

Abstract
Store-operated Ca2+ entry (SOCE) is a conserved mechanism of Ca2+ influx that regulates Ca2+ signaling in many cell types. SOCE is activated by depletion of endoplasmic reticulum (ER) Ca2+ stores in response to physiological agonist stimulation. After it was first postulated by J.W. Putney Jr. in 1986, SOCE has been described in a large number of non-excitable cell types including secretory cells of different exocrine glands. Here we discuss the mechanisms by which SOCE controls salt and fluid secretion in exocrine glands, with a special focus on eccrine sweat glands. In sweat glands, SOCE plays an important, non-redundant role in regulating the function of Ca2+-activated Cl− channels (CaCC), Cl− secretion and sweat production. In the absence of key regulators of SOCE such as the CRAC channel pore subunit ORAI1 and its activator STIM1, the Ca2+-activated chloride channel TMEM16A is inactive and fails to secrete Cl−, resulting in anhidrosis in mice and human patients.
Keywords: CaCC, fluid secretion, ORAI1, STIM1, sweat glands, TMEM16A
Graphical Abstract
1. Introduction
Store-operated Ca2+ entry (SOCE) is a conserved mechanism for Ca2+ influx in a large variety of cells [1]. Initially termed capacitative Ca2+ entry, SOCE was postulated 30 years ago in a seminal review published by J.W. Putney Jr. based on observations in non-excitable salivary gland cells and hepatocytes [2]. At the time, Ca2+ influx was known to be critical for secretion by different exocrine glands after stimulation with secretagogues or neurotransmitters such as acetylcholine (ACh) or norepinephrine [3–8]. The concept of SOCE as a specialized mode of Ca2+ influx and means to refill intracellular Ca2+ stores was confirmed by many labs [5, 9–11], but the molecular players including the Ca2+ channel mediating SOCE and the mechanisms of SOCE activation remained elusive. The prototypical SOCE channel is the Ca2+ release-activated Ca2+ (CRAC) channel, which opens after depletion of Ca2+ from intracellular stores, mainly the ER. Active store depletion is induced by receptor agonists or passively by drugs that cause leakage of Ca2+ from the ER or prevent Ca2+ reuptake by sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) pumps. Agonists that trigger SOCE include ligands of G protein-coupled receptors (GPCRs) and antigens that bind to immunoreceptors in the plasma membrane (PM). Receptor binding results in activation of phospholipase C, production of inositol 1,4,5-trisphosphate (IP3), and opening of IP3 receptors (IP3Rs), which are non-selective Ca2+ channels in the ER membrane that mediate the release of Ca2+ from that organelle [12]. How depletion of Ca2+ from the ER activates SOCE remained elusive until the discovery of stromal interaction molecule 1 (STIM1) and STIM2 [13–15]. Both homologues are single pass transmembrane proteins located in the ER. Their ER luminal N-termini contain EF hand motifs that are bound by Ca2+ in non-stimulated cells. After cell stimulation and Ca2+ store depletion, Ca2+ dissociates from the EF hands, resulting in extensive conformational changes in STIM1 and STIM2 and their translocation to ER-PM junctions. There they bind to and open the CRAC channel, which mediates sustained Ca2+ influx from the extracellular space [1, 16]. CRAC currents were first recorded in immune cells and subsequently in many other cell types including secretory cells, but their molecular composition remained enigmatic until the discovery of ORAI1 as their pore-forming subunit [17–20]. ORAI1 and its homologues ORAI2 and ORAI3 are tetraspanning PM proteins that are unrelated to other ion channels. They form hexameric channel complexes that have very low unitary conductance but are highly Ca2+ selective [21–23]. Genetic deletion of ORAI1, STIM1 and/or STIM2 in mice and other model organisms and mutations in human ORAI1 and STIM1 genes that result in CRAC channelopathy have shed important light on the physiological role of SOCE [1, 24–27]. In this review, we discuss the role of SOCE in exocrine gland function and our recent findings in eccrine sweat glands, in which SOCE regulates Cl− secretion and sweat production.
2. Basic mechanisms of salt and fluid secretion
Exocrine glands produce and secrete a large variety of fluids that are important for digestion, lubrication, lactation and thermoregulation [28–32]. Examples of exocrine glands include sweat, salivary, lacrimal, and mammary glands. These glands are composed of layers of secretory epithelial cells that absorb water, ions and hydrophilic macromolecules from the blood, and secrete them onto epithelial surfaces through a duct. The mechanism of salt and fluid secretion by exocrine glands is based on the active transcellular and paracellular transport of these substances from the basolateral to the apical membrane of epithelial secretory cells. Active transcellular transport requires the function of ion channels and transporters that allow the permeation of charged ions and hydrophilic molecules across the PM of polarized epithelial cells (Figure 1). Cl− secretion across the luminal membrane of acinar cells generates an electrical gradient that drives transepithelial Na+ transport, and this salt enrichment in the lumen of secretory gland acini establishes an osmotic driving force for water flow via paracellular and/or transcellular routes [33–35]. The ion composition of the isotonic primary secretion is further modified by the ductal epithelium, which can reabsorb Na+ and Cl− ions and secrete HCO3− dependent on the type of gland [35–38]. Salivary gland ducts both secrete (HCO3− and K+) and reabsorb (Na+ and Cl−) ions with net reabsorption to give a hypotonic final saliva as the water permeability of the duct is very low. In contrast in the pancreas, the vast majority of the secreted fluid is a result of ductal function due to Cl−/HCO3− exchange resulting in Na+ and HCO3− rich secretion [34, 38].
Figure 1. Basic mechanism of salt and fluid secretion in exocrine glands.

