisplatin resistance in SCC25 cells. High expression of ERCC1 and YAP are associated with cispantin resistance in locally advanced squamous cell carcinoma of the head and neck (18,33). Our results demonstrated that overexpression of PAK1 promoted ERCC1 and YAP protein expression in the SCC25 cells.
miR-485-5p has been reported as a potential tumor suppressor in gastric cancer, breast cancer, hepatocellular carcinoma and ovarian cancer, but its expression, cellular function and clinical features in OSCC are not known (21–24). Consistent with the roles of miR-485-5p in other cancers, we showed that its overexpression inhibited the proliferation of SCC25 cells. PAK1 is a target gene of miR-485-5p in SCC25. But we found that PAK1 did not inhibit the proliferation of SCC25 cells, implying that miR-485-5p inhibits proliferation by regulating other genes. It has been reported that sensitivity to anticancer drugs is confined to the 'epithelial' subset, and the sensitivity to anticancer drugs could be re-established by microRNA-mediated molecular reversal of EMT (34). Consistent with this report, we found that miR-485-5p reversed EMT and cisplatin-resistance in SCC25-CR cells. Thus, an miR-485-5p-mediated molecular mechanism is responsible for cisplatin resistance. It can block the oncogenic role of PAK1 and hence restoration of miR-485-5p has therapeutic potential in PAK1-positive OSCC.
References
- 1.Warnakulasuriya S. Global epidemiology of oral and oropharyngeal cancer. Oral Oncol. 2009;45:309–316. doi: 10.1016/j.oraloncology.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 2.Lo WL, Kao SY, Chi LY, Wong YK, Chang RC. Outcomes of oral squamous cell carcinoma in Taiwan after surgical therapy: Factors affecting survival. J Oral Maxillofac Surg. 2003;61:751–758. doi: 10.1016/S0278-2391(03)00149-6. [DOI] [PubMed] [Google Scholar]
- 3.Pérez-Sayáns M, Somoza-Martín JM, Barros-Angueira F, Diz PG, Rey JM, García-García A. Multidrug resistance in oral squamous cell carcinoma: the role of vacuolar ATPases. Cancer Lett. 2010;295:135–143. doi: 10.1016/j.canlet.2010.03.019. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Q, Shi S, Yen Y, Brown J, Ta JQ, Le AD. A subpopulation of CD133(+) cancer stem-like cells characterized in human oral squamous cell carcinoma confer resistance to chemotherapy. Cancer Lett. 2010;289:151–160. doi: 10.1016/j.canlet.2009.08.010. [DOI] [PubMed] [Google Scholar]
- 5.Nieto MA. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu Rev Cell Dev Biol. 2011;27:347–376. doi: 10.1146/annurev-cellbio-092910-154036. [DOI] [PubMed] [Google Scholar]
- 6.Savagner P, Yamada KM, Thiery JP. The zinc-finger protein slug causes desmosome dissociation, an initial and necessary step for growth factor-induced epithelial-mesenchymal transition. J Cell Biol. 1997;137:1403–1419. doi: 10.1083/jcb.137.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 8.Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–273. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 9.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 10.Kumar R, Gururaj AE, Barnes CJ. 21-activated kinases in cancer. Nat Rev Cancer. 2006;6:459–471. doi: 10.1038/nrc1892. [DOI] [PubMed] [Google Scholar]
- 11.Parvathy M, Sreeja S, Kumar R, Pillai MR. Potential role of p21 activated kinase 1 (PAK1) in the invasion and motility of oral cancer cells. BMC Cancer. 2016;16(Suppl 1):293. doi: 10.1186/s12885-016-2263-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liao XH, Lu DL, Wang N, Liu LY, Wang Y, Li YQ, Yan TB, Sun XG, Hu P, Zhang TC. Estrogen receptor α mediates proliferation of breast cancer MCF-7 cells via a p21/PCNA/E2F1-dependent pathway. FEBS J. 2014;281:927–942. doi: 10.1111/febs.12658. [DOI] [PubMed] [Google Scholar]
- 13.Li H, Xiang Y, Fan LJ, Zhang XY, Li JP, Yu CX, Bao LY, Cao DS, Xing WB, Liao XH, Zhang TC. Myocardin inhibited the gap protein connexin 43 via promoted miR-206 to regulate vascular smooth muscle cell phenotypic switch. Gene. 2017;616:22–30. doi: 10.1016/j.gene.2017.03.029. [DOI] [PubMed] [Google Scholar]
- 14.Liao XH, Xiang Y, Yu CX, Li JP, Li H, Nie Q, Hu P, Zhou J, Zhang TC. STAT3 is required for MiR-17-5p-mediated sensitization to chemotherapy-induced apoptosis in breast cancer cells. Oncotarget. 2017;8:15763–15774. doi: 10.18632/oncotarget.15000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiang Y, Lu DL, Li JP, Yu CX, Zheng DL, Huang X, Wang ZY, Hu P, Liao XH, Zhang TC. Myocardin inhibits estrogen receptor alpha-mediated proliferation of human breast cancer MCF-7 cells via regulating MicroRNA expression. IUBMB Life. 2016;68:477–487. doi: 10.1002/iub.1507. [DOI] [PubMed] [Google Scholar]
