

Abstract
Lysine hydroxylation of type I collagen telopeptides varies from tissue to tissue and these distinct hydroxylation patterns modulate collagen crosslinking to generate a unique extracellular matrix. Abnormalities in these patterns contribute to pathologies that include osteogenesis imperfecta (OI), fibrosis and cancer. Telopeptide procollagen modifications are carried out by lysyl hydroxylase 2 (LH2), however, little is known regarding how this enzyme regulates hydroxylation patterns. We identified an ER complex of resident chaperones that includes HSP47, FKBP65 and BiP regulating the activity of LH2. Our findings show that FKBP65 and HSP47 modulate the activity of LH2 to either favor or repress its activity. BiP was also identified as a member of the complex, playing a role in enhancing the formation of the complex. This newly identified ER chaperone complex contributes to our understanding of how LH2 regulates lysyl hydroxylation of type I collagen C-telopeptides to affect the quality of connective tissues.
Introduction
Posttranslational modifications are essential for proper synthesis and folding of type I procollagen. One key modification, lysyl hydroxylation of the type I procollagen telopeptides, is of particular importance as it is necessary for proper crosslinking and polymerization of mature collagen fibrils. Hydroxylated lysines allow for the formation of pyridinoline and pyrrole cross-links with other collagen molecules, which increases the stability of the fibrils (1-2). The long form of lysyl hydroxylase LH2 (LH2b encoded by PLOD2) is thought to be the enzyme responsible for hydroxylation of type I procollagen residues Kα2-7N (COL1A2 p.K84), Kα1-9N (COL1A1 p.K170) (NH2 telopeptide) and Kα1+16C (COL1A1 p.K1208) (COOH telopeptide). LH2 activity is tightly regulated to generate tissue specific hydroxylation patterns that modulate crosslinking, and thereby the properties of the extracellular matrix in each connective tissue (1-3). Alterations of these patterns lead to pathological conditions that include the skeletal disorders osteogenesis imperfecta (OI) and Bruck syndrome, fibrotic conditions, or changes associated with cancer metastasis (4-7). Recessively inherited loss of function mutations in PLOD2 (LH2) cause Bruck syndrome, a type of OI characterized by fractures, brittle bones, and congenital contractures (8). Tissues from Bruck syndrome patients show that lack of LH2 activity leads to decreased lysyl hydroxylation of type I collagen telopeptides (8,9), yet little is known regarding how LH2 activity is regulated. Further clues regarding regulation of lysyl hydroxylation derive from studies in patients and animal models with OI, Bruck and Kuskokwim syndromes due to recessively inherited mutations in FKBP10, which encodes the procollagen chaperone FKBP65 (10-14). Loss of FKBP65 function leads to decreased telopeptide lysyl hydroxylation similar to that seen in Bruck syndrome due to PLOD2 mutations. This correlation suggested the hypothesis that FKBP65 might regulate LH2 activity (12,15). Supporting this idea, recent work has shown that FKBP65 assists in the dimerization of LH2, providing a potential mechanism for LH2 regulation (16).
Recent studies have also shown that FKBP65 interacts with HSP47 (encoded by SERPINH1), another established type I procollagen chaperone (17). However, contrary to the consequences of FKBP65 and LH2 molecular defects that produce diminished telopeptide lysyl hydroxylation, homozygosity for a HSP47 missense mutation that reduced HSP47 protein levels in a dachshund OI model showed increased lysyl hydroxylation of type I collagen telopeptides (18). These data suggest that HSP47 might also regulate LH2 activity, but in the opposite direction relative to FKBP65.
Immunoglobulin heavy-chain-binding protein (BiP), also known as glucose-regulated protein 78 (GRP78), is another protein chaperone that interacts with type I procollagen during its maturation in the ER. As a chaperone, BiP facilitates correct folding of nascent ER polypeptides and regulates the unfolded protein response (UPR), ensuring protection of the cell from abnormally folded proteins and ultimately apoptosis (19-21). Induction of the UPR, including the expression of BiP, is proportional to the amount of defective protein in the ER, and can range from increased expression of the folding machinery to cellular apoptosis when the system is overwhelmed. Increased ER stress and up regulation of BiP protein levels have been observed in cases of OI due to structural mutations in the type I procollagen genes, such as those that fall in the C-propeptide domain (22) and BiP has been shown to bind to misfolded, structurally abnormal type I procollagen resulting from mutations that produce OI (23). However, whether BiP interacts with the other type I procollagen chaperones is unknown.
In this study, we determined that LH2, FKBP65, HSP47 and BiP form an ER chaperone complex that regulates type I procollagen telopeptide lysyl hydroxylation. Defects affecting each of these respective complex members resulted in changes in the telopeptide hydroxylation pattern, either by enhancing or diminishing lysine hydroxylation. The data suggest that BiP participates in the formation of the overall complex, increasing the affinity of the components for type I procollagen, and that the balance between FKBP65 and HSP47 regulate the extent of telopeptide lysyl hydroxylation. The results identify a mechanism for regulation of type I collagen crosslinking and further our understanding of disease processes associated with abnormal lysyl hydroxylation, particularly OI.
Materials and methods
Human subjects
Informed consent was obtained from all patients under an approved human subjects protocol. All experimental protocols and methods were carried out in accordance with institutional guidelines and regulations and approved by the UCLA Institutional Biosafety Committee.
Cell culture
Dermal fibroblast cultures were established from explanted skin biopsies from the cases (International Skeletal Dysplasia Registry reference numbers R92-020A and R93-138A). Osteoblasts were collected from explanted fetal calvaria from an unaffected individual at 20 weeks gestational age. Cells were grown in Dulbecco-Vogt Modified Eagle Medium supplemented with 10% FBS. For lysyl hydroxylation studies in fibroblasts, confluent cells were supplemented with 100 ug/ml ascorbic acid for 4 days. For protein analyses, cells were collected in RIPA buffer supplemented with proteinase inhibitors. Serum free media was collected for mass spectrometry analysis.
Rescue experiments were performed by electroporation of cells with a vector containing the FKBP10 coding sequence tagged with DDK-flag (Origene). Knockdown experiments were performed using Ambion Silencer Select Validated siRNA for PLOD2 (112475 and 110523; 8 and 11% mRNA remaining) and HSPA5 (s6979 and s6980; 10 and 20% protein remaining). A siRNA control experiment was used as a negative control siRNA (Thermo Fisher Scientific). Electroporation was performed in a Nucleofector X system using Amaxa P1 primary cell kit and program DS-150.
Western blot
For Western blot analyses, proteins lysates were separated by electrophoresis on 10% SDS-polyacrylamide gels, transferred to PVDF membranes, blocked in 5% milk and probed sequentially with primary antibodies (anti-Hsp47 antibody, 1:500 Enzo Life Sciences M16.10A1; rabbit anti-FKBP65 1:5000, Proteintech 12172-1-AP; rabbit anti-GAPDH 1:2000 Cell Signaling 2118S, Anti-Plod2 1:500 Abcam 72939, Anti-BiP 1:1000 Cell signaling C50B12) and viewed simultaneously. Peroxidase-conjugated secondary antibodies (Cell Signaling 7071 and 7072) were used and immunocomplexes identified using the ECL detection reagent (Cell Signaling 7003). FIJI was used to quantify bands following Gel Analysis recommendations from ImageJ (24) and the Mann-Whitney test was performed for statistical analysis using Prism software. Three different cell lines were used as primary fibroblast controls and experiments were replicated at least three times in order to perform statistical analysis.