(A) Acinar cells of exocrine glands secrete Cl− to the acinar lumen, which generates an electrical gradient that drives paracellular Na+ transport. The hypertonic NaCl solution drives the osmotic transport of water via trans- and paracellular mechanisms. The isotonic primary fluid is subsequently modified, for instance by Na+ and Cl− reabsorption and bicarbonate secretion, as it flows through the ducts before is secreted as a hypotonic solution. (B) Main signaling pathways that govern salt and fluid secretion in epithelial cells. The activation of G protein coupled receptors (GPCRs) at the basolateral membrane triggers Ca2+ signaling and activation of Ca2+-activated Cl− channels (CaCCs) and Cl− secretion as well as production of cAMP, protein kinase A (PKA) function and activation of cystic fibrosis transmembrane conductance regulator (CFTR), resulting in Cl− secretion. For details see text.
Fluid and salt secretion in epithelial cells is triggered by many pathways including cyclic adenosine monophosphate (cAMP) and phosphatidylinositol-Ca2+ signaling (Figure 1). The binding of extracellular ligands to GPCRs such as the β-adrenoreceptor and some hormone receptors triggers adenylyl cyclase activation and the generation of cAMP from ATP. Increasing levels of cAMP activate protein kinase A (PKA), which in turn phosphorylates and gates the cystic fibrosis transmembrane conductance regulator (CFTR) anion channel [39, 40]. CFTR activation leads to Cl− secretion by secretory epithelial cells in salivary glands, the pancreas and the lung as well as Cl− reabsorption and secretion by ductal cells [41–47]. The phosphatidylinositol-Ca2+ pathway is activated by extracellular ligands that bind GPCRs (e.g. cholinergic and α-adrenergic receptors) located at the basolateral PM of epithelial cells. GPCR engagement activates phospholipase C and results in the subsequent formation of diacylglycerol (DAG) and IP3. The diffusion of IP3 in the cytosol activates IP3R channels triggering the release of Ca2+ from ER stores. Ca2+ store depletion by IP3Rs determines the site of initiation and the pattern of Ca2+ signals in the cell, and it has been implicated in fluid secretion in the liver, pancreas, salivary and sweat glands [33, 48–52]. Ca2+ signals resulting from store release can potentially stimulate Ca2+-activated Cl− channels (CaCCs) in the PM that mediate the secretion of Cl− from the cell. However, the finite amount of Ca2+ in the ER is likely not sufficient to maintain sustained CaCC activation required for the secretion of large amounts of fluid by secretory epithelial cells. As discussed in more detail further below, we recently demonstrated that Ca2+ influx via SOCE is necessary for sustained CaCC activation, Cl− secretion and sweat production in eccrine sweat glands, whereas Ca2+ release from ER stores alone was not sufficient to mediate this process [53]. These findings are consistent with reports showing that Ca2+ influx from the extracellular space is required for fluid secretion by different exocrine glands [5, 7, 54].
3. Mechanism of salt and fluid secretion in eccrine sweat glands
Eccrine sweat glands are one of the simplest exocrine gland systems [46]. Each eccrine sweat gland unit consists of two portions, the secretory coil and the duct, which differ in morphology and function (Figure 2A) [55, 56]. The secretory coil contains acinar epithelial cells (that can be divided further into clear and dark cells) surrounded by myoepithelial cells, whereas the duct is composed by two layers of cuboidal epithelium [55]. The main physiological function of eccrine sweat glands in humans is thermoregulation. Millions of eccrine sweat glands are spread over almost the entire skin surface to secrete sweat, which evaporates and generates the cooling effect. Sweat secretion follows the basic mechanism of salt and fluid secretion described in section 2. Sympathetic nerve terminals surrounding the secretory coil of sweat glands release the neurotransmitter ACh, which binds to muscarinic ACh receptors expressed in secretory epithelial cells [57, 58]. ACh triggers the production of IP3, Ca2+ store release and SOCE, which is required for CaCC activation and Cl− secretion (see below) [53]. To generate the vectorial transport of Cl− across the polarized secretory sweat gland epithelial cells, Cl− influx is mediated by the Na+-K+-2Cl− co-transporter NKCC1 located in the basolateral membrane [59–62]. Other channels and pumps in the basolateral membrane are required to support Cl− transport by NKCC1. These include the Na+/K+ ATPase pump [62, 63] and Ca2+-activated K+ channels [29, 55, 64, 65] that establish the membrane potential and maintain the driving force for Cl− secretion. The main channels mediating Cl− secretion at the luminal membrane of sweat gland acinar cells are CaCCs [53, 66, 67]. Recent studies have shed light on the molecular nature of the CaCCs in murine and human sweat glands [53, 61, 66, 68, 69]. Bestrophin 2 (BEST2) is a CaCC that is necessary for sweat secretion as Best2−/− mice showed a complete defect in spontaneous sweat production [61]. BEST2 expression was shown to be restricted to dark cells in murine eccrine sweat glands [61], whose functional role, unlike that of clear cells, in sweat secretion is not well characterized [29, 55, 70–72]. Whether BEST2 plays a role in mediating sweat secretion in response to cholinergic stimulation in mice or in humans remains to be investigated. In the human eccrine sweat gland cell line NCL-SG3, another CaCC, TMEM16A (also known as anoctamin 1, ANO1) was shown to be expressed at the transcriptional level [66]. Overexpression of the canonical TMEM16A isoform or one of its splice variants in NCL-SG3 cells mediated Cl− secretion, suggesting that TMEM16A may also contribute to Cl− secretion in primary human sweat glands [66]. We confirmed expression of TMEM16A mRNA and protein in human NCL-SG3 cells and primary sweat gland cells in skin biopsies [53]. In addition, TMEM16A was also expressed in primary murine sweat glands [53]. Importantly, we demonstrate that TMEM16A is essential for Cl− secretion in human eccrine sweat gland cells. Stable deletion of TMEM16A expression in NCL-SG3 cells by shRNAs abolished Cl− secretion and Cl− currents activated following direct intracellular administration of Ca2+ via the patch pipette or induction of SOCE by stimulation of cells with ionomycin or the PAR2 agonist trypsin. By contrast, untransduced or shBEST2 transduced NCL-SG3 cells showed robust CaCC currents, which had properties consistent with those described for TMEM16A [53, 73, 74]. While TMEM16A clearly mediates Cl− currents and Cl− secretion in human sweat gland cells, its role in primary eccrine glands of mice and humans remains to be further studied. To date, no mutations in TMEM16A (or BEST2) have been reported in patients with anhidrosis.
