

Abstract
Rheumatoid arthritis (RA) is a debilitating inflammatory autoimmune disease with no known cure. Recently, we identified the immunomodulatory enzyme indoleamine-2,3-dioxygenase 2 (IDO2) as an essential mediator of autoreactive B and T cell responses driving RA. However, therapeutically targeting IDO2 has been challenging given the lack of small molecules that specifically inhibit IDO2 without also affecting the closely related IDO1. In this study, we develop a novel monoclonal antibody (mAb)-based approach to therapeutically target IDO2. Treatment with IDO2-specific mAb alleviated arthritis in two independent preclinical arthritis models, reducing autoreactive T and B cell activation and recapitulating the strong anti-arthritic effect of genetic IDO2 deficiency. Mechanistic investigations identified FcγRIIb as necessary for mAb internalization, allowing targeting of an intracellular antigen traditionally considered inaccessible to mAb therapy. Taken together, our results offer preclinical proof of concept for antibody-mediated targeting of IDO2 as a new therapeutic strategy to treat RA and other autoantibody-mediated diseases.
Keywords: experimental arthritis, antibody therapy, IDO, murine model, B cells
Graphical Abstract
1.0 Introduction
Rheumatoid arthritis (RA) is a debilitating inflammatory joint disease that affects approximately 1.3 million Americans and has no known cure [1, 2]. Even with the development of new biologic therapies, current treatment strategies for RA still primarily focus on controlling inflammatory symptoms. Standard treatment regimens include non-steroidal anti-inflammatory drugs (NSAIDs), corticosteroids, disease-modifying anti-rheumatic drugs (DMARDs) and biologic response-modifying drugs [3–5], but none are fully effective. Moreover, alleviation of disease symptoms is often associated with severe side effects, including liver toxicity, bone marrow suppression and increased susceptibility to infections and cancer [6–11]. Thus, improved strategies to specifically target the pathogenic mechanisms underlying autoimmunity are still urgently needed.
Recently, we have explored the indoleamine 2,3-dioxygenases IDO1 and IDO2 as candidate therapeutic targets for the development of new treatments for inflammatory autoimmunity [12–18]. IDO mediates the first and rate-limiting step in the catabolism of tryptophan to kynurenine [19]. Immune modulatory effects of the IDO pathway first described in maternal tolerance to fetal tissue [20] were later extended to many disease settings [21–23], particularly in cancer where IDO1 was implicated as a pivotal mediator of immune escape by tumors [24, 25]. Although primarily considered immunosuppressive in cancer, the role the IDO pathway plays in autoimmunity is less clear. Human autoimmune patients exhibit elevated tryptophan catabolism in their blood and urine that correlates with disease pathogenesis, indicating an immune activating role for the IDO pathway [26–28]. In murine models of autoimmunity, some studies have suggested that the IDO pathway is immune suppressive [29–32], whereas others have demonstrated that it is immune activating and supportive to pathogenesis [17, 33–35], a result confounded by the use of the nonspecific IDO pathway inhibitor 1-methyl-tryptophan (1MT) to assess IDO function [15, 36]. In addressing this conundrum, we recently presented genetic evidence showing how IDO2 differs from IDO1 in contributing a pathogenic immune activating function specific for the establishment and development of autoimmunity [15, 16]. Mechanistic investigations revealed that IDO2, but not IDO1, was needed to activate CD4+ T cells, generate pathogenic autoantibodies, and drive arthritis development [15]. Notably, IDO2 expression in cognate, antigen-specific B cells was both necessary and sufficient to mediate disease, defining a key pathogenic role for IDO2 specifically in B cells through their interaction with T cells [16]. In contrast, the role IDO1 plays in the autoimmune response is unclear, with some studies suggesting a regulatory function [29–31, 37], while others suggest a pro-inflammatory role [33–35] or no role at all [15, 38]. These results suggest an explanation for the seemingly opposing roles of the IDO pathway in autoimmune pathogenesis by illuminating a unique function for IDO2 in driving inflammatory autoimmunity. A therapeutic strategy that specifically targets IDO2, rather than the broader IDO pathway, could be of great benefit to clinical disease management
In this study, we present a preclinical proof-of-concept for use of a monoclonal antibody approach to specifically target IDO2 and treat autoimmunity. Monoclonal antibodies (mAb) are important modalities for treating cancer and autoimmune disease [39, 40], acting to eliminate target-expressing cells or their secreted products [41–46] or, more recently, to disrupt regulatory factors that direct immune responses [47, 48]. While mAb therapies developed to date have targeted cell surface or secreted antigens, recent preclinical studies demonstrate that certain mAb can also target intracellular antigens traditionally considered inaccessible [49–52]. Here, we show that administering a cell-penetrating mAb to selectively target intracellular IDO2 inhibits autoreactive T and B cell responses and alleviates joint inflammation in the KRN preclinical model of autoimmune arthritis, fully recapitulating the effect of genetic IDO2 deficiency in this model. We validate the effect of IDO2 Ig in alleviating joint inflammation in a second, mechanistically independent arthritis model, collagen induced arthritis (CIA). Mechanistic studies indicate that IDO2 Ig is able to access its intracellular target to exert its anti-arthritic effect by internalization via FcγRIIb specifically on B cells. On the basis of these findings, we propose that antibody targeting of IDO2 offers a feasible and disease-selective approach to treat RA and possibly other autoantibody- mediated autoimmune disorders.
2.0 Materials and Methods
2.1 Mice
Arthritic KRN IDO wt, IDO1 ko, and IDO2 ko mice on a C57BL/6 background carrying the MHC Class II I-Ag7 allele have been described [15]. DBA/1J and FcγRIIb ko C57BL/6 mice were purchased from Jackson Laboratories. C57BL/6 IDO2 ko mice lacking the TCR alpha chain (TCRα ko) and carrying a single copy of the MHC Class II I-Ag7 allele (TCRα ko IDO2 ko B6.g7/b) were generated as recipient mice for adoptive transfer of KRN T cells. T cell donor mice were IDO2 ko KRN TCR tg C57BL/6 mice carrying 2 copies of the I-Ab allele. All mice were bred and housed under specific pathogen free conditions in the animal facility at the Lankenau Institute for Medical Research. Studies were performed in accordance with National Institutes of Health and Association for Assessment and Accreditation of Laboratory Animal Care guidelines with approval from the LIMR Institutional Animal Care and Use Committee.
2.2 Generation and purification of monoclonal IDO2 Ig
IDO1 ko BALB/c mice were immunized with KLH-conjugated IDO2 peptide318-338 in complete Freund's adjuvant, boosted twice with the peptide-KLH conjugate in incomplete Freund's adjuvant, and then injected with the peptide-KLH conjugate in PBS. Spleen cells from the immunized mice were fused with Sp2/0-Ag14 myeloma cells, plated in methylcellulose (Stem Cell Technologies) and screened by ELISA and Western blot. A clonal hybridoma, designated 4-3 (mouse IgG1/κ), was subcloned by sorting single cells on a BD FACSAria III cell sorter. The resulting subclone (termed 4-3.8) was cultured in Hybridoma-SFM (Gibco, Life Technologies) containing 1% hybridoma enhancing supplement (Sigma) in CELLine bioreactor flasks (Wheaton) and IgG from supernatants purified over protein G.
2.3 IDO2 Ig treatment
Male and female KRN.g7, IDO1 ko KRN.g7, and IDO2 ko KRN.g7 mice were injected with 0.5 mg IDO2 Ig (clone 4-3.8) or control mouse Ig (Jackson Immunoresearch) at 21 days of age (pre-arthritis regimen) or 28 days of age (post-arthritis regimen). CIA mice were injected weekly with 0.5 mg IDO2 Ig or control mouse Ig starting the day before primary immunization. Adoptive transfer mice were injected the day before cell transfer with 0.5 mg IDO2 Ig or control mouse Ig.