Co-immunoprecipitation
Proteins from MC3T3 cells were extracted with RIPA buffer and endogenous proteins were immunoprecipitated with anti-HSP47 (Enzo Life Sciences M16.10A1) or anti-PLOD2 (Abcam 72939) antibodies. Antibodies were immobilized on agarose beads using a Pierce Co-immunoprecipitation Kit (26149). Immunoprecipitates were washed in PBS, collected and separated by SDS-PAGE. For co-immunoprecipitation of tagged proteins, DDK fusion vectors and an anti-DDK Magnetic Immunoprecipitation and detection kit (AR100024 Origene) were used. Control beads without anti-DDK or GFP were used from each kit according to the manufacturers' protocol. For tandem mass spectrometry, gels were stained with Coomassie Blue and parallel Western blots were probed with FKBP65, HSP47, PLOD2 and BiP antibodies to confirm the immunoprecipitation. The corresponding bands stained with Coomassie blue were used for trypsin digestion and analysis by Liquid Chromatography Mass Spectrometry (LC-MS) as described (25).
Protein identification by Mass Spectrometry
Gel slices were de-stained in 200mM ammonium bicarbonate in 40% acetonitril, SpeedVac dried and proteomic grade Trypsin (Sigma) was added to a final concentration of 20μg/mL. After overnight incubation at 37°C, peptides were isolated using ZipTip C18 tips (Merck-Millipore) according to the manufacturers' protocol. MS analysis was performed on a NanoAcquity UPLC (Waters) on-line coupled to an ESI Q-Tof Premier (Waters) Mass spectrometer. Peptides were separated on a BEH300 C18 analytical column (75μm i.d. × 150mm length, particle size 1.7μm, reverse-phase; Waters, UK) with a linearly increasing gradient of solvent B from initial 3% (v/v) to 40% B (v/v) in 30 minutes; solvent A was 0.1% (v/v) formic acid in water and solvent B was 0.1% (v/v) formic acid in acetonitril. Peptides eluted from the column during the gradient flowed directly into the ESI source. Raw data was acquired in data independent MŜe Identity (Waters) mode. Precursor ion spectra were acquired with collision energy 5V and fragment ion spectra with a collision energy 20-35V ramp in alternating 1 second scans. Additionally, data dependent analysis mode was used. Peptide spectra were acquired with collision energy 5V and peptides with charge states of +2, +3 and +4 were selected for MS/MS analysis. Fragment spectra were collected with a collision energy 20-40V ramp. Using both modes, peptide spectra and fragment spectra were acquired with 2ppm and 5ppm tolerance, respectively. Raw data processed and spectra subjected to a database search using species-specific mouse databases (downloaded from the NCBI and Uniprot sites) by the PLGS2.3 software (Waters).
Proximity Ligation Assay
In situ interactions were detected using the Duolink Proximity Ligation Assay kit (SIGMA, DUO92101) (26,27). Cultured fibroblasts were fixed and permeabilized as described for immunofluorescence. Antibody incubation and probe amplification were performed according to the manufacturer's instructions. Elimination of one of the primary antibodies, as well as null allele cells for FKBP65 were used as negative controls.
Immunofluorescence
Immunofluorescence experiments were performed using a Zeiss 780 laser scanning confocal microscope. Cultured fibroblasts were fixed in 4% PFA in PBS, then washed and permeabilized with 0.5% Triton X100 for 5 minutes, followed by blocking in 10% goat serum for 1 hour. The primary antibody was incubated overnight at 4°C (mouse anti-HSP47, 1:100 Enzo Life Sciences M16.10A1; rabbit anti-FKBP65, 1:100 Proteintech 12172-1-AP, Anti-Plod2 1:500 Abcam 72939, Anti-BiP 1:1000 Cell signaling C50B12). The fluorescent secondary antibody was incubated at a 1:1000 dilution for 1 hour at room temperature (Alexa-fluor goat anti-mouse 488 and goat anti-rabbit 568 for all the samples), and DAPI at a 1:1000 dilution was applied for 5 minutes at room temperature before mounting.
Lysyl hydroxylation Mass spectrometry analysis
Collagen was collected from culture media from control and OI patient fibroblasts treated with 100ug/ml ascorbic acid for 4 days. Type I procollagen chains were extracted by heat denaturation at 90°C in SDS–PAGE sample buffer, resolved on 6% SDS-PAGE gels (28), and digested with trypsin in gel (29). Subsequently, collagenase-generated peptides were separated by reverse-phase HPLC and hydroxylation of N- and C-telopeptides were analyzed individually (C8, Brownlee Aquapore RP-300, 4.6 mm × 25 cm) with a linear gradient of acetonitrile:n-propanol (3:1 v/v) in aqueous 0.1% (v/v) trifluoroacetic acid (30). Individual fractions were analyzed by LC–MS. Peptides were analyzed by electrospray LC-MS using an LTQ XL ion-trap mass spectrometer (Thermo Scientific) equipped with in-line liquid chromatography using a C4 5 mm capillary column (300 mm × 150 mm; Higgins Analytical RS-15M3-W045) eluted at 4.5 ml/min. The LC mobile phase consisted of buffer A (0.1% formic acid in MilliQ water) and buffer B (0.1% formic acid in 3:1 acetonitrile:n-propanol, v:v). An electrospray ionization source introduced the LC sample stream into the mass spectrometer with a spray voltage of 3 kV. Proteome Discoverer search software (Thermo Scientific) was used for peptide identification using the NCBI protein database.
Plasmon surface resonance
Binding studies were performed on a Biacore 3000 instrument (Biacore AB, Uppsala, Sweden). Proteins were immobilized on CM5 sensor chips by amine coupling. The solution phase analytes were dissolved in HBS-N buffer, which contained 0.15 M NaCl, 10 mM HEPES, pH 7.4, 3 mM EDTA, and 0.005% polysorbate 20, with addition of 5mM Ca++, 1mM Zn++. The solutions traversed the sensors at a flow rate of 50μl/minute, unless otherwise noted. Costar low-retention polypropylene tubes (catalog number 3207) were used throughout. Binding results were expressed in resonance units. Kinetic studies were analyzed with BIAevaluation Software Version 4.1. Purified proteins were purchased from Origene.
Results
Defects in HSP47 and FKBP65 destabilize LH2
Based on the altered telopeptide crosslinking observed in OI cases with defects in FKBP65 or HSP47 (15,18), we tested the hypothesis that recessively inherited mutations in the genes encoding these proteins would affect LH2 protein levels. We measured LH2 synthesized by cultured fibroblasts derived from an OI patient homozygous for the FKBP10 loss-of-function mutation p.Gly278ArgfsX295 (11) and from a patient homozygous for the p.Met237Thr SERPINH1 mutation (17). Both mutations are associated with severe forms of OI. Fibroblasts were treated with ascorbic acid to increase type I procollagen synthesis and produce a lysyl hydroxylation pattern more similar to that seen in osteoblasts (9). Under these conditions, FKBP65 deficient cells showed reduced levels of LH2 (Fig. 1A, B) while HSP47M237T/M237T mutant cells showed an increase in LH2 levels with induction of a protein band corresponding to the shorter isoform, LH2a (Fig. 1A, B)(31,32). These data show that FKBP65 -/- and HSP47M237T/M237T defects have opposing effects on LH2 proteins levels. The data are concordant with findings derived from OI bone, showing that loss of FKBP65 leads to diminished lysyl hydroxylation, that diminished HSP47 results in increased lysyl hydroxylation (15,18). Thus HSP47 and FKBP65 influence LH2 activity, at least in part, by regulation of LH2 levels.
Fig. 1.