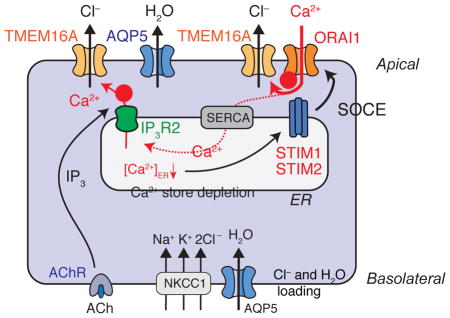
Figure 2. Mechanisms of Ca2+-dependent Cl− and fluid secretion in eccrine sweat glands.

(A) Morphology of an eccrine sweat gland in the skin. Modified from [56]. The enlargement on the right shows a crossection through the secretory coil portion of an eccrine sweat gland. CC, clear cells; DC, dark cells; MC, myoepithelial cells. (B) Schematic representation of Ca2+ signaling in eccrine sweat glands after cholinergic stimulation. ACh binding to the ACh receptor (AChR) induces IP3-mediated release of Ca2+ from ER stores through IP3Rs. Ca2+ released from the ER may directly activate the Ca2+-activated Cl− channel TMEM16A and Cl− secretion (indicated by ➊). Ca2+ release results in a decrease of the [Ca2+] in the ER and activation of stromal interaction molecule 1 (STIM1) and STIM2, which bind to and open ORAI1 channels in the plasma membrane. The resulting store-operated Ca2+ entry (SOCE) is required for sustained activation of TMEM16A and Cl− secretion (indicated by ➋). The dotted line indicates that SOCE mediates refilling of ER Ca2+ stores via Ca2+ uptake through sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) pumps. Aquaporin 5 (AQP5) mediates water secretion. NKCC1, Na+-K+-2Cl− co-transporter. For details see text.
4. Ca2+ release from ER stores and SOCE in eccrine sweat gland function
While Cl− secretion in NCL-SG3 cells is well known to be dependent on increases in cytoplasmic [Ca2+] [66, 67, 75], the source of Ca2+ required for CaCC activation is unclear. Early studies have shown that Ca2+ influx from the extracellular space is essential for sweating in response to cholinergic and α-adrenergic stimulation [7]. The Ca2+ channels mediating this influx in secretory sweat gland cells, however, remained unknown [76]. Alternatively, Ca2+ release from intracellular stores may activate CaCCs and Cl− secretion. However, the Ca2+ content of ER stores is finite and, as discussed above, may not be sufficient for the sustained activation of CaCCs and Cl− secretion. Arguing in favor of an important role of ER stores as the source of Ca2+ for CaCC activation is the hypohidrotic phenotype of patients with a missense mutation in the ITPR2 gene, which encodes IP3R2, and Itpr2−/− mice [51]. Sweat gland cells isolated from Itpr2−/− mice had reduced ACh-induced Ca2+ signals and sweating of Itpr2−/− mice after cholinergic stimulation was significantly impaired [51]. IP3Rs were shown to colocalize with CaCCs in several cell types [77–80] and it therefore seemed plausible that Ca2+ release from the ER through IP3R2 mediates CaCC activation, Cl− secretion and sweat production. Importantly, however, Ca2+ release via IP3R2 not only increases intracellular [Ca2+] but also decreases [Ca2+] in the ER, which activates store-operated CRAC channels. Genetic deletion or mutation of IP3R2 in mice or patients, respectively, will therefore also impair SOCE. We showed that human patients with loss-of-function mutations in ORAI1 or STIM1 that abolish CRAC channel function and SOCE are anhidrotic despite the presence of eccrine sweat gland cells in their dermis [53]. Likewise, mice with conditional deletion of Orai1 or both Stim1 and Stim2 in ectoderm-derived tissues fail to sweat upon cholinergic stimulation, although sweat glands are present and express TMEM16A and BEST2 channels [53]. Furthermore, primary sweat gland cells of Orai1-deficient mice and human NCL-SG3 sweat gland cells lacking ORAI1 failed to secrete Cl− due to abolished SOCE. ORAI1-deficient NCL-SG3 cells also lacked CaCC currents activated by ionomycin stimulation and the PAR2 agonist trypsin [53]. These data demonstrate that SOCE mediated by CRAC channels is required for activation of CaCCs, Cl− secretion and ultimately sweat production by eccrine sweat glands. They also argue against Ca2+ release from the ER as a sufficient Ca2+ signal for CaCC activation and Cl− secretion. This conclusion is supported by the normal Ca2+ content of ER stores in primary sweat gland cells from mice lacking Orai1 or both Stim1 and Stim2, and cells from patients with mutations in ORAI1 or STIM1 genes [53]. Depletion of ER Ca2+ stores in ORAI1-deficient cells by ionomycin or trypsin stimulation resulted in a transient increase in intracellular [Ca2+], which was not sufficient for sustained Cl− secretion and activation of CaCC currents. How can the anhidrosis in IP3R2-deficient patients and mice on one hand, and ORAI1 and STIM1-deficient patients and mice on the other be reconciled? While SOCE is clearly essential for activation of CaCCs and Cl− secretion in human and mouse sweat glands, it is not fully understood whether Ca2+ entering via ORAI1 directly activates CaCCs, facilitated by colocalization of ORAI1 channels with CaCCs in the plasma membrane, or whether SOCE is primarily needed to refill ER Ca2+ stores (Figure 2B). In the latter model, Ca2+ release from the ER through IP3Rs would provide the Ca2+ signal required for CaCC activation and Cl− secretion. IP3Rs have been shown to colocalize with CaCCs in other cell types such as Xenopus oocytes and nociceptive sensory neurons and may therefore create microdomains with enhanced cytoplasmic [Ca2+] that mediate CaCC activation [77, 79–81]. Such a model is supported by studies in Xenopus oocytes that suggest that Ca2+ entering the cytosol through CRAC channels is taken up by the ER via SERCA pumps and released by IP3Rs at different sites to activate CaCCs [77]. Whether a similar mechanism plays a role in eccrine sweat gland cells remains to be investigated. Taken together, SOCE mediated by STIM1/ORAI1 either directly provides the Ca2+ signal that is required for CaCC activation, Cl− secretion and sweat production or allows cells to refill their Ca2+ stores, the depletion of which via IP3R2 results in Ca2+ dependent activation of CaCCs (Figure 2B) [53, 77].