2.4 Western Blot Analysis
Liver tissue from wt, IDO1 ko, and IDO2 ko C57BL/6 mice was harvested and homogenized in a polytron blender in the presence of RIPA buffer containing protease and phosphatase inhibitors. Tissue lysates were centrifuged and protein concentrations determined. Equal protein per sample (30 μg/lane) was fractioned using standard SDS-PAGE and blotted to Immobilon-NC membranes (Millipore, USA). After blocking, blots were incubated at 4 C overnight with primary antibody followed by incubation with an HRP-conjugated secondary antibody. Blots were developed with HYGLO Quickspray chemiluminescent HRP reagent (Denville Scientific) and analyzed using a ChemiDoc System with Image Lab Software (Biorad). Primary antibodies to the following antigens were used: IDO2 (Clone 4-3.8); α-tubulin (Santa Cruz Biotechnology); and pFcγRIIb (Cell Signaling Technology). HRP-conjugated anti-mouse Igκ (Jackson Immunoresearch) or anti-rabbit IgG (Cell Signaling Technology) were used as secondary antibodies.
2.5 Arthritis incidence
The two rear ankles of experimental KRN.g7 mice were measured starting at weaning (3 weeks of age). Measurement of ankle thickness was made above the footpad axially across the ankle joint using a Fowler Metric Pocket Thickness Gauge. Ankle thickness was rounded off to the nearest 0.05mm. At the termination of the experiment, ankles were fixed in 10% buffered formalin for 48 hrs, decalcified in 14% EDTA for 2 wks, embedded in paraffin, sectioned, and stained with H&E. Histology sections were imaged using a Zeiss Axioplan microscope with a Zeiss Plan-Apochromat 10×/0.32 objective and Zeiss AxioCam HRC camera using AxioVision 4.7.1 software. The images were then processed using Adobe Photoshop CS2 software.
2.6 Induction of Collagen Induced Arthritis (CIA)
CIA was induced in 8wk old male and female DBA/1J mice by an intradermal injection (tailbase) of 100μg bovine collagen II (CII) in complete Freund’s adjuvant (CFA), followed 21 days later by a booster injection of 100μg CII in incomplete Freund’s adjuvant (IFA) according to standard protocols [53]. Clinical scoring was evaluated on a scale of 0–4 for each paw for a maximum score of 16 per mouse: (0) no evidence of erythema and swelling, (1) erythema and mild swelling confined to the tarsals or ankle joint, (2) erythema and mild swelling extending from ankle to the tarsals, (3) erythema and moderate swelling extending from the ankle to the metatarsal joints, (4) erythema and severe swelling encompassing the ankle, foot, and digits, or ankylosis of the limb [53]. Front and rear paws were imaged using a Nikon D5300 camera and processed using Adobe Photoshop CS2 software.
2.7 1MT treatment
Mice were given 400 mg/kg/dose (100μl total volume) D/L-1MT (Sigma) diluted in Methocel/Tween (0.5% Tween 80, 0.5% methylcellulose (v/v in water; Sigma) twice daily by oral gavage (p.o.) using a curved feeding needle (20G ×1 ½ in; Fisher) as described [17]. 1MT treatment was started the day before primary injection of collagen (day 0) and continued for the duration of the experiment.
2.8 Analysis of T helper subsets
Joint draining LN cells from 6 week old control mouse Ig or IDO2 Ig-treated KRN.g7 mice were harvested and stained for CD4+ T cells (BioLegend clone GK1.5) and the following markers to distinguish Th subsets: bcl6 (Tfh, BD Pharmingen Clone K112g1), foxP3 (Treg, Biolegend clone 150D), gata3 (Th2, eBioscience clone TWAJ), rorγt (Th17, eBioscience clone AFKJS-9), T-bet (Th1, eBioscience clone 4B10). The samples were acquired on a BDFACSCanto II flow cytometer using FACSDiva Software (BD Bioscience) and analyzed using FlowJo Software (TreeStar).
2.9 Intracellular IL-21
Cells from the joint draining LNs of 6 week old control or IDO2 Ig-treated KRN.g7 mice were harvested and cultured for 4 hours with 50 ng/ml PMA, 500 ng/ml ionomycin, and 3 μg/ml Brefeldin A. After 4 hours, cells were harvested, surface stained for CD4 and CD8 (eBioscience), fixed and permeabilized (IC Fixation and Permeabilization Buffer, eBioscience), then stained for intracellular IL-21 or isotype control. The samples were acquired on a BDFACSCanto II flow cytometer using FACSDiva software and analyzed with FlowJo software.
2.10 ELISPOT assay
Cells from the joint draining LN (axillary, brachial, and popliteal LNs) from 6 week-old control mouse Ig or IDO2 Ig-treated KRN.g7 mice were plated at 4 × 105 cells per well and diluted serially 1:4 in Multiscreen HA mixed cellulose ester membrane plates (Millipore) coated with GPI-his (10μg/ml). The cells were incubated on the Ag-coated plates for 4hr at 37ºC. The Ig secreted by the plated cells was detected by Alkaline Phosphatase-conjugated goat anti-mouse total Ig secondary Ab (Southern Biotechnology Associates) and visualized using NBT/BCIP substrate (nitroblue tetrazolium / 5-bromo-4-chloro-3-indolyl phosphate; Sigma).
2.11 Internalization of IDO2 Ig
B cells from spleens of wt or FcγRIIb ko C57BL/6 mice were purified by anti-CD43 negative selection with MACS beads (Miltenyi). B cell purity was routinely ~97%. Purified B cells were cultured in vitro for 24h with anti-CD40 (eBioscience, 2μg/ml) + IL-21 (eBioscience, 100ng/ml). IDO2 Ig-PE or isotype control-PE was incubated with the cells for the last 2h of culture. Internalized IDO2 was measured by flow cytometry on a BD FACSCanto II and analyzed using FlowJo Software (TreeStar).
2.12 FcγRIIb stimulation assay
B cells from spleens of wt or FcγRIIb ko C57BL/6 mice were purified by anti-CD43 negative selection with MACS beads (Miltenyi). Purified B cells were stimulated for 10 min. with whole IgM (10μg/ml, Jackson Immunoresearch), anti-FcγRIIb (10μg/ml, clone AT130-5, AbD Serotec), or IDO2 Ig (clone 4-3.8, 10μg/ml). Cells were lysed in RIPA buffer containing protease and phosphatase inhibitors and analyzed by Western Blotting as described in section 2.4.
2.13 Adoptive transfer model of arthritis
Spleen and lymph node tissue was harvested from IDO2 ko KRN TCR tg (IDO2 ko KRN B6) mice. CD4+ T cells were purified by positive selection with anti-CD4 mouse MACS microbeads (Miltyeni Biotec). For T cell purification, elutant was purified over a second column to achieve higher purity (~90%). B cells from spleens of wt or FcγRIIb ko I-Ag7/b were purified by anti-CD43 negative selection with MACS beads (Miltenyi Biotec). Following purification, 3.5×105 CD4+ T cells and 1×106 B cells were adoptively transferred i.v. into TCR ko IDO2 ko B6.g7 hosts. Arthritis was measured starting at the day of adoptive transfer, as described in section 2.5. Mice were sacrificed after 2 weeks.
2.14 Statistical Analysis
Statistical significance was determined using one way-ANOVAs followed by comparison of means with Tukey's post-hoc multiple comparison correction or Kruskal-Wallis non-parametric ANOVA with Dunn's multiple comparison correction as indicated using Prism6 (GraphPad Software, Inc).
3.0 Results
3.1 IDO2 Ig inhibits arthritis in preclinical models of autoimmune arthritis
Genetic deficiency in IDO2 leads to an attenuated level of disease in the KRN.g7 preclinical model of autoimmune arthritis, implicating IDO2 as a therapeutic target to treat RA [15]. As a strategy to specifically target IDO2, we developed a monoclonal antibody (IDO2 Ig, clone 4-3.8) that recognizes IDO2, but not the closely related IDO1 enzyme (Fig. 1). KRN.g7 mice treated with IDO2 Ig starting before the onset of arthritis developed arthritis later and with reduced overall severity (Fig. 2a) compared to those treated with control Ig, recapitulating the phenotype of genetic loss of IDO2 in this model. A similar reduction in arthritis was obtained when IDO2 Ig was started after the onset of arthritis indicating IDO2 Ig is effective in either a preventive or therapeutic experimental design (Fig. 2a). Reduced arthritis in IDO2 Ig-treated mice was confirmed histologically by a decrease in immune cell infiltrates, pannus formation, synovial hyperplasia, and cartilage and bone destruction, compared to control mouse Ig-treated (Fig. 2b) or untreated mice [15]. To validate IDO2 as the target for IDO2 Ig in vivo, arthritis was evaluated in IDO1 ko KRN.g7 and IDO2 ko KRN.g7 mice treated with IDO2 Ig. Treatment with IDO2 Ig reduced joint inflammation in IDO1 ko KRN.g7 mice (Fig. 2c), but did not affect the residual disease displayed in IDO2 ko KRN.g7 mice (Fig. 2d), providing a genetic validation for the in vivo specificity of the antibody.