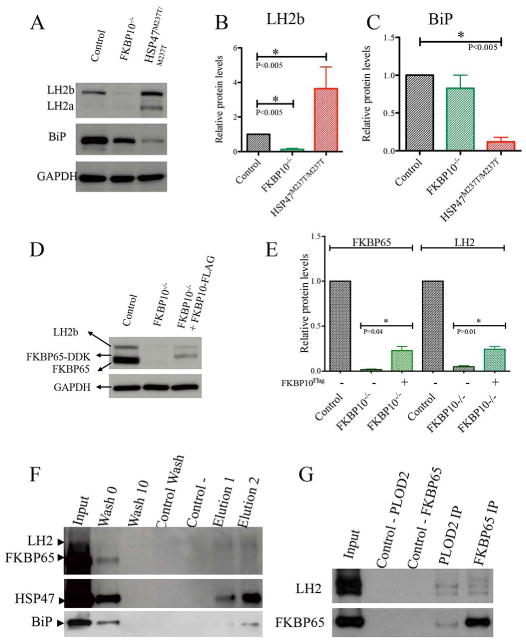
LH2, FKBP65, HSP47 and BiP interact. A-C. Protein levels of LH2 were altered in cells with mutations in FKBP10 and SERPINH1. BiP levels were decreased in HSP47 defective cells. D-E. Rescue experiment using FKBP10-/- cells transfected with FKBP10 tagged vector. LH2 levels were rescued by expression of wildtype FKBP65 protein. F. Co-immunoprecipitation of endogenous LH2, FKBP65, HSP47 and BiP in human osteoblasts (OB) with specific antibodies. LH2 immunoprecipitated HSP47 and BiP. G. Co-immunoprecipitation of tagged FKBP65 and LH2 in OB cells. LH2 and FKBP65 immunoprecipitated each other, including both LH2 isoforms, respectively.
To further test how the FKBP65 chaperone defects contribute to the changes in LH2 levels, rescue experiments were conducted by constitutive expression of a tagged version of FKBP65 in the FKBP10-/- cells. As shown in Figures 1D and E, the levels of LH2 increased about 20% when transfection of a FKBP10 construct restored about 20% of WT FKBP65 in FKBP10-/- cells, indicating that increasing FKBP65 partially corrected the reduced amount of LH2.
Type I procollagen chaperones HSP47 and FKBP65 form a complex with LH2
Previous work has shown that FKBP65 and HSP47 form a complex (Supplemental Fig. 1), and that FKBP65 is required for LH2 dimerization (16,17) and the data presented above show that both molecules regulate LH2 isoform protein levels. This information suggested that FKBP65, HSP47 and LH2 collaborate during the synthesis of type I procollagen, perhaps forming a complex. Three independent approaches were used to test this hypothesis. First, using antibodies against LH2, HSP47 and protein lysates derived from wild-type mouse osteoblasts precursor (MC3T3), we performed co-immunoprecipitation followed by tandem mass spectrometry (MS). Both antibodies were used in order to capture as many interactions as possible. Using anti-LH2 antibody, we identified an interaction between LH2 and HSP47 (Table 1). Using anti-HSP47 antibody, we detected an interaction between HSP47 and BiP (Table 1). Second, to further test these interactions, immunoprecipitation experiments followed by Western blots were performed using human osteoblast (hOB) lysates, taking advantage of the high level of type I procollagen synthesis by these cells. Using anti-PLOD2 antibody, HSP47 and BiP clearly co-immunoprecipitated (Fig. 1F lanes 6, 7), confirming the interactions between LH2 with HSP47 and BiP identified by MS. However, the results for FKBP65 were not as conclusive. Because loss of FKBP65 affected LH2 protein levels, and reintroduction of FKBP65 rescued LH2 protein levels, we furthered pursued the potential interaction using expression of tagged proteins. Co-immunoprecipitation of co-expressed FKBP65 and LH2 tagged proteins in hOBs identified an interaction between FKBP65 and LH2 (Fig. 1G), whether antibodies were directed against LH2 or FKBP65. The difficulty of detecting the FKBP65-LH2 interaction using LH2 antibody might reflect that the interaction is too weak to be captured in the extracts or that the exposed epitopes interacted poorly with the LH2 antibody. Third, we performed proximity ligation assays (PLA) that identified the interaction between FKBP65 and LH2 (Fig. 2) in cells. Surprisingly, we detected a significant increase in the FKBP65-LH2 PLA signal in HSP47M237T/M237T mutant cells (Fig. 2C, D), suggesting that reduced HSP47 function may enhance the interaction between FKBP65 and LH2.
Table 1. Proteins identified by Immunoprecipitation followed by Mass Spectrometry.
| Immunoprecipitated proteins | Identified proteins by Mass Spectrometry |
|---|---|
| LH2 | COL1A1 |
| HSP47 | |
| HSP47 | COL1A1 |
| BiP |
Fig. 2. In situ.
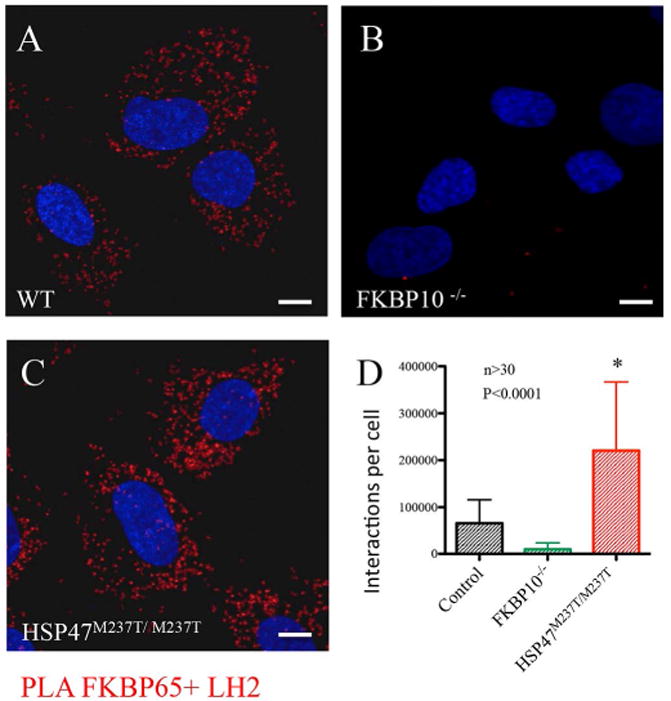
interaction of FKBP65 and LH2 by proximity ligation assay. A. FKBP65 and LH2 interacted in WT cells (red): B. FKBP10 mutant cells did not show interaction between LH2 and FKBP65. C-D. Interaction was increased in HSP47 defective cells.
BiP is a component of the HSP47-FKBP65-LH2 complex
BiP (GRP78) is an ER resident chaperone, which recognizes and assists in the folding of trimeric type I procollagen (33). BiP levels can be upregulated in forms of OI resulting from structurally abnormal type I procollagen that is retained in the ER (22). By tandem MS, interactions were identified between BiP and HSP47 (Table 1), and BiP could be co-immunoprecipitated with LH2 antibody (Fig. 1F). In HSP47M237T/M237T mutant cells, the level of BiP was markedly reduced (Fig. 1A-C), indicating that BiP was destabilized by loss of wild type HSP47. In FKBP10-/- cells, the level of BiP was also reduced, but not as dramatically as in HSP47 mutant cells. Taken together, these data show that BiP directly interacts with HSP47 and is part of the FKBP65/HSP47/LH2 complex.