5. Effects of mutations in human genes that impair Ca2+ release and SOCE on sweat gland function
Human patients with CRAC channelopathy due to autosomal recessive loss-of-function and null mutations in ORAI1 and STIM1 genes present with a complex clinical syndrome that is characterized by anhidrotic ectodermal dysplasia (EDA) with severe immunodeficiency (ID), autoimmunity and skeletal myopathy [24]. The immunodeficiency is defined by recurrent, life-threatening bacterial and viral infections. To date, patients with 15 different mutations in ORAI1 and STIM1 have been observed that either abolish protein expression or impair ORAI1 or STIM1 function. In both cases, SOCE is severely impaired resulting in similar clinical phenotypes [53]. Ectodermal dysplasia in ORAI1 and STIM1-deficient patients is defined by defects in tooth development, specifically the calcification of dental enamel (amelogenesis imperfecta) and anhidrosis [24, 25, 53]. All patients with CRAC channelopathy reported to date uniformly have impaired sweat production when tested by pilocarpine iontophoresis [53, 82–85]. In many of these patients, anhidrosis or reduced sweat production (hypohidrosis) is apparent clinically and results in elevated body temperatures (hyperthermia), especially at high ambient temperature in hot summer months, and even in the absence of infections caused by the patients’ immunodeficiency. It is noteworthy, that CRAC channel deficiency does not result in apparent development defects of ectodermal structures besides dental enamel that occurs in other forms of EDA [86, 87]. Sweat glands were present in the dermis of ORAI1 and STIM1-deficient patients and had a normal morphology except reduced diameters of the sweat gland lumen that is presumably due to abolished sweat production [53]. CRAC channelopathy thus represents a new form of EDA associated with immunodeficiency (EDA-ID) that has a distinctive pathophysiology compared to another form of EDA-ID caused by mutations in IKBKG and NFKBIA genes, encoding IKKγ and Iκ-Bα, respectively. Both proteins are essential components of the canonical NF-κB signaling pathway and their mutation results in impaired sweat gland development as the cause of anhidrosis in those patients [88–91].
Isolated hypohidrosis without other clinical features of EDA was observed in patients homozygous for a novel missense mutation in ITPR2, c.7492G>A, which results in a glycine-to-serine (p.G2498S) substitution [51]. All five affected family members failed to sweat when exposed to heat (45°C) and suffered from severe heat intolerance. Skin biopsies showed normal morphology and number of eccrine glands. The ITPR2 mutation was not associated with other symptoms characteristic of EDA such as skin, hair or tooth defects or immunodeficiency as in patients with CRAC channelopathy. The milder clinical phenotype in ITPR2-deficient patients (i.e. isolated hypohidrosis) compared to ORAI1 or STIM1-deficient patients (anhidrosis and additional symptoms of EDA-ID) may be due to, for instance, the more moderate reduction in Ca2+ signals or compensatory effects by IP3R1 and/or IP3R3.
6. SOCE functions in other exocrine glands
Besides eccrine sweat glands, CRAC currents and SOCE have been reported in many exocrine gland systems including pancreas, lacrimal, salivary and mammary glands where they regulate cell function [28, 33, 92–96]. For instance, deletion of Orai1 in mice abolishes SOCE in lacrimal glands and results in diminished lacrimal fluid secretion in response to muscarinic-cholinergic stimulation [28, 94]. In mammary glands, impaired SOCE in Orai1−/− mice abolishes lactation due to impaired milk ejection from mammary gland secretory alveoli, which was caused by decreased myoepithelial cell contraction [93]. In salivary glands, SOCE mediated by ORAI1 has been proposed to facilitate the recruitment of the non-selective transient receptor potential channel TRPC1 to the PM, where it amplifies the Ca2+ signal initiated by SOCE [92]. Knockdown of ORAI1 or STIM1 in human salivary gland adenocarcinoma HSG cells abolishes SOCE, whereas knockdown of TRPC1 impaired Ca2+ influx by only ~ 50% [97–99]. Isolated salivary gland acinar cells from Trpc1−/− mice, however, have significantly reduced Ca2+ influx upon agonist stimulation, likely accounting for the reduction in neurotransmitter-induced salivary fluid secretion in Trpc1−/− mice [100]. In salivary gland cells from patients with Sjoegren’s syndrome, the loss of salivary fluid secretion was associated with reduced expression of IP3R2 and IP3R3, which mediate Ca2+ release from ER stores and thus indirectly affect SOCE [52]. A direct role for ORAI and STIM proteins in salivary gland function of mice and humans is likely but requires further study. In pancreatic acinar cells, ORAI1 was found to be expressed at the apical membrane where it colocalized with IP3Rs [95, 101], and – in one study – at the basolateral membrane where it colocalized with STIM1 [95]. A recently developed inhibitor of ORAI1 abolished SOCE in pancreatic acinar cells in response to stimulation with thapsigargin [102, 103], cholecystokinin 8 (CCK8) [103] and the bile acid taurolithocholic acid 3-sulfate (TLCS). The inhibitor also reduced TLCS-induced cell death, indicating that ORAI1 mediates SOCE in pancreatic acinar cells [102, 103]. Importantly, SOCE inhibition attenuated the severity of acute pancreatitis in three different mouse models, strongly suggesting that ORAI1 is responsible for the pathological Ca2+ influx that leads to Ca2+ overload of pancreatic acinar cells and tissue injury [103].