Figure 1. Monoclonal antibody specifically recognizes IDO2.

Liver protein lysates (10μg) from IDO1 ko, IDO2 ko, or wt C57BL/6 mice were immunoblotted with monoclonal IDO2 Ig (clone 4–3.8) and detected with anti-mouse Igκ-HRP. Blots were then probed with anti-αtubulin, followed by anti-rabbit-HRP as a loading control. (A) Representative blot of 3 total. (B) Graph shows the mean ratio of IDO2 / αtubulin ± SEM for n=3 blots. P-values were calculated by one way-ANOVA followed by comparison of means with Tukey's post-hoc multiple comparison correction. * p < 0.05, n.s. = not significant
Figure 2. IDO2 Ig inhibits joint inflammation in KRN.g7 mice.

(A,B) KRN.g7 mice were injected once with 0.5mg control mouse Ig or IDO2 Ig before (21 days of age) or after (28 days of age) the onset of arthritis. (C) IDO1 ko or (D) IDO2 ko KRN.g7 mice were injected once with 0.5 mg control mouse Ig or IDO2 Ig at 21 days of age. (A,C,D) Rear ankles were measured as an indication of arthritis and represented as mean ankle thickness ± SEM for n=9 KRN.g7, n=10 IDO1 ko KRN.g7, and n=8 IDO2 ko KRN.g7 mice per treatment group. (B) Metatarsal joint from KRN.g7 mice treated with control Ig, IDO2 Ig (pre-arthritis), or IDO2 Ig (post-arthritis). Representative sections from a total of n=8 mice per treatment group. Scale bar = 100μm. (A) P-values were calculated by one way-ANOVA followed by comparison of means with Tukey's post-hoc multiple comparison correction. (C,D) P-values were calculated using a 2-tailed unpaired t-test. *** p < 0.001, n.s. = not significant
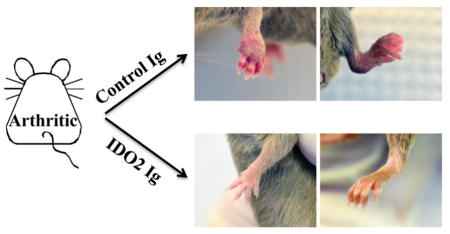
To determine if IDO2 Ig could inhibit joint inflammation outside of the context of the KRN model, we also tested its effects in the well-characterized collagen- induced arthritis (CIA) model. In this model, arthritis was induced in DBA/1J mice by immunization with heterologous collagen, as previously described [53]. As a control group, a separate cohort of mice were treated with the nonselective IDO pathway inhibitor 1MT. Treatment with IDO2 Ig or 1MT was started at the time of the first immunization and continued through the duration of the experiment. Control-treated DBA/1J mice developed robust arthritis within one week of secondary immunization (Fig. 3a, b). Administration of IDO2 Ig significantly attenuated arthritis (Fig. 3a), with some mice developing only mild arthritis and others no arthritis at all (Fig. 3b). Furthermore, the limited arthritis that did develop was more isolated (mouse Ig: 3.2 ± 0.2; IDO2 Ig: 1.9 ± 0.3 involved paws, p=0.004). Similar to one published report [54], but not others [31, 55], we found that 1MT did not affect the frequency or severity of arthritis induced by collagen immunization (Fig. 3c). While it is unclear why 1MT affects CIA in some studies, but not others, the lack of consistent results using 1MT may reflect in part the complications inherent in using non-selective pathway inhibitors. Taken together, our results support the conclusion that IDO2 drives arthritis in two independent preclinical models and that its targeting with an IDO2-specific antibody is sufficient to alleviate disease.
Figure 3. IDO2 Ig inhibits joint inflammation in second model of arthritis, CIA.

CIA was induced in DBA/1J mice by immunization with collagen + CFA (day 0) and collagen + IFA (day 21, arrows). Mice were treated with (A,B) 0.5mg IDO2 Ig or mouse Ig (weekly) or (C) carrier or 400mg/kg 1MT (b.i.d) starting on day -1. (A,C) Arthritis is indicated as a clinical score (0–4 per paw for a maximum of 16 per mouse). n=18 mouse Ig, n=18 IDO2 Ig, n=8 carrier, and n=8 1MT-treated mice. P-values were calculated using a 2-tailed unpaired t-test. (B) Representative front and rear paws from CIA mice treated with mouse Ig or IDO2 Ig on day 49 post-immunization. Mouse Ig-treated mice developed robust arthritis (left panel), whereas IDO2 Ig-treated mice developed only mild arthritis (right panel) or no arthritis at all (middle panel). n=9 mice per group. *** p < 0.001, n.s. = not significant
3.2 IDO2 Ig inhibits autoreactive T and B cell responses
We next evaluated the parameters of the immune response affected by targeting IDO2. To do this, we again made use of the KRN model, where it was possible to specifically follow autoreactive T and B cell responses. We reported previously that the most dramatic differences in IDO2 ko KRN.g7 mice were their significant reductions in CD4+ helper T cell subsets and numbers of autoantibody-secreting B cells, compared to those with wild-type (wt) IDO2 [15]. To determine if IDO2 Ig administration could phenocopy these effects, we measured percentages of differentiated T and B cells in KRN.g7 mice treated with a single injection of IDO2 Ig in the same manner as before. Differentiated T helper subsets were examined by measuring the expression of intracellular transcription factors using flow cytometry. KRN.g7 mice treated with IDO2 Ig, either before or after the onset arthritis, exhibited reduced percentages of T-bet+ (Th1), Gata-3+ (Th2), RoRγt+ (Th17), and Bcl-6+ (Tfh) subsets compared to mice treated with control Ig (Fig. 4a). The percentage of FoxP3+ (Treg) cells was not different between the treatment groups; however percentages of cells that co-expressed FoxP3 and T-bet, Gata-3, or RoRγt, so-called effector T-reg cells [56] were reduced in IDO2 Ig treated mice (Fig. 4b). Likewise, the small population of FoxP3+Bcl-6+ T follicular regulatory cells was also diminished in IDO2 Ig treated mice (Fig. 4b). Levels of IL-21, a cytokine that upregulates IDO2 expression [16] and is essential for T cell-mediated antibody production [57] and induction of arthritis [58], was likewise decreased (Fig. 4c). T helper function is required to induce the pathogenic autoantibody response in KRN.g7 mice. Consistent with the reduction in T cell helper function, numbers of autoantibody-secreting cells were decreased in IDO2 Ig-treated mice (Fig. 4d). Therefore, IDO2 Ig is able to recapitulate the reduced arthritic response observed in mice that were genetically deficient for IDO2, at the level of both T cells and B cells.
Figure 4. IDO2 Ig alters autoreactive T and B cell differentiation.

(A,B) Frequency of CD4+ T helper cell subpopulations were measured by flow cytometry by intracellular staining for the transcription factors T-bet (Th1), GATA-3 (Th2), RORγt (Th17), Bcl-6 (Tfh), and FoxP3 (Treg). Percentages are shown for CD4+ cells expressing (A) a single transcription factor or (B) co-expressing FoxP3 with a second effector transcription factor. Graphs show mean % ± SEM of each subpopulation out of total CD4+ T cells for n=9 mouse Ig, n=5 IDO2 Ig (pre-arthritis), and n=9 IDO2 Ig (post arthritis) treated KRN.g7 mice, pooled from 3 independent experiments. (C) Cells from the joint dLNs were cultured for 4h in PMA (50ng/ml) + ionomycin (500ng/ml) + 3μg/ml Brefeldin A. Intracellular IL-21 was measured by flow cytometry. Graph shows % IL-21+ cells ± SEM out of total CD4+ T cell populations from n=12 mouse Ig, n=9 IDO2 Ig (pre-arthritis), and n=9 IDO2 Ig (post arthritis) treated KRN.g7 mice, pooled from 4 independent experiments. P-values were calculated by one way-ANOVA followed by comparison of means with Tukey's post-hoc multiple comparison correction. (D) Number of anti-GPI ASCs was measured by ELISpot. Graph shows mean number anti-GPI ASCs ± SEM for n=15 control Ig, n=11 IDO2 Ig (pre-arthritis), and n=9 IDO2 Ig (post-arthritis). P-values were calculated with Kruskal-Wallis non-parametric ANOVA followed by comparison of means with Dunn's multiple comparison correction. * p< 0.05, ** p < 0.01, *** p < 0.001, n.s. not significant.