Abnormal LH2 localization in cells with mutations in FKBP65 and HSP47
Cells with defects in either FKBP65 or HSP47 show abnormal trafficking of type I procollagen and the associated chaperones, preventing their export from the ER to the Golgi and segregating the complex into vesicles (17,34). To analyze the intracellular relationship between LH2 and this type I procollagen chaperone complex, we studied the cellular localization of LH2 in control and mutant fibroblasts. In control cells, LH2 expression was localized to the ER (Fig. 3A) and co-localized with the ER chaperone FKBP65 (Fig. 3D). In FKBP10-/- cells there was a consistently diminished LH2 signal in the ER (Fig. 3B, E), concordant with the results of Western blots shown in Fig. 1A. HSP47M237T/M237T cells showed near normal ER LH2 expression but there was also accumulation of FKBP65 and LH2 in the abnormal vesicles known to be present in these cells (Fig. 3C, F). Previous work (17) showed that these vesicles included type I procollagen and mutant HSP47, consistent with the idea that some of FKBP65/HSP47/LH2/type I procollagen complex is abnormally trafficked into vesicles when HSP47 is defective.
Fig. 3.
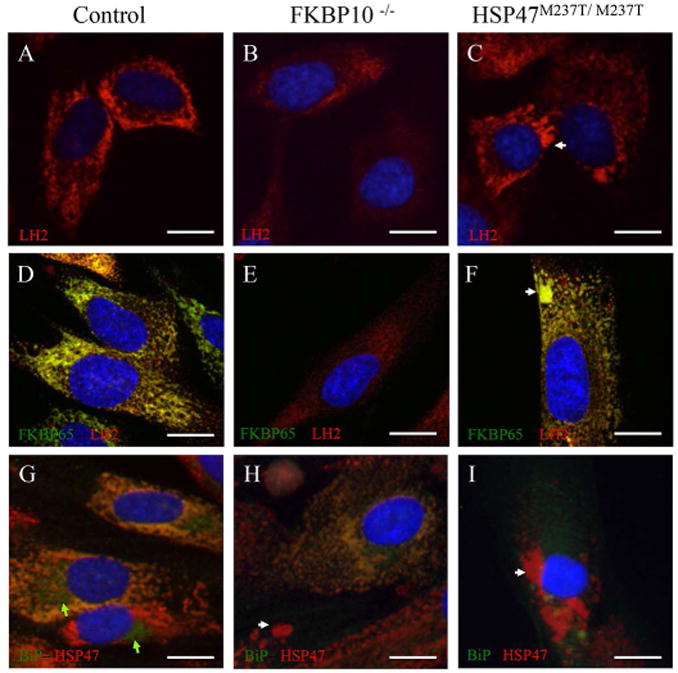
Cellular localization of LH2 and BiP. Intracellular immunolocalization of LH2 and BiP in human fibroblasts. A-F: LH2 is shown in red and FKBP65 in green. G-I: BiP is shown in green and HSP47 in red. control cells (A, D and G); FKBP10-/- cells (B, E and H); and HSP47M237T/M237T cells (C, F and I). LH2 colocalized with FKBP65 in the ER (D-F), including within abnormal vesicles in mutant cells (arrows). BiP colocalized partially with HSP47 in the ER but not in either the Golgi or abnormal vesicles (arrow). Nuclei were stained with DAPI (in blue). Green arrows identify the Golgi compartment.
Similar subcellular localization studies were performed to assess BiP localization. In control cells, BiP was found in both the ER, where it co-localized with HSP47 and in the Golgi (Fig. 3G, green arrows). This result suggests that the complex of type I procollagen, BiP, FKBP65, HSP47 and LH2 forms within the ER. However, unlike the results seen for HSP47, FKBP65 and LH2 in the mutant cells, BiP was not apparent in the abnormal intracellular vesicles, indicating that BiP is not involved in abnormal vesicle formation (Fig. 3H-I).
Defects in FKBP65, HSP47 and BiP alter type I collagen lysyl hydroxylation
Defects in FKBP65 and HSP47 that produce OI have been previously described to alter the hydroxylation pattern of type I collagen N- and C- telopeptides (15,18, 40), however, the mutations in each respective gene had differing effects regarding LH2 activity and telopeptide lysyl hydroxylation. To determine how loss of each complex member affects telopeptide hydroxylation we used mass spectrometry to quantify the level of hydroxylated lysines in telopeptides of secreted type I collagen from cultured fibroblasts with FKBP10 and SERPINH1 mutations. We also employed siRNA to inhibit expression of PLOD2 and HSPA5 (which encodes BiP) in wild-type fibroblasts and measured lysyl hydroxylation. We found altered hydroxylation of lysine 1208, corresponding to a hydroxylated residue in the carboxyl-terminal telopeptide (Figure 4). FKBP10-/- fibroblasts showed markedly decreased lysyl hydroxylation at lysine 1208 relative to control, similar to cells treated with PLOD2 siRNA, suggesting that this chaperone positively affects the bioactivity of LH2 at the site. Interestingly, SERPINH1 mutant cells and cells treated with HSPA5 siRNA showed increased levels of hydroxylation at lysine 1208, suggesting an opposing regulatory function that normally limits the activity of LH2. In ascorbic acid treated cultured fibroblasts, hydroxylation levels of the N-telopeptide lysine 170 did not show significant changes (Supplemental Fig. 2).
Fig. 4.
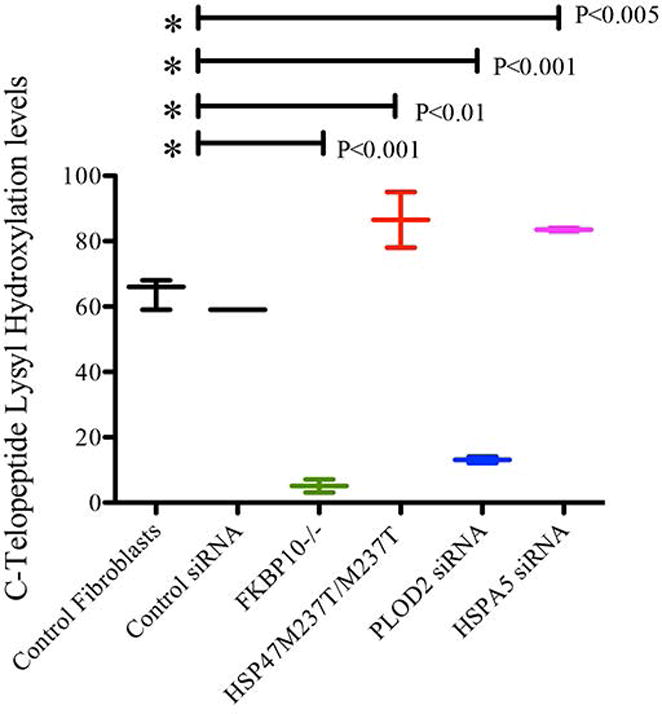
Lysyl Hydroxylation of type I collagen C-telopeptides. Percentage of hydroxylation of lysine 1208 in secreted type I collagen by fibroblasts treated with ascorbic acid. The control represents the average of three different fibroblast cell lines from unaffected persons. Control siRNA represents same control fibroblasts transfected with non-target siRNA. FKBP10-/- and HSP47M237T/M237T are patient fibroblast. PLOD2 and HSPA5 siRNA are knockdown fibroblast by use of validated iRNA sequences. FKBP10-/-, and PLOD2 knockdown cells showed a significant decrease in hydroxylation. HSP47M237T/M237T and HSPA5 knockdown cells showed a significant increase in hydroxylation.
HSP47 and FKBP65 compete for binding to LH2 and type I procollagen
To further investigate the apparently opposing actions of FKBP65 and HSP47 on site-specific LH2 activity, Surface Plasmon Resonance (SPR) was performed to assess direct interactions of the chaperones and/or LH2 with type I procollagen. LH2b was immobilized on the sensing chip and, both serially and in combination, type I procollagen, FKBP65 and HSP47 (Fig. 5A) were added to define the association and dissociation rates (kinetics) and binding strength (affinity) of the interactions. First, we found that the chaperones were not able to bind individually to LH2; the complex members only interacted in the presence of type I procollagen (data not shown). This finding revealed a dependence of complex association on trimeric procollagen and suggested that in the ER the complex forms on folding or folded type I procollagen molecules. Second, the strength of the LH2-type I procollagen association and its stability increased when FKBP65 or HSP47 were added to the mix individually (Fig. 5A), confirming the binding of these chaperones with LH2. However, when FKBP65 and HSP47 were added simultaneously to the mix, the association rate of the components decreased, suggesting that FKBP65 and HSP47 compete for binding, consistent with antagonistic effects that ultimately could affect LH2 activity on its type I procollagen substrate (Fig. 5A). Similar results were obtained when HSP47 was used as the immobilized component in the analysis (Supplemental Fig. 3,4).