Although SOCE have been reported in many exocrine gland systems as discussed above, the only symptom related to exocrine gland dysfunction in CRAC channel-deficient patients is anhidrosis, whereas the function of the exocrine pancreas, salivary and lacrimal glands appears to be normal [24, 25]. This discrepancy may be due to species differences (as many physiological studies of SOCE in exocrine glands were conducted in mice), redundancies among ORAI and STIM isoforms, and/or compensation for impaired SOCE by other Ca2+ channels. It is also possible that Ca2+ independent Cl− channels and transport mechanisms compensate for the impaired activation of CaCCs and mediate Cl− secretion in CRAC channel-deficient secretory gland cells [104].
Conclusions
SOCE is a unique and specialized mechanism for Ca2+ influx in many cell types including secretory cells. In exocrine glands, secretory epithelial cells use intracellular Ca2+ signals to control salt and fluid secretion across the luminal membrane. SOCE mediated by ORAI and STIM proteins provides secretory cells with a sustained increase in intracellular [Ca2+] required to activate CaCCs and to induce Cl− and fluid secretion. In eccrine sweat glands, SOCE is indispensable for CaCC function, Cl− secretion and sweat production as demonstrated by genetic deletion or mutation of components of the SOCE pathway (i.e. IP3R2, ORAI1, STIM1, and STIM2) in humans and mice that result in sweat gland dysfunction and anhidrosis. Other exocrine glands also use SOCE for Ca2+ influx and genetic deletion or inhibition of ORAI1 in mice significantly impairs lacrimal gland function and prevents toxic Ca2+ influx and overload in pancreatic acinar cells. Inhibition of SOCE for the treatment of acute pancreatitis or other disorders caused by dysfunction of exocrine glands is attractive but requires further studies regarding the pathophysiological role of SOCE in these tissues.
Highlights.
Store-operated Ca2+ entry (SOCE) is required for sweat gland function in humans and mice
Defects in ORAI1 and STIM1 abolish
SOCE and cause anhidrosis SOCE controls activation of Ca2+-activated Cl− channels (CaCC)
CaCC function in human sweat gland cells is mediated by TMEM16A
Acknowledgments
This work was funded by National Institutes of Health (NIH) grant AI097302 to S.F. and a postdoctoral fellowship from the Alfonso Martin Escudero Foundation to A.R.C. The authors thank Dr. David Yule for critically reading the manuscript.
Footnotes
Conflict of interest statement: S.F. is a cofounder of Calcimedica; A.R.C. declares no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Prakriya M, Lewis RS. Store-Operated Calcium Channels. Physiological reviews. 2015;95:1383–1436. doi: 10.1152/physrev.00020.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Putney JW., Jr A model for receptor-regulated calcium entry. Cell calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- 3.Selinger Z, Naim E. The effect of calcium on amylase secretion by rat parotid slices. Biochim Biophys Acta. 1970;203:335–337. doi: 10.1016/0005-2736(70)90148-3. [DOI] [PubMed] [Google Scholar]
- 4.Herman G, Rossignol B. Regulation of protein secretion and metabolism in rat salivary glands. Effects of norepinephrine and carbachol on the glycogenolysis in submaxillary glands. Eur J Biochem. 1975;55:105–110. doi: 10.1111/j.1432-1033.1975.tb02142.x. [DOI] [PubMed] [Google Scholar]
- 5.Putney JW., Jr Muscarinic, alpha-adrenergic and peptide receptors regulate the same calcium influx sites in the parotid gland. The Journal of physiology. 1977;268:139–149. doi: 10.1113/jphysiol.1977.sp011851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prompt CA, Quinton PM. Functions of calcium in sweat secretion. Nature. 1978;272:171–172. doi: 10.1038/272171a0. [DOI] [PubMed] [Google Scholar]
- 7.Sato K, Sato F. Role of calcium in cholinergic and adrenergic mechanisms of eccrine sweat secretion. The American journal of physiology. 1981;241:C113–120. doi: 10.1152/ajpcell.1981.241.3.C113. [DOI] [PubMed] [Google Scholar]
- 8.Mauduit P, Herman G, Rossignol B. Effect of trifluoperazine on 3H-labeled protein secretion induced by pentoxifylline, cholinergic or adrenergic agonists in rat lacrimal gland. A possible role of calmodulin? FEBS Lett. 1983;152:207–211. doi: 10.1016/0014-5793(83)80381-0. [DOI] [PubMed] [Google Scholar]
- 9.Bird GS, Rossier MF, Hughes AR, Shears SB, Armstrong DL, Putney JW., Jr Activation of Ca2+ entry into acinar cells by a non-phosphorylatable inositol trisphosphate. Nature. 1991;352:162–165. doi: 10.1038/352162a0. [DOI] [PubMed] [Google Scholar]