3.3 IDO2 Ig accesses intracellular IDO2 through internalization via FcγRIIb
We next sought to determine the mechanism by which IDO2 Ig is able to access IDO2 in mediating a therapeutic effect. One mechanism by which antibodies can be internalized into cells is via FcγRs, specialized receptors for IgG expressed on a wide variety of cell types, including macrophages, neutrophils, dendritic cells, and B cells [59]. In previous work, we used adoptive transfer experiments to define B cells as the critical IDO2-expressing cell type for mediating arthritis in the KRN model [16]. Therefore, we examined the ability of B cells to internalize the therapeutic antibody using an in vitro antibody internalization assay. Purified B cells were cultured with anti-CD40 + IL-21, a stimulus that upregulates IDO2 [16], and analyzed for their ability to internalize fluorescently labeled IDO2 Ig by flow cytometry. As a control, B cells were incubated with fluorescently labeled isotype control Ig. IDO2 Ig, but not isotype control Ig, was able to access the intracellular compartment of B cells after 2h in culture. However, if the B cells were pre-incubated with an antibody that blocks FcγRs, IDO2 Ig was not internalized (Fig. 5a). To confirm the requirement for FcγRs, we repeated the in vitro internalization assay but used B cells purified from mice that were genetically deficient in FcγRIIb (FcγRIIb ko), the only FcγR expressed by B cells [60]. Notably, IDO2 Ig internalization was abolished completely in the genetic absence of FcγRIIb, confirming that FcgRIIb-mediated internalization was essential for IDO2 Ig to access its intracellular target (Fig. 5a).
Figure 5. IDO2 Ig is internalized, but does not signal via FcγRIIb.

(A) Purified wild-type or FcγRIIb knock-out (ko) B cells were cultured in vitro for 24h with anti-CD40 (2μg/ml) + IL-21 (100ng/ml). IDO2-Ig-PE or isotype control-PE was incubated with the cells for the last 2h of culture. The cells were then analyzed by flow cytometry; graph shows internalization of IDO2-Ig (red) overlaid isotype control Ig (gray). Representative plots or images from 3 independent experiments. (B,C) Purified B cells from wt or FcγRIIb ko B6 mice were stimulated for 15 min. with 10μg/ml anti-FcγRIIb (AT130–5), whole IgM, or IDO2 Ig and analyzed for pFcγRIIb by Western blot with αtubulin as a loading control. (B) Representative blot and (C) ratio of pFCγRIIb / αtubulin from 3 independent experiments. P-values were calculated by one way-ANOVA followed by comparison of means with Tukey's post-hoc multiple comparison correction. ** p< 0.01, *** p < 0.001, n.s. = not significant
In the following experiments we sought to rule out the possibility that FcγRIIb signaling contributed to the therapeutic effect of the IDO2 Ig. Ligation of FcγRIIb on B cells has been shown to inhibit B cell proliferation and antibody production, through a mechanism initiated by phosphorylation of the ITIM found in the cytoplasmic domain of FcγRIIb [61, 62]. Thus, it was possible that some or all of the therapeutic activity of the IDO2 Ig could be due to binding and triggering FcγRIIb signaling, rather than a direct inhibition of IDO2. To rule out this possibility, we purified B cells from wt or FcγRIIb ko mice, cultured them with IDO2 Ig, and then evaluated the phosphorylation status of the cytoplasmic tail of FcγRIIb (pFcγRIIb) in the cells by Western blotting (Fig. 5c,d). Wt B cells cultured with whole IgM or the FcγRIIb-stimulating antibody AT130-5 [63] as positive controls demonstrated strong phosphorylation of FcγRIIb. As expected, no pFcγRIIb was detected in FcγRIIb ko B cells, confirming the specificity of the phospho-specific antibody used to detect the receptor. Importantly, IDO2 Ig did not stimulate phosphorylation of FcγRIIb (Fig. 5c,d). This result argued against FcγRIIb-mediated inhibition as a basis for the therapeutic action of the IDO2 Ig. Overall, we conclude that IDO2 Ig gains access to its intracellular target in B cells through internalization via FcγRIIb, but does not stimulate its signaling pathway.
3.4 FcγRIIb on B cells is required in vivo for IDO2 Ig therapeutic activity
To determine if FcγRIIb expression on B cells was required for the activity of IDO2 Ig in vivo, we made use of an adoptive transfer model of arthritis where contributions of individual receptors and cell types can be discriminated [15, 16]. In this model, adoptive transfer of KRN T cells into T cell deficient mice expressing the I-Ag7 MHC Class II molecule required for antigen presentation to KRN T cells (TCRα ko B6.g7/b) results in robust arthritis 7–10 days post-cell transfer [64]. Using this approach, we previously demonstrated that transfer of IDO2 ko KRN T cells into IDO2 ko recipient mice (IDO2 ko TCRα ko B6.g7/b) results in an attenuated level of arthritis relative to wt recipients, similar to that seen in the IDO2 ko KRN.g7 spontaneous model [15]. Furthermore, addition of wt but not IDO2 ko B cells fully restored arthritis to wt levels, confirming the critical requirement for B cells as the IDO2-expressing cell type needed for robust arthritis in the model [16]. To determine if IDO2 Ig requires FcγRIIb expression on B cells to exert its effect in vivo, we used a similar approach, transferring B cells purified from C57BL/6 mice that either express (wt) or lack FcγRIIb (FcγRIIb ko) into IDO2 ko recipients. Arthritis was induced by transfer of IDO2 ko KRN T cells. The mice were then treated with either IDO2 Ig or control Ig (Fig. 6a). Control treated mice that received wt B cells developed robust arthritis whereas IDO2 Ig treated mice that received wt B cells had attenuated disease (Fig 6b). Control-treated mice that received FcγRIIb ko B cells developed arthritis at a level similar to that induced by wt B cells, demonstrating that FcγRIIb is not required on B cells for arthritis induction. In contrast to its anti-arthritic effect in the wt B cell group, IDO2 Ig had no effect on arthritis development in mice that received FcγRIIb ko B cells (Fig. 6b). Taken together, these results demonstrate that to inhibit arthritis development in vivo, IDO2 Ig requires FcγRIIb on B cells to access intracellular IDO2.
Figure 6. FcγRIIb expression on B cells is required for IDO2 Ig activity in vivo.