Fig. 5.
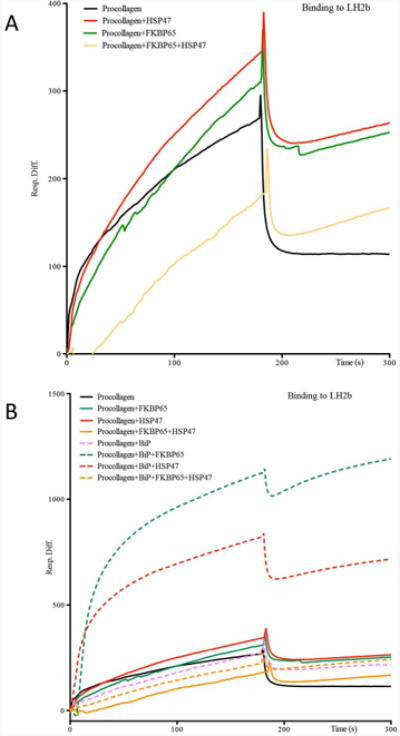
Interaction analysis by surface plasmon resonance. A. Type I procollagen, FKBP65, HSP47 proteins were exposed to immobilized LH2. Both FKBP65 and HSP47 bound to the LH2-procollagen mix but the association was altered when both chaperones were presented simultaneously. B. Same experiment for binding to LH2 and type I procollagen, but including BiP protein in addition. The association curve was observed during addition of proteins during the initial 180 seconds, with a positive slope followed by a peak of affinity around this time and the dissociation curve afterward. Presence of BiP increased the affinity of HSP47 and FKBP65 for Procollagen and LH2 without altering their competition.
BiP increases affinity of the lysyl hydroxylation complex
After determining that FKBP65/HSP47 chaperone complex bound to LH2 in the presence of type I procollagen, the role of BiP, the third chaperone component of the complex, was similarly interrogated. As previously described, the interactions of each chaperone with LH2 and type I procollagen were analyzed by SPR. In a series of experiments, BiP was added alone and then in combination with FKBP65 and/or HSP47. The results showed that BiP alone did not enhance the binding of type I procollagen to LH2 (Fig. 5B, pink). However, the dissociation of the complex was slightly slower in the presence of BiP, suggesting that BiP may enhance complex affinity (Fig. 5B). When FKBP65 or HSP47 were added individually (Fig. 5B green and red) in the presence of BiP, there was a marked increase in the association with LH2. These data further suggest that BiP has an important role in enhancing the formation and stability of the complex. However, association values returned to a baseline levels when FKBP65 and HSP47 were added simultaneously to the mix of LH2, type I procollagen and BiP (Fig. 5B, yellow), most likely reflecting competition between FKBP65 and HSP47 for binding. Similar results were obtained when HSP47 was immobilized instead of LH2 (Supplemental Fig. 3,4), supporting the hypothesis that this chaperone complex regulates the kinetics and affinity of binding to LH2 to modulate its function.
Discussion
FKBP65, HSP47 and BiP are well-recognized ER-resident type I procollagen chaperones, but the precise roles these proteins play in the synthesis and secretion of type I procollagen is incompletely understood. The discovery that defects in FKBP65 and HSP47 result in recessive forms of OI, a disorder primarily due to altered synthesis and secretion of type I procollagen, revealed essential roles for these proteins in the maturation of type I procollagen and ultimately in bone mineralization. While one of the roles ascribed to these proteins is to assist in procollagen folding, there may be additional functions that they perform. The data presented here demonstrate that these chaperones directly interact with LH2, demonstrating that LH2 is part of the recently identified FKBP65-HSP47 complex. In this context, the results show that FKBP65 and HSP47 participate in the regulation of the posttranslational modification activity of LH2. The OI mutations due to defects in these chaperones alter LH2 levels, which is reflected in an altered telopeptide lysine hydroxylation pattern of type I collagen. The altered hydroxylation would be predicted to lead to abnormal type I collagen crosslinking in the extracellular matrix, which in bone leads to abnormal mineralization and increased bone brittleness, ultimately contributing to increased fracture risk and the OI phenotype (35).
The opposite effects on LH2 activity and hydroxylation patterns between FKBP65 and HSP47 deficient cells are consistent with the idea that FKBP65 and HSP47 act as positive and negative regulators of LH2 activity, respectively. The increased posttranslational hydroxylation of telopeptide lysines observed in HSP47 deficient cells and the positive regulatory role of FKBP65 in this process is supported by the increased interaction between FKBP65 and LH2 observed by PLA in HSP47 mutant cells. Additionally, the reduced association of LH2 with type I procollagen when HSP47 and FKBP65 are simultaneously added in the SPR studies suggests that these chaperones competitively regulate lysyl hydroxylation or LH2 activity. Based on these data, we hypothesize that HSP47 attaches at or close to type I procollagen (telopeptide) hydroxylation sites, displacing LH2 from the target lysine to inhibit or limit hydroxylation (Fig. 6A). Alternatively, HSP47 could also act by blocking LH2 dimerization by displacing the molecule that favors LH2 assembly, FKBP65. In tissues exhibiting telopeptide lysine hydroxylation, particularly skeletal tissues such as bone and tendon (35), FKBP65 interacts with LH2 to activate it (Fig. 6B) (16). When FKBP65 is lost, such as in the OI cases due to FKBP10 mutations, hydroxylation cannot be promoted and HSP47 through either binding or effects LH2 dimerization inhibits proper LH2 access to the hydroxylation site (Fig. 6C). In contrast, when HSP47 function is abnormal due to mutation, the balance favors FKBP65, promoting increased access to telopeptide lysines by LH2 dimers and leading to increased hydroxylation of type I procollagen (Fig. 6D). Since HSP47 has a role in formation and/or stabilization of triple helical procollagen, reduced HSP47 activity may permit extended exposure of the procollagen to LH2 activity. Which factors regulate the balance between FKBP65 and HSP47 activity are not understood, but distinct regulatory modules could represent a component of the tissue-specific differences in hydroxylation observed among type I procollagen expressing tissues.
Fig. 6.
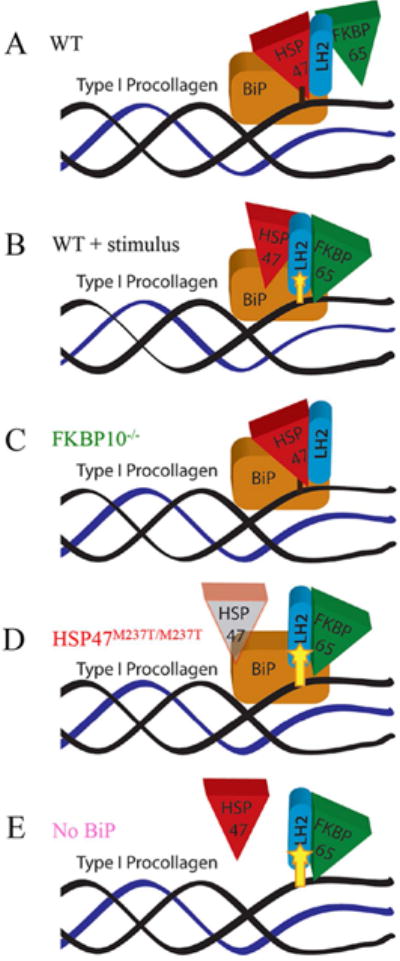
The chaperone complex modulates LH2 activity. A. Without stimulus favoring collagen crosslinking, HSP47 binds to the hydroxylation site, supported by BiP, and impedes LH2 function, resulting in lack of hydroxylation. B. In the presence of a hydroxylation stimulus, FKBP65 supports the binding and activity of LH2 to promote lysyl hydroxylation. C. In cells with loss of FKBP65, LH2 activity cannot be induced, resulting in decreased hydroxylation. D. In cells with altered HSP47, the defective HSP47 cannot compete with FKBP65 and LH2 has unlimited access to the hydroxylation site. E. In the absence of BiP, complex stability is affected, limiting HSP47 access to the hydroxylation site and allowing over hydroxylation by LH2.