- 10.Casteels R, Droogmans G. Exchange characteristics of the noradrenaline-sensitive calcium store in vascular smooth muscle cells or rabbit ear artery. The Journal of physiology. 1981;317:263–279. doi: 10.1113/jphysiol.1981.sp013824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parod RJ, Putney JW., Jr The role of calcium in the receptor mediated control of potassium permeability in the rat lacrimal gland. The Journal of physiology. 1978;281:371–381. doi: 10.1113/jphysiol.1978.sp012428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alzayady KJ, Wang L, Chandrasekhar R, Wagner LE, 2nd, Van Petegem F, Yule DI. Defining the stoichiometry of inositol 1,4,5-trisphosphate binding required to initiate Ca2+ release. Science signaling. 2016;9:ra35. doi: 10.1126/scisignal.aad6281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, Jr, Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Current biology: CB. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Velicelebi G, Stauderman KA. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169:435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shaw PJ, Qu B, Hoth M, Feske S. Molecular regulation of CRAC channels and their role in lymphocyte function. Cellular and molecular life sciences: CMLS. 2013;70:2637–2656. doi: 10.1007/s00018-012-1175-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 18.Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443:230–233. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- 19.Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan-Huberson M, Kraft S, Turner H, Fleig A, Penner R, Kinet JP. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 2006;312:1220–1223. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang SL, Yeromin AV, Zhang XH, Yu Y, Safrina O, Penna A, Roos J, Stauderman KA, Cahalan MD. Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:9357–9362. doi: 10.1073/pnas.0603161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cai X, Zhou Y, Nwokonko RM, Loktionova NA, Wang X, Xin P, Trebak M, Wang Y, Gill DL. The Orai1 Store-operated Calcium Channel Functions as a Hexamer. The Journal of biological chemistry. 2016 doi: 10.1074/jbc.M116.758813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hou X, Pedi L, Diver MM, Long SB. Crystal structure of the calcium release-activated calcium channel Orai. Science. 2012;338:1308–1313. doi: 10.1126/science.1228757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yen M, Lokteva LA, Lewis RS. Functional Analysis of Orai1 Concatemers Supports a Hexameric Stoichiometry for the CRAC Channel. Biophys J. 2016;111:1897–1907. doi: 10.1016/j.bpj.2016.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lacruz RS, Feske S. Diseases caused by mutations in ORAI1 and STIM1. Annals of the New York Academy of Sciences. 2015;1356:45–79. doi: 10.1111/nyas.12938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feske S. CRAC channelopathies. Pflugers Archiv: European journal of physiology. 2010;460:417–435. doi: 10.1007/s00424-009-0777-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lewis RS. Store-operated calcium channels: new perspectives on mechanism and function. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a003970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shaw PJ, Feske S. Physiological and pathophysiological functions of SOCE in the immune system. Front Biosci (Elite Ed) 2012;4:2253–2268. doi: 10.2741/540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Putney JW, Bird GS. Calcium signaling in lacrimal glands. Cell calcium. 2014;55:290–296. doi: 10.1016/j.ceca.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cui CY, Schlessinger D. Eccrine sweat gland development and sweat secretion. Experimental dermatology. 2015;24:644–650. doi: 10.1111/exd.12773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miletich I. Introduction to salivary glands: structure, function and embryonic development. Front Oral Biol. 2010;14:1–20. doi: 10.1159/000313703. [DOI] [PubMed] [Google Scholar]
- 31.Pallagi P, Hegyi P, Rakonczay Z., Jr The Physiology and Pathophysiology of Pancreatic Ductal Secretion: The Background for Clinicians. Pancreas. 2015;44:1211–1233. doi: 10.1097/MPA.0000000000000421. [DOI] [PubMed] [Google Scholar]
- 32.Peng Y, Cui X, Liu Y, Li Y, Liu J, Cheng B. Systematic review focusing on the excretion and protection roles of sweat in the skin. Dermatology. 2014;228:115–120. doi: 10.1159/000357524. [DOI] [PubMed] [Google Scholar]
- 33.Ambudkar IS. Ca(2)(+) signaling and regulation of fluid secretion in salivary gland acinar cells. Cell calcium. 2014;55:297–305. doi: 10.1016/j.ceca.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hong JH, Park S, Shcheynikov N, Muallem S. Mechanism and synergism in epithelial fluid and electrolyte secretion. Pflugers Archiv: European journal of physiology. 2014;466:1487–1499. doi: 10.1007/s00424-013-1390-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee RJ, Foskett JK. Ca(2)(+) signaling and fluid secretion by secretory cells of the airway epithelium. Cell calcium. 2014;55:325–336. doi: 10.1016/j.ceca.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 36.Concepcion AR, Lopez M, Ardura-Fabregat A, Medina JF. Role of AE2 for pHi regulation in biliary epithelial cells. Front Physiol. 2013;4:413. doi: 10.3389/fphys.2013.00413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jung J, Lee MG. Role of calcium signaling in epithelial bicarbonate secretion. Cell calcium. 2014;55:376–384. doi: 10.1016/j.ceca.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 38.Lee MG, Ohana E, Park HW, Yang D, Muallem S. Molecular mechanism of pancreatic and salivary gland fluid and HCO3 secretion. Physiological reviews. 2012;92:39–74. doi: 10.1152/physrev.00011.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tabcharani JA, Chang XB, Riordan JR, Hanrahan JW. Phosphorylation-regulated Cl− channel in CHO cells stably expressing the cystic fibrosis gene. Nature. 1991;352:628–631. doi: 10.1038/352628a0. [DOI] [PubMed] [Google Scholar]