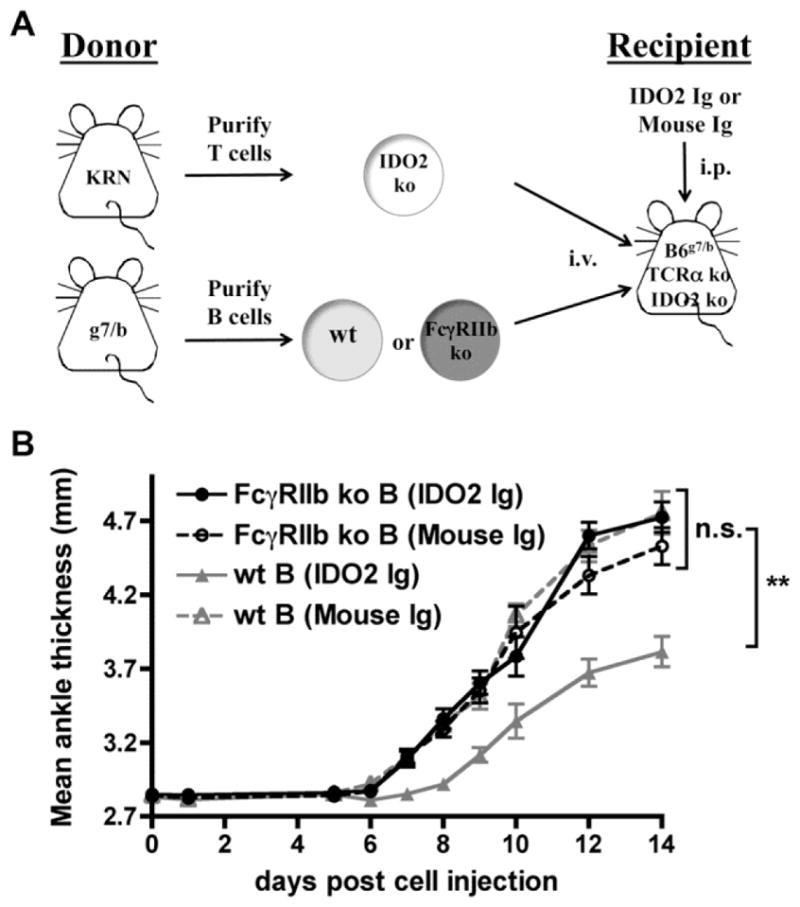
(A) Schematic of adoptive transfer strategy. 3.5 × 105 IDO2 ko KRN CD4+ T cells and 1×106 wt or FcγRIIb ko I-Ag7/b B cells were adoptively transferred into IDO2 ko TCRα ko IDO2 ko C57BL/6 (I-Ag7/b) hosts. The day before cell transfer, host mice were injected with 0.5mg IDO2 Ig or control mouse Ig. (B) Rear ankles were measured as an indication of arthritis and represented as mean ankle thickness ± SEM for n=11 mice that received FcγRIIb ko B cells and n=13 mice that received wt B cells per treatment group, pooled from 3 independent experiments. P-values were calculated by one way-ANOVA followed by comparison of means with Tukey's post-hoc multiple comparison correction. ** p < 0.01, n.s. not significant.
4.0 Discussion
This study establishes the feasibility of antibody-mediated targeting of the intracellular immunomodulatory enzyme IDO2 as a therapeutic strategy to treat autoimmune arthritis. IDO2 Ig treatment inhibits autoreactive T and B cell responses and joint inflammation, recapitulating the phenotype observed in the genetic absence of IDO2. Although monoclonal antibodies are generally used to target cell surface or secreted targets due to easy accessibility, mechanistic studies indicate that IDO2 Ig is able to access its intracellular target to exert its anti-arthritic effect by internalization via FcγRIIb on B cells. Together, this work validates IDO2 as a selective contributor to autoimmune pathogenesis and offers a new approach to therapeutically target IDO2 in the treatment of inflammatory autoimmune disease.
The IDO pathway has been implicated in both suppressive and supportive roles in autoimmune responses. Immunosuppressive activity has been suggested by studies using trinitrobenzene sulfonic acid-induced colitis and experimental autoimmune encephalomyelitis [29, 30]. In contrast, studies in other models, including the KRN model of arthritis [17], as well as models of inflammatory airway disease [33], allergy [34], and contact hypersensitivity [35], have provided evidence that the IDO pathway plays a positive role in inflammatory responses. Even within a single model, CIA, conflicting results regarding the role of the IDO pathway have been reported [31, 54, 55]. Complicating the interpretation of these experiments has been the use of 1MT, which is not a direct inhibitor of IDO1 or IDO2, but an indirect inhibitor of the IDO pathway that functions as a tryptophan mimetic [15, 36]. Recently, studies in IDO1 and IDO2 deficient mice have helped clarify specific contributions of these enzymes to autoimmune responses. We found that IDO2 exerted a specific proinflammatory role in the initiation and progression of autoimmune responses, distinct from IDO1 [15, 16]. Therefore, therapies to specifically inhibit IDO1 and IDO2 individually could provide great therapeutic benefit in the treatment of inflammatory disease.
While small molecule inhibitors that target IDO1 have entered cancer clinical trials [65–68], small molecules that can be used to specifically target IDO2 in vivo have yet to be identified [69, 70]. Our work demonstrates a novel monoclonal antibody strategy to directly target IDO2, without concomitant effects on IDO1. Monoclonal antibody targeting of IDO2 is able to fully recapitulate the strong anti-arthritic effect of genetic IDO2 deficiency. Like genetic IDO2 deficiency, antibody targeting of IDO2 attenuates, but does not completely eliminate arthritis in KRN mice. Previous work in our lab demonstrated that IDO2 acts at the initiation phase of the autoimmune response by affecting T cell help and autoantibody production. Downstream mediators of inflammation were not affected by the absence of IDO2 [15, 16]. In fact, combining IDO pathway inhibition with RA therapeutics that either reinstate the initiation phase (B cell depletion) or target downstream inflammation (e.g. MTX) was more effective than either treatment alone at alleviating arthritis [12, 13]. Together, these data suggest that an antibody-based approach to specifically target IDO2 offers a rational direction for drug development either by itself or as a co-therapeutic strategy combined with current RA treatments.
Targeting strategies using monoclonal Abs have generated effective therapeutics for both autoimmunity and cancer due to their exquisite specificity [39, 71]. In fact, two of the most promising new therapies for RA are monoclonal antibodies directed against the B cell surface receptor CD20 and secreted inflammatory cytokine TNFα or its surface receptor TNFRI [40, 72–75]. Although predominantly used against cell surface and secreted Ags, recent evidence suggests monoclonal antibodies can also be effective therapeutics for targeting intracellular Ags [49, 76]. Proof-of-concept studies demonstrated that monoclonal antibody treatment or vaccination directed against intracellular tumor antigens neutralized tumor formation in murine models of metastatic melanoma and breast cancer [49]. Our findings demonstrate monoclonal antibody targeting of intracellular IDO2 inhibits inflammation in two independent preclinical models of arthritis and highlight the enormous and, as yet, unexplored potential of monoclonal antibodies as a therapeutic strategy to target intracellular antigens in the treatment of autoimmune diseases.
We found that FcγRIIb expression on B cells was necessary for anti-arthritic activity of the therapeutic IDO2 Ig. Binding to FcγRs is important for the therapeutic activity of other mAb therapies by several different mechanisms, including FcγR-dependent stimulation of macrophages, NK cells, or neutrophils [46], antibody crosslinking to enhance target cell killing by antibody-dependent cellular cytotoxicity (ADCC) [77], and induction of immunomodulatory activity [78, 79]. Our work demonstrated a role for FcγRIIb in mediating the uptake and therapeutic activity of IDO2 Ig by allowing access to its intracellular target within the B cell where IDO2 exerts its pathogenic function. Internalization via FcγRIIb also occurs for anti-CD20 antibodies; however, internalization reduced cell surface expression of the target antigen, diminishing therapeutic efficacy [80, 81]. For an intracellular target antigen like IDO2, FcγRIIb-mediated internalization in the absence of signaling is beneficial, and could be exploited to help mediate its therapeutic properties.
5.0 Conclusions
In summary, our data demonstrate that targeting IDO2 with a monoclonal antibody inhibits autoreactive B and T cell activation and alleviates joint inflammation in two well-characterized preclinical models of arthritis. The B cell surface molecule FcγRIIb is necessary for mAb internalization, allowing it to target an intracellular antigen traditionally considered inaccessible to mAb therapy. Together, these results demonstrate that cell-permeable small molecules are not the only therapeutic options for intracellular antigens and open up a new strategy for specific targeting of IDO2 and other intracellular mediators of autoimmunity.
Highlights.
IDO2-specific Ig treatment alleviates arthritis in two preclinical arthritis models
IDO2 Ig inhibits autoreactive T and B cells, recapitulating genetic IDO2 deficiency
IDO2 Ig uses FcγRIIb to internalize and access its intracellular target
FcγRIIb on B cells is necessary for IDO2 Ig function in vivo
Acknowledgments
The authors would like to thank Jon Messerschmidt for technical assistance with the Western blot assays, Summer Sedano for the initial purification of the 4–3 hybridoma, and Gwen Gilliard for sectioning and staining the arthritic ankles.