In the context of OI, defects in either FKBP65 or HSP47 both produce severe forms of OI with bone deformity and fractures, even though the respective mutations lead to decreased or increased hydroxylation and altered cross-linking. This suggests that the effect of either abnormality may affect bone matrix in similar ways to result in brittle bones. Based on the clinical phenotype, the reduced hydroxylation observed in Bruck syndrome, whether indirectly due to loss of FKBP65 or directly due to PLOD2 mutations, results in tendon/ligament abnormalities and joint contractures (36). Conversely, increased hydroxylation in these tissues resulting from HSP47 deficiency has been reported to lead to articular hypermobility (17). Thus, there may be different functional consequences of abnormal hydroxylation and crosslinking on type I procollagen expressing tissues other than bone that may help explain the phenotypic differences among different forms of OI associated with chaperone complex members. A previous study suggested that HSP47 might act as a negative regulator of other posttranslational modifications, such as 4-prolyl-hydroxylation (37), which supports the idea that HSP47 may be a more general negative regulator of several posttranslational modifications during the formation of the triple helix, suggesting that phenotypic consequences of HSP47 mutations may result from perturbation of multiple cellular events.
A recent study demonstrated that FKBP65 favors the dimerization of LH2 (15). Whether FKBP65 defective cells alter LH2 function through disruption of dimerization is still unclear. FKBP65 may be inducing LH2 dimerization during the formation of the complex and HSP47 could be acting against LH2 dimerization through competing with FKBP65 for access to LH2. Our previous results showed that HSP47 defective cells have slightly reduced levels of FKBP65, however, LH2 activity is still increased in this mutant, suggesting that this minor reduction of FKBP65 levels is not significant for LH2 function or dimerization. The precise levels of FKBP65 necessary for appropriate LH2 activity is not known, however there is no evidence that patients who carry one mutant FKBP65 allele have a phenotype. This suggests that there is a FKBP65 critical threshold below which LH2 activity and crosslinking is affected.
By co-immunoprecipitation and mass spectrometry, BiP was shown to be part of the type I procollagen chaperone/posttranslational hydroxylation complex. Interestingly, Nakai et al., (38), using varying crosslinking reagents, also suggested that HSP47 and BiP may form a complex in the ER. This result was underscored by the observation that defects in HSP47 that produce OI led to reduction of BiP levels, consistent with a direct interaction between these proteins. Enhanced binding of both FKBP65 and HSP47 to LH2 and type I procollagen by BiP suggests that it promotes formation and/or stabilization of the chaperone-lysyl hydroxylation complex. While BiP does play a role in complex stabilization, it does not alter the competition for binding between FKBP65 and HSP47. Furthermore, because BiP knockdown also led to increased hydroxylation at the carboxyl-terminal telopeptide hydroxylation site, BiP appears to contribute to control of posttranslational hydroxylation of type I collagen telopeptide lysines in a similar fashion to HSP47. This suggests that BiP not only stabilizes the complex during procollagen trimerization but also plays a specific role in its interaction with HSP47. BiP is well described as an ER stress mediator, and increased BiP expression is an essential response to the large amount of collagen synthesis by skeletal cells, including osteoblasts, osteocytes or tenocytes, all of which synthesize extensive extracellular matrix. (19,20) Thus, changes in BiP levels could modulate the affinity of the chaperone complex to enhance or reduce the LH2 activity (Fig. 6E).
We found that LH2 activity is positively regulated by FKBP65 and negatively regulated by HSP47, consistent with the changes observed biochemically by crosslinking, in OI patients with mutations in the genes encoding these proteins. The studies suggest that a proper balance between positive and negative regulators allows for appropriate occupancy of LH2 at its substrates and is essential for achieving the proper pattern of hydroxylation. In addition, we found that BiP regulates the formation of the overall complex, increasing the affinity among the components, allowing HSP47 repression and connecting the hydroxylation machinery to ER stress. Identification of this ER chaperone complex and its role in regulating telopeptide type I collagen lysyl hydroxylation, and ultimately tissue specific crosslinking, suggests mechanisms by which regulation of tissue specific extracellular matrix formation may be determined.
In summary, the findings uncover some of the complex mechanisms that may modulate LH2 function to generate specific hydroxylation patterns in type I collagen telopeptides. One mechanism is based on a newly identified chaperone complex that includes FKBP65, HSP47 and BiP that regulates the occupancy and/or activity of LH2 on telopeptide lysines. The findings also support the conclusion that HSP47 interacts preferentially with multiple sites in the triple helical domain of procollagen molecules (39), which suggests that the chaperone complex may also act on procollagen during trimerization to modulate LH2 function. It remains possible that this complex contains other ER resident chaperones and molecules, and each component may have other partners that contribute to regulation of distinct ER activities beyond lysyl hydroxylation. Genetic studies in humans and animal models have aided in uncovering the components of this chaperone complex. Interesting questions remain on how both under- and over-hydroxylation of telopeptide lysines lead to phenotypically similar forms of OI, and precisely how these abnormalities lead to altered collagen crosslinking and effects on the quality of bone. The data also suggest the potential for improving the appropriate balance of LH2 activity through modulation of the complex, which could result in new effective treatments for pathological states that involve altered collagen crosslinking.
Supplementary Material
Supplemental Fig. 1. Co-immunoprecipitation between FKBP65 and HSP47. Specific antibodies against FKBP65 immunoprecipitated HSP47 in mouse embryonic fibroblasts (MEFs). This interaction was not present in Fkpb10 knock-out MEFs.
Supplemental Fig. 2. Lysyl Hydroxylation of type I collagen N-telopeptides. Percentage of hydroxylation of lysine 170 in secreted type I collagen by fibroblasts treated with ascorbic acid. The control represents the average of three different cell lines. Control siRNA represents wild type fibroblasts transfected with non-target siRNA. Under these conditions, no significant changes in the hydroxylation of this residue were observed.
Supplemental Fig. 3. Affinity and kinetics of the chaperone complex by SPR. A. Association and dissociation of type I procollagen to immobilized LH2 (long form) or HSP47 proteins. Association (Ka) and Dissociation (Kd) constants are shown for each interaction. B. Interaction curves for components of the complex to immobilized HSP47. C. Injection of procollagen at two timepoints and then FKBP65 or LH2 and binding to immobilized HSP47, showing independent association peaks for each component (second injection time marked with red arrow). D. Prolonged curve of association to HSP47 of procollagen alone and procollagen with FKBP65, showing increased stability of the complex with both procollagen and FKBP65 present.
Supplemental Fig. 4. Affinity and kinetics of the chaperone complex to HSP47 by SPR. Interaction study of all of the chaperone complex components and their combinations to immobilized HSP47 as a separate experiment.