- 40.Howell LD, Borchardt R, Kole J, Kaz AM, Randak C, Cohn JA. Protein kinase A regulates ATP hydrolysis and dimerization by a CFTR (cystic fibrosis transmembrane conductance regulator) domain. The Biochemical journal. 2004;378:151–159. doi: 10.1042/BJ20021428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zabner J, Smith JJ, Karp PH, Widdicombe JH, Welsh MJ. Loss of CFTR chloride channels alters salt absorption by cystic fibrosis airway epithelia in vitro. Mol Cell. 1998;2:397–403. doi: 10.1016/s1097-2765(00)80284-1. [DOI] [PubMed] [Google Scholar]
- 42.Reddy MM, Quinton PM. ENaC activity requires CFTR channel function independently of phosphorylation in sweat duct. J Membr Biol. 2005;207:23–33. doi: 10.1007/s00232-005-0798-8. [DOI] [PubMed] [Google Scholar]
- 43.Banales JM, Prieto J, Medina JF. Cholangiocyte anion exchange and biliary bicarbonate excretion. World J Gastroenterol. 2006;12:3496–3511. doi: 10.3748/wjg.v12.i22.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang D, Shcheynikov N, Zeng W, Ohana E, So I, Ando H, Mizutani A, Mikoshiba K, Muallem S. IRBIT coordinates epithelial fluid and HCO3- secretion by stimulating the transporters pNBC1 and CFTR in the murine pancreatic duct. The Journal of clinical investigation. 2009;119:193–202. doi: 10.1172/JCI36983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frizzell RA, Hanrahan JW. Physiology of epithelial chloride and fluid secretion. Cold Spring Harbor perspectives in medicine. 2012;2:a009563. doi: 10.1101/cshperspect.a009563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reddy MM, Stutts MJ. Status of fluid and electrolyte absorption in cystic fibrosis. Cold Spring Harbor perspectives in medicine. 2013;3:a009555. doi: 10.1101/cshperspect.a009555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zeng W, Lee MG, Yan M, Diaz J, Benjamin I, Marino CR, Kopito R, Freedman S, Cotton C, Muallem S, Thomas P. Immuno and functional characterization of CFTR in submandibular and pancreatic acinar and duct cells. The American journal of physiology. 1997;273:C442–455. doi: 10.1152/ajpcell.1997.273.2.C442. [DOI] [PubMed] [Google Scholar]
- 48.Petersen OH, Tepikin AV. Polarized calcium signaling in exocrine gland cells. Annu Rev Physiol. 2008;70:273–299. doi: 10.1146/annurev.physiol.70.113006.100618. [DOI] [PubMed] [Google Scholar]
- 49.Hirata K, Pusl T, O’Neill AF, Dranoff JA, Nathanson MH. The type II inositol 1,4,5-trisphosphate receptor can trigger Ca2+ waves in rat hepatocytes. Gastroenterology. 2002;122:1088–1100. doi: 10.1053/gast.2002.32363. [DOI] [PubMed] [Google Scholar]
- 50.Orabi AI, Luo Y, Ahmad MU, Shah AU, Mannan Z, Wang D, Sarwar S, Muili KA, Shugrue C, Kolodecik TR, Singh VP, Lowe ME, Thrower E, Chen J, Husain SZ. IP3 receptor type 2 deficiency is associated with a secretory defect in the pancreatic acinar cell and an accumulation of zymogen granules. PLoS One. 2012;7:e48465. doi: 10.1371/journal.pone.0048465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klar J, Hisatsune C, Baig SM, Tariq M, Johansson AC, Rasool M, Malik NA, Ameur A, Sugiura K, Feuk L, Mikoshiba K, Dahl N. Abolished InsP3R2 function inhibits sweat secretion in both humans and mice. The Journal of clinical investigation. 2014;124:4773–4780. doi: 10.1172/JCI70720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Teos LY, Zhang Y, Cotrim AP, Swaim W, Won JH, Ambrus J, Shen L, Bebris L, Grisius M, Jang SI, Yule DI, Ambudkar IS, Alevizos I. IP3R deficit underlies loss of salivary fluid secretion in Sjogren’s Syndrome. Scientific reports. 2015;5:13953. doi: 10.1038/srep13953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Concepcion AR, Vaeth M, Wagner LE, 2nd, Eckstein M, Hecht L, Yang J, Crottes D, Seidl M, Shin HP, Weidinger C, Cameron S, Turvey SE, Issekutz T, Meyts I, Lacruz RS, Cuk M, Yule DI, Feske S. Store-operated Ca2+ entry regulates Ca2+-activated chloride channels and eccrine sweat gland function. The Journal of clinical investigation. 2016;126:4303–4318. doi: 10.1172/JCI89056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Melvin JE, Koek L, Zhang GH. A capacitative Ca2+ influx is required for sustained fluid secretion in sublingual mucous acini. The American journal of physiology. 1991;261:G1043–1050. doi: 10.1152/ajpgi.1991.261.6.G1043. [DOI] [PubMed] [Google Scholar]
- 55.Sato K, Kang WH, Saga K, Sato KT. Biology of sweat glands and their disorders. I. Normal sweat gland function. Journal of the American Academy of Dermatology. 1989;20:537–563. doi: 10.1016/s0190-9622(89)70063-3. [DOI] [PubMed] [Google Scholar]
- 56.Kurata R, Futaki S, Nakano I, Tanemura A, Murota H, Katayama I, Sekiguchi K. Isolation and characterization of sweat gland myoepithelial cells from human skin. Cell Struct Funct. 2014;39:101–112. doi: 10.1247/csf.14009. [DOI] [PubMed] [Google Scholar]
- 57.Torres NE, Zollman PJ, Low PA. Characterization of muscarinic receptor subtype of rat eccrine sweat gland by autoradiography. Brain research. 1991;550:129–132. doi: 10.1016/0006-8993(91)90415-r. [DOI] [PubMed] [Google Scholar]
- 58.Shibasaki M, Wilson TE, Crandall CG. Neural control and mechanisms of eccrine sweating during heat stress and exercise. Journal of applied physiology. 2006;100:1692–1701. doi: 10.1152/japplphysiol.01124.2005. [DOI] [PubMed] [Google Scholar]