Funding:
This work was supported in part by a grant from the Lupus Research Alliance. (LM-N.), a grant from the Women's Board of Lankenau Medical Center (LM-N), NIH grants R01 AR057847 (LM-N) and R21 CA159337 (GCP) and by support from the Zuckerman Family Autoimmune Disorder Research Fund at Lankenau Medical Center, the Lankenau Medical Center Foundation, and Main Line Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Helmick CG, Felson DT, Lawrence RC, Gabriel S, Hirsch R, Kwoh CK, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part I. Arthritis Rheum. 2008;58:15–25. doi: 10.1002/art.23177. [DOI] [PubMed] [Google Scholar]
- 2.Gibofsky A. Epidemiology, pathophysiology, and diagnosis of rheumatoid arthritis: A Synopsis. Am J Manag Care. 2014;20:s128–45. [PubMed] [Google Scholar]
- 3.Curtis JR, Singh JA. Use of biologics in rheumatoid arthritis: current and emerging paradigms of care. Clin Ther. 2011;33:679–707. doi: 10.1016/j.clinthera.2011.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh JA, Furst DE, Bharat A, Curtis JR, Kavanaugh AF, Kremer JM, et al. 2012 update of the 2008 American College of Rheumatology recommendations for the use of disease-modifying antirheumatic drugs and biologic agents in the treatment of rheumatoid arthritis. Arthritis Care Res (Hoboken) 2012;64:625–39. doi: 10.1002/acr.21641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alghasham A, Rasheed Z. Therapeutic targets for rheumatoid arthritis: Progress and promises. Autoimmunity. 2014 doi: 10.3109/08916934.2013.873413. [DOI] [PubMed] [Google Scholar]
- 6.Tian H, Cronstein BN. Understanding the mechanisms of action of methotrexate: implications for the treatment of rheumatoid arthritis. Bulletin of the NYU hospital for joint diseases. 2007;65:168–73. [PubMed] [Google Scholar]
- 7.Albrecht K, Muller-Ladner U. Side effects and management of side effects of methotrexate in rheumatoid arthritis. Clin Exp Rheumatol. 2010;28:S95–101. [PubMed] [Google Scholar]
- 8.Buch MH, Smolen JS, Betteridge N, Breedveld FC, Burmester G, Dorner T, et al. Updated consensus statement on the use of rituximab in patients with rheumatoid arthritis. Ann Rheum Dis. 2011;70:909–20. doi: 10.1136/ard.2010.144998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cooper N, Arnold DM. The effect of rituximab on humoral and cell mediated immunity and infection in the treatment of autoimmune diseases. Br J Haematol. 2010;149:3–13. doi: 10.1111/j.1365-2141.2010.08076.x. [DOI] [PubMed] [Google Scholar]
- 10.Solomon DH, Kremer JM, Fisher M, Curtis JR, Furer V, Harrold LR, et al. Comparative cancer risk associated with methotrexate, other non-biologic and biologic disease-modifying anti-rheumatic drugs. Semin Arthritis Rheum. 2014;43:489–97. doi: 10.1016/j.semarthrit.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 11.Curtis JR, Yang S, Patkar NM, Chen L, Singh JA, Cannon GW, et al. Risk of hospitalized bacterial infections associated with biologic treatment among US veterans with rheumatoid arthritis. Arthritis Care Res (Hoboken) 2014;66:990–7. doi: 10.1002/acr.22281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pigott E, Duhadaway JB, Muller AJ, Gilmour S, Prendergast GC, Mandik-Nayak L. 1-Methyl-tryptophan synergizes with methotrexate to alleviate arthritis in a mouse model of arthritis. Autoimmunity. 2014 doi: 10.3109/08916934.2014.914507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pigott E, Mandik-Nayak L. Addition of an indoleamine 2,3,-dioxygenase inhibitor to B cell-depletion therapy blocks autoreactive B cell activation and recurrence of arthritis in K/BxN mice. Arthritis Rheum. 2012;64:2169–78. doi: 10.1002/art.34406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muller AJ, Mandik-Nayak L. Prendergast GC, Beyond immunosuppression: reconsidering indoleamine 2,3-dioxygenase as a pathogenic element of chronic inflammation. Immunotherapy. 2010;2:293–7. doi: 10.2217/imt.10.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Merlo LM, Pigott E, Duhadaway JB, Grabler S, Metz R, Prendergast GC, et al. IDO2 Is a Critical Mediator of Autoantibody Production and Inflammatory Pathogenesis in a Mouse Model of Autoimmune Arthritis. J Immunol. 2014;192:2082–90. doi: 10.4049/jimmunol.1303012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Merlo LM, DuHadaway J, Grabler S, Prendergast GC, Muller AJ, Mandik-Nayak L. IDO2 modulates T cell-dependent autoimmune responses through a B cell-intrinsic mechanism. J Immunol. 2016;196:4487–97. doi: 10.4049/jimmunol.1600141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scott GN, DuHadaway J, Pigott E, Ridge N, Prendergast GC, Muller AJ, et al. The immunoregulatory enzyme IDO paradoxically drives B cell-mediated autoimmunity. J Immunol. 2009;182:7509–17. doi: 10.4049/jimmunol.0804328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prendergast GC, Metz R, Muller AJ, Merlo LM, Mandik-Nayak L. IDO2 in Immunomodulation and Autoimmune Disease. Front Immunol. 2014;5:585. doi: 10.3389/fimmu.2014.00585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taylor MW, Feng GS. Relationship between interferon-gamma, indoleamine 2,3-dioxygenase, and tryptophan catabolism. FASEB J. 1991;5:2516–22. [PubMed] [Google Scholar]
- 20.Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281:1191–3. doi: 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- 21.Elovainio M, Hurme M, Jokela M, Pulkki-Raback L, Kivimaki M, Hintsanen M, et al. Indoleamine 2,3-dioxygenase activation and depressive symptoms: results from the Young Finns Study. Psychosom Med. 2012;74:675–81. doi: 10.1097/PSY.0b013e318266d0f5. [DOI] [PubMed] [Google Scholar]
- 22.Oxenkrug GF. Tryptophan kynurenine metabolism as a common mediator of genetic and environmental impacts in major depressive disorder: the serotonin hypothesis revisited 40 years later. Isr J Psychiatry Relat Sci. 2010;47:56–63. [PMC free article] [PubMed] [Google Scholar]
- 23.Xiao Y, Christou H, Liu L, Visner G, Mitsialis SA, Kourembanas S, et al. Endothelial Indoleamine 2,3-Dioxygenase Protects against Development of Pulmonary Hypertension. Am J Respir Crit Care Med. 2013;188:482–91. doi: 10.1164/rccm.201304-0700OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prendergast GC, Chang MY, Mandik-Nayak L, Metz R, Muller AJ. Indoleamine 2,3-dioxygenase as a modifier of pathogenic inflammation in cancer and other inflammation-associated diseases. Curr Med Chem. 2011;18:2257–62. doi: 10.2174/092986711795656072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muller AJ, Scherle PA. Targeting the mechanisms of tumoral immune tolerance with small-molecule inhibitors. Nat Rev Cancer. 2006;6:613–25. doi: 10.1038/nrc1929. [DOI] [PubMed] [Google Scholar]
- 26.Schroecksnadel K, Winkler C, Duftner C, Wirleitner B, Schirmer M, Fuchs D. Tryptophan degradation increases with stage in patients with rheumatoid arthritis. Clin Rheumatol. 2006;25:334–7. doi: 10.1007/s10067-005-0056-6. [DOI] [PubMed] [Google Scholar]