Supplemental Fig. 5. Proximity ligation assay negative controls. A. PLA assay without rabbit anti-FKBP10 antibody. B. PLA assay without mouse anti-PLOD2 antibody.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health (RO1 AR062651 and P01 HD070394), a Geisman award from the Osteogenesis Imperfecta Foundation and funds from the Orthopaedic Hospital Research Center at UCLA, the Orthopaedic Institute for Children, and the March of Dimes Foundation, NIH P01 HD070394, F31DE022483, the BCM Intellectual and Developmental Disabilities Research Center (HD024064), the BCM Advanced Technology Cores NIH (AI036211, CA125123, and RR024574), the Rolanette and Berdon Lawrence Bone Disease Program of Texas.
Authors roles: ID designed and performed the experiments and wrote the manuscript. JM optimized and performed experiments; MW, and DE performed hydroxylation analysis. PK(6) and PK(7) performed the tandem MS identification. BL(4) generated essential information for hypothesis generation. CL and BL(8) performed FKBP65 Co-immunoprecipitation experiments. YA identified cases, determined their phenotypic features and referred them to the International Skeletal Dysplasia Registry at UCLA; DHC participated in experimental design and writing and DK participated in experimental design, writing and supervised the study.
Footnotes
Disclosures: The authors state that they have no conflicts of interest.
References
- 1.Yamauchi M, Sricholpech M. Lysine post-translational modifications of collagen. Essays Biochem. 2012;52:113–133. doi: 10.1042/bse0520113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walker LC, Overstreet MA, Yeowell HN. Tissue-specific expression and regulation of the alternatively-spliced forms of lysyl hydroxylase 2 (LH2) in human kidney cells and skin fibroblasts. Matrix Biol. 2005 Jan;23(8):515–523. doi: 10.1016/j.matbio.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 3.Hyry M, Lantto J, Myllyharju J. Missense mutations that cause Bruck syndrome affect enzymatic activity, folding, and oligomerization of lysyl hydroxylase 2. Journal of Biological Chemistrty. 2009 Nov 6;284(45):30917–30924. doi: 10.1074/jbc.M109.021238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van der Slot AJ, Zuurmond AM, van den Bogaerdt AJ, Ulrich MMW, Middelkoop E, Boers W, et al. Increased formation of pyridinoline cross-links due to higher telopeptide lysyl hydroxylase levels is a general fibrotic phenomenon. Matrix Biol. 2004 Jul;23(4):251–257. doi: 10.1016/j.matbio.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 5.Remst DFG, Blaney Davidson EN, Vitters EL, Blom AB, Stoop R, Snabel JM, et al. Osteoarthritis-related fibrosis is associated with both elevated pyridinoline cross-link formation and lysyl hydroxylase 2b expression. Osteoarthr Cartil. 2013 Jan;21(1):157–164. doi: 10.1016/j.joca.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 6.Pankova D, Chen Y, Terajima M, Schliekelman MJ, Baird BN, Fahrenholtz M, et al. Cancer-associated Fibroblasts Induce a Collagen Cross-link Switch in Tumor Stroma. Mol Cancer Res. 2015 Dec 2; doi: 10.1158/1541-7786.MCR-15-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marini JC, Reich A, Smith SM. Osteogenesis imperfecta due to mutations in non-collagenous genes: lessons in the biology of bone formation. Curr Opin Pediatr. 2014 Aug;26(4):500–507. doi: 10.1097/MOP.0000000000000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ha-Vinh R, Alanay Y, Bank RA, Campos-Xavier AB, Zankl A, Superti-Furga A, et al. Phenotypic and molecular characterization of Bruck syndrome (osteogenesis imperfecta with contractures of the large joints) caused by a recessive mutation in LOD2. Am J Med Genet. 2004 Dec 1;131(2):115–120. doi: 10.1002/ajmg.a.30231. [DOI] [PubMed] [Google Scholar]
- 9.van der Slot AJ, Zuurmond AM, Bardoel AFJ, Wijmenga C, Pruijs HEH, Sillence DO, et al. Identification of LOD2 as telopeptide lysyl hydroxylase, an important enzyme in fibrosis. J Biol Chem. 2003 Oct 17;278(42):40967–40972. doi: 10.1074/jbc.M307380200. [DOI] [PubMed] [Google Scholar]
- 10.Barnes AM, Duncan G, Weis M, Paton W, Cabral WA, Mertz EL, et al. Kuskokwim Syndrome, a Recessive Congenital Contracture Disorder, Extends the Phenotype of FKBP10 Mutations. Human Mutation. 2013;34(9):1279–1288. doi: 10.1002/humu.22362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alanay Y, Avaygan H, Camacho N, Utine GE, Boduroglu K, Aktas D, et al. Mutations in the Gene Encoding the RER Protein FKBP65 Cause Autosomal-Recessive Osteogenesis Imperfecta. The American Journal of Human Genetics. 2010 Apr 9;86(4):551–559. doi: 10.1016/j.ajhg.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kelley BP, Malfait F, Bonafé L, Baldridge D, Homan E, Symoens S, et al. Mutations in FKBP10 cause recessive osteogenesis imperfecta and bruck syndrome. J Bone Miner Res. 2011 Feb 18;26(3):666–672. doi: 10.1002/jbmr.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shaheen R, Al-Owain M, Faqeih E, Al-Hashmi N, Awaji A, Al-Zayed Z, et al. Mutations in FKBP10 cause both Bruck syndrome and isolated osteogenesis imperfecta in humans. Am J Med Genet. 2011 May 12;155(6):1448–1452. doi: 10.1002/ajmg.a.34025. [DOI] [PubMed] [Google Scholar]
- 14.Setijowati ED, Van Dijk FS, Cobben JM, van Rijn RR, Sistermans EA, Faradz SMH, et al. A novel homozygous 5 bp deletion in FKBP10 causes clinically Bruck syndrome in an Indonesian patient. Eur J Med Genet. 2012 Jan;55(1):17–21. doi: 10.1016/j.ejmg.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 15.Schwarze U, Cundy T, Pyott SM, Christiansen HE, Hegde MR, Bank RA, et al. Mutations in FKBP10, which result in Bruck syndrome and recessive forms of osteogenesis imperfecta, inhibit the hydroxylation of telopeptide lysines in bone collagen. Human Molecular Genetics. 2012 Dec 14;22(1):1–17. doi: 10.1093/hmg/dds371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gjaltema RAF, van der Stoel MM, Boersema M, Bank RA. Disentangling mechanisms involved in collagen pyridinoline cross-linking: The immunophilin FKBP65 is critical for dimerization of lysyl hydroxylase 2. Proceedings of the National Academy of Sciences. 2016 Jun 28;113(26):7142–7147. doi: 10.1073/pnas.1600074113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duran I, Nevarez L, Sarukhanov A, Wu S, Lee K, Krejci P, et al. HSP47 and FKBP65 cooperate in the synthesis of type I procollagen. Human Molecular Genetics. 2014 Dec 15; doi: 10.1093/hmg/ddu608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lindert U, Weis MA, Rai J, Seeliger F, Hausser I, Leeb T, et al. Molecular Consequences of the SERPINH1/HSP47 Mutation in the Dachshund Natural Model of Osteogenesis Imperfecta. Journal of Biological Chemistry. 2015 Jul 17;290(29):17679–17689. doi: 10.1074/jbc.M115.661025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Behnke J, Feige MJ, Hendershot LM. BiP and Its Nucleotide Exchange Factors Grp170 and Sil1: Mechanisms of Action and Biological Functions. Journal of Molecular Biology. 2015 Apr 10;427(7):1589–1608. doi: 10.1016/j.jmb.2015.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gardner BM, Pincus D, Gotthardt K, Gallagher CM, Walter P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harbor Perspectives in Biology. 2013 Mar;5(3):a013169. doi: 10.1101/cshperspect.a013169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carrara M, Prischi F, Nowak PR, Kopp MC, Ali MM. Noncanonical binding of BiP ATPase domain to Ire1 and Perk is dissociated by unfolded protein CH1 to initiate ER stress signaling. Elife. 2015;4 doi: 10.7554/eLife.03522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chessler SD, Byers PH. BiP binds type I procollagen pro alpha chains with mutations in the carboxyl-terminal propeptide synthesized by cells from patients with osteogenesis imperfecta. J Biol Chem. 1993 Aug 25;268(24):18226–18233. [PubMed] [Google Scholar]