- 16.Lu Y, Chopp M, Zheng X, Katakowski M, Buller B, Jiang F. MiR-145 reduces ADAM17 expression and inhibits in vitro migration and invasion of glioma cells. Oncol Rep. 2013;29:67–72. doi: 10.3892/or.2012.2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liao XH, Zheng L, He HP, Zheng DL, Wei ZQ, Wang N, Dong J, Ma WJ, Zhang TC. STAT3 regulated ATR via microRNA-383 to control DNA damage to affect apoptosis in A431 cells. Cell Signal. 2015;27:2285–2295. doi: 10.1016/j.cellsig.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 18.Yoshikawa K, Noguchi K, Nakano Y, Yamamura M, Takaoka K, Hashimoto-Tamaoki T, Kishimoto H. The Hippo pathway transcriptional co-activator, YAP, confers resistance to cisplatin in human oral squamous cell carcinoma. Int J Oncol. 2015;46:2364–2370. doi: 10.3892/ijo.2015.2948. [DOI] [PubMed] [Google Scholar]
- 19.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-Y. [DOI] [PubMed] [Google Scholar]
- 20.Pasquinelli AE, Reinhart BJ, Slack F, Martindale MQ, Kuroda MI, Maller B, Hayward DC, Ball EE, Degnan B, Müller P, et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature. 2000;408:86–89. doi: 10.1038/35040556. [DOI] [PubMed] [Google Scholar]
- 21.Kang M, Ren MP, Zhao L, Li CP, Deng MM. miR-485-5p acts as a negative regulator in gastric cancer progression by targeting flotillin-1. Am J Transl Res. 2015;7:2212–2222. [PMC free article] [PubMed] [Google Scholar]
- 22.Jing LL, Mo XM. Reduced miR-485-5p expression predicts poor prognosis in patients with gastric cancer. Eur Rev Med Pharmacol Sci. 2016;20:1516–1520. [PubMed] [Google Scholar]
- 23.Kim TH, Kim YK, Kwon Y, Heo JH, Kang H, Kim G, An HJ. Deregulation of miR-519a, 153, and 485-5p and its clinicopathological relevance in ovarian epithelial tumours. Histopathology. 2010;57:734–743. doi: 10.1111/j.1365-2559.2010.03686.x. [DOI] [PubMed] [Google Scholar]
- 24.Lou C, Xiao M, Cheng S, Lu X, Jia S, Ren Y, Li Z. MiR-485-3p and miR-485-5p suppress breast cancer cell metastasis by inhibiting PGC-1α expression. Cell Death Dis. 2016;7:e2159. doi: 10.1038/cddis.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Vita V, Jr, Hellman S, Rosenberg S, Markoe AM. Cancer: Principles and practice of oncology. Am J Clin Oncol. 1986;9:90. doi: 10.1097/00000421-198602000-00020. [DOI] [Google Scholar]
- 26.Birchmeier W, Hulsken J, Behrens J. E-cadherin as an invasion suppressor. Ciba Found Symp. 1995;189:124–141. 174–176. doi: 10.1002/9780470514719.ch10. [DOI] [PubMed] [Google Scholar]
- 27.Vleminckx K, Vakaet L, Jr, Mareel M, Fiers W, van Roy F. Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell. 1991;66:107–119. doi: 10.1016/0092-8674(91)90143-M. [DOI] [PubMed] [Google Scholar]
- 28.Cano A, Pérez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 29.Batlle E, Sancho E, Francí C, Domínguez D, Monfar M, Baulida J, García De Herreros A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2:84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 30.Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D, van Roy F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell. 2001;7:1267–1278. doi: 10.1016/S1097-2765(01)00260-X. [DOI] [PubMed] [Google Scholar]
- 31.Woodhouse EC, Chuaqui RF, Liotta LA. General mechanisms of metastasis. Cancer. 1997;80(Suppl 8):1529–1537. doi: 10.1002/(SICI)1097-0142(19971015)80:8+<1529::AID-CNCR2>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 32.Zhang W, Feng M, Zheng G, Chen Y, Wang X, Pen B, Yin J, Yu Y, He Z. Chemoresistance to 5-fluorouracil induces epithelial-mesenchymal transition via up-regulation of Snail in MCF7 human breast cancer cells. Biochem Biophys Res Commun. 2012;417:679–685. doi: 10.1016/j.bbrc.2011.11.142. [DOI] [PubMed] [Google Scholar]
- 33.Jun HJ, Ahn MJ, Kim HS, Yi SY, Han J, Lee SK, Ahn YC, Jeong HS, Son YI, Baek JH, et al. ERCC1 expression as a predictive marker of squamous cell carcinoma of the head and neck treated with cisplatin-based concurrent chemoradiation. Br J Cancer. 2008;99:167–172. doi: 10.1038/sj.bjc.6604464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McConkey DJ, Choi W, Marquis L, Martin F, Williams MB, Shah J, Svatek R, Das A, Adam L, Kamat A, et al. Role of epithelial-to-mesenchymal transition (EMT) in drug sensitivity and metastasis in bladder cancer. Cancer Metastasis Rev. 2009;28:335–344. doi: 10.1007/s10555-009-9194-7. [DOI] [PMC free article] [PubMed] [Google Scholar]