- 27.Pertovaara M, Hasan T, Raitala A, Oja SS, Yli-Kerttula U, Korpela M, et al. Indoleamine 2,3-dioxygenase activity is increased in patients with systemic lupus erythematosus and predicts disease activation in the sunny season. Clin Exp Immunol. 2007;150:274–8. doi: 10.1111/j.1365-2249.2007.03480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Furuzawa-Carballeda J, Lima G, Jakez-Ocampo J, Llorente L. Indoleamine 2,3-dioxygenase-expressing peripheral cells in rheumatoid arthritis and systemic lupus erythematosus: a cross-sectional study. Eur J Clin Invest. 2011;41:1037–46. doi: 10.1111/j.1365-2362.2011.02491.x. [DOI] [PubMed] [Google Scholar]
- 29.Gurtner GJ, Newberry RD, Schloemann SR, McDonald KG, Stenson WF. Inhibition of indoleamine 2,3-dioxygenase augments trinitrobenzene sulfonic acid colitis in mice. Gastroenterology. 2003;125:1762–73. doi: 10.1053/j.gastro.2003.08.031. [DOI] [PubMed] [Google Scholar]
- 30.Sakurai K, Zou JP, Tschetter JR, Ward JM, Shearer GM. Effect of indoleamine 2,3-dioxygenase on induction of experimental autoimmune encephalomyelitis. J Neuroimmunol. 2002;129:186–96. doi: 10.1016/s0165-5728(02)00176-5. [DOI] [PubMed] [Google Scholar]
- 31.Szanto S, Koreny T, Mikecz K, Glant TT, Szekanecz Z, Varga J. Inhibition of indoleamine 2,3-dioxygenase- mediated tryptophan catabolism accelerates collagen-induced arthritis in mice. Arthritis research & therapy. 2007;9:R50. doi: 10.1186/ar2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fallarino F, Volpi C, Zelante T, Vacca C, Calvitti M, Fioretti MC, et al. IDO mediates TLR9-driven protection from experimental autoimmune diabetes. J Immunol. 2009;183:6303–12. doi: 10.4049/jimmunol.0901577. [DOI] [PubMed] [Google Scholar]
- 33.Xu H, Oriss TB, Fei M, Henry AC, Melgert BN, Chen L, et al. Indoleamine 2,3-dioxygenase in lung dendritic cells promotes Th2 responses and allergic inflammation. Proc Natl Acad Sci U S A. 2008;105:6690–5. doi: 10.1073/pnas.0708809105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.von Bubnoff D, Bieber T. The indoleamine 2,3-dioxygenase (IDO) pathway controls allergy. Allergy. 2012;67:718–25. doi: 10.1111/j.1398-9995.2012.02830.x. [DOI] [PubMed] [Google Scholar]
- 35.Metz R, Smith C, DuHadaway JB, Chandler P, Baban B, Merlo LMF, et al. IDO2 is critical for IDO1-mediated T cell regulation and exerts a non-redundant function in inflammation. Int Immunol. 2014;26:357–67. doi: 10.1093/intimm/dxt073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Metz R, Rust S, Duhadaway JB, Mautino MR, Munn DH, Vahanian NN, et al. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: A novel IDO effector pathway targeted by D-1-methyl-tryptophan. Oncoimmunology. 2012;1:1460–8. doi: 10.4161/onci.21716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ravishankar B, Liu H, Shinde R, Chandler P, Baban B, Tanaka M, et al. Tolerance to apoptotic cells is regulated by indoleamine 2,3-dioxygenase. Proc Natl Acad Sci U S A. 2012;109:3909–14. doi: 10.1073/pnas.1117736109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Put K, Brisse E, Avau A, Imbrechts M, Mitera T, Janssens R, et al. IDO1 Deficiency Does Not Affect Disease in Mouse Models of Systemic Juvenile Idiopathic Arthritis and Secondary Hemophagocytic Lymphohistiocytosis. PloS one. 2016;11:e0150075. doi: 10.1371/journal.pone.0150075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Imai K, Takaoka A. Comparing antibody and small-molecule therapies for cancer. Nat Rev Cancer. 2006;6:714–27. doi: 10.1038/nrc1913. [DOI] [PubMed] [Google Scholar]
- 40.Townsend MJ, Monroe JG, Chan AC. B-cell targeted therapies in human autoimmune diseases: an updated perspective. Immunol Rev. 2010;237:264–83. doi: 10.1111/j.1600-065X.2010.00945.x. [DOI] [PubMed] [Google Scholar]
- 41.Cantini F, Niccoli L, Nannini C, Cassara E, Kaloudi O, Giulio Favalli E, et al. Tailored first-line biologic therapy in patients with rheumatoid arthritis, spondyloarthritis, and psoriatic arthritis. Semin Arthritis Rheum. 2016;45:519–32. doi: 10.1016/j.semarthrit.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 42.Dinarello CA, Joosten LA. Inflammation in rheumatology in 2015: New tools to tackle inflammatory arthritis. Nat Rev Rheumatol. 2016;12:78–80. doi: 10.1038/nrrheum.2015.180. [DOI] [PubMed] [Google Scholar]
- 43.Kamenarska ZG, Hristova MH, Vinkov AI, Dourmishev LA. Monoclonal Antibody Drugs for Systemic Lupus Erythematosus. Folia Med (Plovdiv) 2015;57:89–92. doi: 10.1515/folmed-2015-0025. [DOI] [PubMed] [Google Scholar]
- 44.Milo R. Therapeutic strategies targeting B-cells in multiple sclerosis. Autoimmunity reviews. 2016;15:714–8. doi: 10.1016/j.autrev.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 45.Safdari Y, Ahmadzadeh V, Farajnia S. CD20-targeting in B-cell malignancies: novel prospects for antibodies and combination therapies. Invest New Drugs. 2016;34:497–512. doi: 10.1007/s10637-016-0349-4. [DOI] [PubMed] [Google Scholar]
- 46.Nimmerjahn F, Ravetch JV. Translating basic mechanisms of IgG effector activity into next generation cancer therapies. Cancer Immun. 2012;12:13. [PMC free article] [PubMed] [Google Scholar]
- 47.Tsirigotis P, Savani BN, Nagler A. Programmed death-1 immune checkpoint blockade in the treatment of hematological malignancies. Ann Med. 2016:1–12. doi: 10.1080/07853890.2016.1186827. [DOI] [PubMed] [Google Scholar]
- 48.Farkona S, Diamandis EP, Blasutig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73. doi: 10.1186/s12916-016-0623-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guo K, Li J, Tang JP, Tan CP, Hong CW, Al-Aidaroos AQ, et al. Targeting intracellular oncoproteins with antibody therapy or vaccination. Sci Transl Med. 2011;3:99ra85. doi: 10.1126/scitranslmed.3002296. [DOI] [PubMed] [Google Scholar]
- 50.Noble PW, Chan G, Young MR, Weisbart RH, Hansen JE. Optimizing a lupus autoantibody for targeted cancer therapy. Cancer Res. 2015;75:2285–91. doi: 10.1158/0008-5472.CAN-14-2278. [DOI] [PubMed] [Google Scholar]
- 51.Dao T, Yan S, Veomett N, Pankov D, Zhou L, Korontsvit T, et al. Targeting the intracellular WT1 oncogene product with a therapeutic human antibody. Sci Transl Med. 2013;5:176ra33. doi: 10.1126/scitranslmed.3005661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gondi G, Mysliwietz J, Hulikova A, Jen JP, Swietach P, Kremmer E, et al. Antitumor efficacy of a monoclonal antibody that inhibits the activity of cancer-associated carbonic anhydrase XII. Cancer Res. 2013;73:6494–503. doi: 10.1158/0008-5472.CAN-13-1110. [DOI] [PubMed] [Google Scholar]
- 53.Brand DD, Latham KA, Rosloniec EF. Collagen- induced arthritis. Nature protocols. 2007;2:1269–75. doi: 10.1038/nprot.2007.173. [DOI] [PubMed] [Google Scholar]
- 54.Seo SK, Choi JH, Kim YH, Kang WJ, Park HY, Suh JH, et al. 4–1BB-mediated immunotherapy of rheumatoid arthritis. Nat Med. 2004;10:1088–94. doi: 10.1038/nm1107. [DOI] [PubMed] [Google Scholar]
- 55.Criado G, Simelyte E, Inglis JJ, Essex D, Williams RO. Indoleamine 2,3 dioxygenase-mediated tryptophan catabolism regulates accumulation of Th1/Th17 cells in the joint in collagen- induced arthritis. Arthritis Rheum. 2009;60:1342–51. doi: 10.1002/art.24446. [DOI] [PubMed] [Google Scholar]