- 23.Lamandé SR, Chessler SD, Golub SB, Byers PH, Chan D, Cole WG, et al. Endoplasmic reticulum-mediated quality control of type I collagen production by cells from osteogenesis imperfecta patients with mutations in the pro alpha 1 (I) chain carboxyl-terminal propeptide which impair subunit assembly. J Biol Chem. 1995 Apr 14;270(15):8642–8649. doi: 10.1074/jbc.270.15.8642. [DOI] [PubMed] [Google Scholar]
- 24.Gassmann M, Grenacher B, Rohde B, Vogel J. Quantifying Western blots: pitfalls of densitometry. Electrophoresis. 2009 Jun;30(11):1845–1855. doi: 10.1002/elps.200800720. [DOI] [PubMed] [Google Scholar]
- 25.Morello R, Bertin TK, Chen Y, Hicks J, Tonachini L, Monticone M, et al. CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. 2006 Oct 20;127(2):291–304. doi: 10.1016/j.cell.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 26.Söderberg O, Gullberg M, Jarvius M, Ridderstråle K, Leuchowius KJ, Jarvius J, et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006 Dec;3(12):995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- 27.Weibrecht I, Leuchowius KJ, Clausson CM, Conze T, Jarvius M, Howell WM, et al. Proximity ligation assays: a recent addition to the proteomics toolbox. Expert Rev Proteomics. 2010 Jun;7(3):401–409. doi: 10.1586/epr.10.10. [DOI] [PubMed] [Google Scholar]
- 28.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970 Aug 15;227(5259):680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 29.Hanna SL, Sherman NE, Kinter MT, Goldberg JB. Comparison of proteins expressed by Pseudomonas aeruginosa strains representing initial and chronic isolates from a cystic fibrosis patient: an analysis by 2-D gel electrophoresis and capillary column liquid chromatography-tandem mass spectrometry. Microbiology (Reading, Engl) 2000 Oct;146(Pt 10):2495–2508. doi: 10.1099/00221287-146-10-2495. [DOI] [PubMed] [Google Scholar]
- 30.Wu JJ, Woods PE, Eyre DR. Identification of cross-linking sites in bovine cartilage type IX collagen reveals an antiparallel type II-type IX molecular relationship and type IX to type IX bonding. J Biol Chem. 1992 Nov 15;267(32):23007–23014. [PubMed] [Google Scholar]
- 31.van der Slot AJ, van Dura EA, de Wit EC, De Groot J, Huizinga TWJ, Bank RA, et al. Elevated formation of pyridinoline cross-links by profibrotic cytokines is associated with enhanced lysyl hydroxylase 2b levels. Biochim Biophys Acta. 2005 Jun 30;1741(1):95–102. doi: 10.1016/j.bbadis.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 32.Pornprasertsuk S, Duarte WR, Mochida Y, Yamauchi M. Overexpression of lysyl hydroxylase-2b leads to defective collagen fibrillogenesis and matrix mineralization. J Bone Miner Res. 2005 Jan;20(1):81–87. doi: 10.1359/JBMR.041026. [DOI] [PubMed] [Google Scholar]
- 33.Ferreira LR, Norris K, Smith T, Hebert C, Sauk JJ. Association of Hsp47, Grp78, and Grp94 with procollagen supports the successive or coupled action of molecular chaperones. J Cell Biochem. 1994 Dec;56(4):518–526. doi: 10.1002/jcb.240560412. [DOI] [PubMed] [Google Scholar]
- 34.Ito S, Nagata K. Mutants of collagen-specific molecular chaperone Hsp47 causing osteogenesis imperfecta are structurally unstable with weak binding affinity to collagen. Biochemical and Biophysical Research Communications. 2016 Jan 15;469(3):437–442. doi: 10.1016/j.bbrc.2015.12.028. [DOI] [PubMed] [Google Scholar]
- 35.Eyre DR, Weis MA. Bone collagen: new clues to its mineralization mechanism from recessive osteogenesis imperfecta. Calcif Tissue Int. 2013 Oct;93(4):338–347. doi: 10.1007/s00223-013-9723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barnes AM, Cabral WA, Weis M, Makareeva E, Mertz EL, Leikin S, et al. Absence of FKBP10in recessive type XI osteogenesis imperfecta leads to diminished collagen cross-linking and reduced collagen deposition in extracellular matrix. Human Mutation. 2012 Jul 16;33(11):1589–1598. doi: 10.1002/humu.22139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Asada S, Koide T, Yasui H, Nagata K. Effect of HSP47 on prolyl 4-hydroxylation of collagen model peptides. Cell Struct Funct. 1999 Aug;24(4):187–196. doi: 10.1247/csf.24.187. [DOI] [PubMed] [Google Scholar]
- 38.Nakai A, Satoh M, Hirayoshi K, Nagata K. Involvement of the stress protein HSP47 in procollagen processing in the endoplasmic reticulum. The Journal of Cell Biology. 1992 May;117(4):903–914. doi: 10.1083/jcb.117.4.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tasab M, Batten MR, Bulleid NJ. Hsp47: a molecular chaperone that interacts with and stabilizes correctly-folded procollagen. EMBO J. 2000 May 15;19(10):2204–2211. doi: 10.1093/emboj/19.10.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lietman C, Rajagopal A, Homan EP, Munivez E, Jiang MM, Bertin TK, et al. Connective tissue alterations in Fkbp10-/- mice. Human Molecular Genetics. 2014 Sep;15(23):4822–31. doi: 10.1093/hmg/ddu197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. 1. Co-immunoprecipitation between FKBP65 and HSP47. Specific antibodies against FKBP65 immunoprecipitated HSP47 in mouse embryonic fibroblasts (MEFs). This interaction was not present in Fkpb10 knock-out MEFs.
Supplemental Fig. 2. Lysyl Hydroxylation of type I collagen N-telopeptides. Percentage of hydroxylation of lysine 170 in secreted type I collagen by fibroblasts treated with ascorbic acid. The control represents the average of three different cell lines. Control siRNA represents wild type fibroblasts transfected with non-target siRNA. Under these conditions, no significant changes in the hydroxylation of this residue were observed.
Supplemental Fig. 3. Affinity and kinetics of the chaperone complex by SPR. A. Association and dissociation of type I procollagen to immobilized LH2 (long form) or HSP47 proteins. Association (Ka) and Dissociation (Kd) constants are shown for each interaction. B. Interaction curves for components of the complex to immobilized HSP47. C. Injection of procollagen at two timepoints and then FKBP65 or LH2 and binding to immobilized HSP47, showing independent association peaks for each component (second injection time marked with red arrow). D. Prolonged curve of association to HSP47 of procollagen alone and procollagen with FKBP65, showing increased stability of the complex with both procollagen and FKBP65 present.
Supplemental Fig. 4. Affinity and kinetics of the chaperone complex to HSP47 by SPR. Interaction study of all of the chaperone complex components and their combinations to immobilized HSP47 as a separate experiment.
Supplemental Fig. 5. Proximity ligation assay negative controls. A. PLA assay without rabbit anti-FKBP10 antibody. B. PLA assay without mouse anti-PLOD2 antibody.