- 56.Krebs CF, Steinmetz OM. CD4+ T Cell Fate in Glomerulonephritis: A Tale of Th1, Th17, and Novel Treg Subtypes. Mediators Inflamm. 2016;2016:5393894. doi: 10.1155/2016/5393894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ettinger R, Kuchen S, Lipsky PE. The role of IL-21 in regulating B-cell function in health and disease. Immunol Rev. 2008;223:60–86. doi: 10.1111/j.1600-065X.2008.00631.x. [DOI] [PubMed] [Google Scholar]
- 58.Jang E, Cho SH, Park H, Paik DJ, Kim JM, Youn J. A positive feedback loop of IL-21 signaling provoked by homeostatic CD4+CD25- T cell expansion is essential for the development of arthritis in autoimmune K/BxN mice. J Immunol. 2009;182:4649–56. doi: 10.4049/jimmunol.0804350. [DOI] [PubMed] [Google Scholar]
- 59.Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nature reviews. 2008;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 60.Takai T, Li M, Sylvestre D, Clynes R, Ravetch JV. FcR gamma chain deletion results in pleiotrophic effector cell defects. Cell. 1994;76:519–29. doi: 10.1016/0092-8674(94)90115-5. [DOI] [PubMed] [Google Scholar]
- 61.Amigorena S, Bonnerot C, Drake JR, Choquet D, Hunziker W, Guillet JG, et al. Cytoplasmic domain heterogeneity and functions of IgG Fc receptors in B lymphocytes. Science. 1992;256:1808–12. doi: 10.1126/science.1535455. [DOI] [PubMed] [Google Scholar]
- 62.Muta T, Kurosaki T, Misulovin Z, Sanchez M, Nussenzweig MC, Ravetch JV. A 13-amino-acid motif in the cytoplasmic domain of Fc gamma RIIB modulates B-cell receptor signalling. Nature. 1994;369:340. doi: 10.1038/369340a0. [DOI] [PubMed] [Google Scholar]
- 63.Williams EL, Tutt AL, French RR, Chan HT, Lau B, Penfold CA, et al. Development and characterisation of monoclonal antibodies specific for the murine inhibitory FcgammaRIIB (CD32B) Eur J Immunol. 2012;42:2109–20. doi: 10.1002/eji.201142302. [DOI] [PubMed] [Google Scholar]
- 64.LaBranche TP, Hickman-Brecks CL, Meyer DM, Storer CE, Jesson MI, Shevlin KM, et al. Characterization of the KRN cell transfer model of rheumatoid arthritis (KRN-CTM), a chronic yet synchronized version of the K/BxN mouse. Am J Pathol. 2010;177:1388–96. doi: 10.2353/ajpath.2010.100195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Soliman H, Mediavilla-Varela M, Antonia S. Indoleamine 2,3-dioxygenase: is it an immune suppressor? Cancer journal (Sudbury, Mass) 2010;16:354–9. doi: 10.1097/PPO.0b013e3181eb3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Soliman HH, S A, Sullivan D, Vanahanian N, Link C. Overcoming tumor antigen anergy in human malignancies using the novel indoleamine 2,3-dioxygenase (IDO) enzyme inhibitor, 1-methyl-D-tryptophan (1MT) J Clin Oncol. 2009;27 Abstract 3004. [Google Scholar]
- 67.Soliman HH, Minton SE, Han HS, Ismail-Khan R, Mahipal A, Janssen W, et al. A phase I study of ad.p53 DC vaccine in combination with indoximod in metastatic solid tumors. J Clin Oncol. 2013 suppl abstract 3069. [Google Scholar]
- 68.Soliman HH, Jackson E, Neuger T, Dees EC, Harvey RD, Han H, et al. A first in man phase I trial of the oral immunomodulator, indoximod, combined with docetaxel in patients with metastatic solid tumors. Oncotarget. 2014;5:8136–46. doi: 10.18632/oncotarget.2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Austin CJ, Mailu BM, Maghzal GJ, Sanchez-Perez A, Rahlfs S, Zocher K, et al. Biochemical characteristics and inhibitor selectivity of mouse indoleamine 2,3-dioxygenase-2. Amino Acids. 2010;39:565–78. doi: 10.1007/s00726-010-0475-9. [DOI] [PubMed] [Google Scholar]
- 70.Bakmiwewa SM, Fatokun AA, Tran A, Payne RJ, Hunt NH, Ball HJ. Identification of selective inhibitors of indoleamine 2,3-dioxygenase 2. Bioorg Med Chem Lett. 2012;22:7641–6. doi: 10.1016/j.bmcl.2012.10.010. [DOI] [PubMed] [Google Scholar]
- 71.Wang D, Li Y, Liu Y, Shi G. The use of biologic therapies in the treatment of rheumatoid arthritis. Curr Pharm Biotechnol. 2014;15:542–8. doi: 10.2174/138920101506140910150612. [DOI] [PubMed] [Google Scholar]
- 72.Cohen SB, Emery P, Greenwald MW, Dougados M, Furie RA, Genovese MC, et al. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: Results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006;54:2793–806. doi: 10.1002/art.22025. [DOI] [PubMed] [Google Scholar]
- 73.Lipsky PE, van der Heijde DM, St Clair EW, Furst DE, Breedveld FC, Kalden JR, et al. Infliximab and methotrexate in the treatment of rheumatoid arthritis. Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group. N Engl J Med. 2000;343:1594–602. doi: 10.1056/NEJM200011303432202. [DOI] [PubMed] [Google Scholar]
- 74.Maini RN, Breedveld FC, Kalden JR, Smolen JS, Davis D, Macfarlane JD, et al. Therapeutic efficacy of multiple intravenous infusions of anti-tumor necrosis factor alpha monoclonal antibody combined with low-dose weekly methotrexate in rheumatoid arthritis. Arthritis Rheum. 1998;41:1552–63. doi: 10.1002/1529-0131(199809)41:9<1552::AID-ART5>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 75.Elliott MJ, Maini RN, Feldmann M, Kalden JR, Antoni C, Smolen JS, et al. Randomised double-blind comparison of chimeric monoclonal antibody to tumour necrosis factor alpha (cA2) versus placebo in rheumatoid arthritis. Lancet. 1994;344:1105–10. doi: 10.1016/s0140-6736(94)90628-9. [DOI] [PubMed] [Google Scholar]
- 76.Guo K, Tang JP, Tan CP, Wang H, Zeng Q. Monoclonal antibodies target intracellular PRL phosphatases to inhibit cancer metastases in mice. Cancer Biol Ther. 2008;7:750–7. doi: 10.4161/cbt.7.5.5764. [DOI] [PubMed] [Google Scholar]
- 77.Haynes NM, Hawkins ED, Li M, McLaughlin NM, Hammerling GJ, Schwendener R, et al. CD11c+ dendritic cells and B cells contribute to the tumoricidal activity of anti-DR5 antibody therapy in established tumors. J Immunol. 2010;185:532–41. doi: 10.4049/jimmunol.0903624. [DOI] [PubMed] [Google Scholar]
- 78.Li F, Ravetch JV. Inhibitory Fcgamma receptor engagement drives adjuvant and anti-tumor activities of agonistic CD40 antibodies. Science. 2011;333:1030–4. doi: 10.1126/science.1206954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.White AL, Chan HT, Roghanian A, French RR, Mockridge CI, Tutt AL, et al. Interaction with FcgammaRIIB is critical for the agonistic activity of anti-CD40 monoclonal antibody. J Immunol. 2011;187:1754–63. doi: 10.4049/jimmunol.1101135. [DOI] [PubMed] [Google Scholar]
- 80.Lim SH, Vaughan AT, Ashton-Key M, Williams EL, Dixon SV, Chan HT, et al. Fc gamma receptor IIb on target B cells promotes rituximab internalization and reduces clinical efficacy. Blood. 2011;118:2530–40. doi: 10.1182/blood-2011-01-330357. [DOI] [PubMed] [Google Scholar]
- 81.Beers SA, French RR, Chan HT, Lim SH, Jarrett TC, Vidal RM, et al. Antigenic modulation limits the efficacy of anti-CD20 antibodies: implications for antibody selection. Blood. 2010;115:5191–201. doi: 10.1182/blood-2010-01-263533. [DOI] [PubMed] [Google Scholar]