- 59.Sato F, Sato K. Effect of periglandular ionic composition and transport inhibitors on rhesus monkey eccrine sweat gland function in vitro. The Journal of physiology. 1987;393:195–212. doi: 10.1113/jphysiol.1987.sp016819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nejsum LN, Praetorius J, Nielsen S. NKCC1 and NHE1 are abundantly expressed in the basolateral plasma membrane of secretory coil cells in rat, mouse, and human sweat glands. American journal of physiology Cell physiology. 2005;289:C333–340. doi: 10.1152/ajpcell.00228.2004. [DOI] [PubMed] [Google Scholar]
- 61.Cui CY, Childress V, Piao Y, Michel M, Johnson AA, Kunisada M, Ko MS, Kaestner KH, Marmorstein AD, Schlessinger D. Forkhead transcription factor FoxA1 regulates sweat secretion through Bestrophin 2 anion channel and Na-K-Cl cotransporter 1. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:1199–1203. doi: 10.1073/pnas.1117213109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang M, Zeng S, Zhang L, Li H, Chen L, Zhang X, Li X, Lin C, Shu S, Xie S, He Y, Mao X, Peng L, Shi L, Yang L, Tang S, Fu X. Localization of Na(+)-K(+)-ATPase alpha/beta, Na(+)-K(+)-2Cl-cotransporter 1 and aquaporin-5 in human eccrine sweat glands. Acta histochemica. 2014;116:1374–1381. doi: 10.1016/j.acthis.2014.08.010. [DOI] [PubMed] [Google Scholar]
- 63.Saga K. Structure and function of human sweat glands studied with histochemistry and cytochemistry. Progress in histochemistry and cytochemistry. 2002;37:323–386. doi: 10.1016/s0079-6336(02)80005-5. [DOI] [PubMed] [Google Scholar]
- 64.Henderson RM, Cuthbert AW. A high-conductance Ca(2+)-activated K+ channel in cultured human eccrine sweat gland cells. Pflugers Archiv: European journal of physiology. 1991;418:271–275. doi: 10.1007/BF00370526. [DOI] [PubMed] [Google Scholar]
- 65.Cui CY, Sima J, Yin M, Michel M, Kunisada M, Schlessinger D. Identification of potassium and chloride channels in eccrine sweat glands. Journal of dermatological science. 2016;81:129–131. doi: 10.1016/j.jdermsci.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ertongur-Fauth T, Hochheimer A, Buescher JM, Rapprich S, Krohn M. A novel TMEM16A splice variant lacking the dimerization domain contributes to calcium-activated chloride secretion in human sweat gland epithelial cells. Experimental dermatology. 2014;23:825–831. doi: 10.1111/exd.12543. [DOI] [PubMed] [Google Scholar]
- 67.Servetnyk Z, Roomans GM. Chloride transport in NCL-SG3 sweat gland cells: channels involved. Experimental and molecular pathology. 2007;83:47–53. doi: 10.1016/j.yexmp.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 68.Inoue R, Sohara E, Rai T, Satoh T, Yokozeki H, Sasaki S, Uchida S. Immunolocalization and translocation of aquaporin-5 water channel in sweat glands. Journal of dermatological science. 2013;70:26–33. doi: 10.1016/j.jdermsci.2013.01.013. [DOI] [PubMed] [Google Scholar]
- 69.Goto K. The role of DOG1 immunohistochemistry in dermatopathology. J Cutan Pathol. 2016;43:974–983. doi: 10.1111/cup.12787. [DOI] [PubMed] [Google Scholar]
- 70.Bovell DL, MacDonald A, Meyer BA, Corbett AD, MacLaren WM, Holmes SL, Harker M. The secretory clear cell of the eccrine sweat gland as the probable source of excess sweat production in hyperhidrosis. Experimental dermatology. 2011;20:1017–1020. doi: 10.1111/j.1600-0625.2011.01361.x. [DOI] [PubMed] [Google Scholar]
- 71.Bovell DL. The human eccrine sweat gland: Structure, function and disorders. Journal of Local and Global Health Science. 2015;5:1–16. [Google Scholar]
- 72.Lu C, Fuchs E. Sweat gland progenitors in development, homeostasis, and wound repair. Cold Spring Harbor perspectives in medicine. 2014;4 doi: 10.1101/cshperspect.a015222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ferrera L, Caputo A, Ubby I, Bussani E, Zegarra-Moran O, Ravazzolo R, Pagani F, Galietta LJ. Regulation of TMEM16A chloride channel properties by alternative splicing. The Journal of biological chemistry. 2009;284:33360–33368. doi: 10.1074/jbc.M109.046607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Romanenko VG, Catalan MA, Brown DA, Putzier I, Hartzell HC, Marmorstein AD, Gonzalez-Begne M, Rock JR, Harfe BD, Melvin JE. Tmem16A encodes the Ca2+-activated Cl− channel in mouse submandibular salivary gland acinar cells. The Journal of biological chemistry. 2010;285:12990–13001. doi: 10.1074/jbc.M109.068544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ring A, Mork AC, Roomans GM. Calcium-activated chloride fluxes in cultured NCL-SG3 sweat gland cells. Cell biology international. 1995;19:265–278. doi: 10.1006/cbir.1995.1069. [DOI] [PubMed] [Google Scholar]
- 76.Metzler-Wilson K, Sammons DL, Ossim MA, Metzger NR, Jurovcik AJ, Krause BA, Wilson TE. Extracellular calcium chelation and attenuation of calcium entry decrease in vivo cholinergic-induced eccrine sweating sensitivity in humans. Experimental physiology. 2014;99:393–402. doi: 10.1113/expphysiol.2013.076547. [DOI] [PubMed] [Google Scholar]
- 77.Courjaret R, Machaca K. Mid-range Ca2+ signalling mediated by functional coupling between store-operated Ca2+ entry and IP3-dependent Ca2+ release. Nature communications. 2014;5:3916. doi: 10.1038/ncomms4916. [DOI] [PubMed] [Google Scholar]
- 78.Barro-Soria R, Aldehni F, Almaca J, Witzgall R, Schreiber R, Kunzelmann K. ER-localized bestrophin 1 activates Ca2+-dependent ion channels TMEM16A and SK4 possibly by acting as a counterion channel. Pflugers Archiv: European journal of physiology. 2010;459:485–497. doi: 10.1007/s00424-009-0745-0. [DOI] [PubMed] [Google Scholar]
- 79.Jin X, Shah S, Du X, Zhang H, Gamper N. Activation of Ca(2+) -activated Cl(−) channel ANO1 by localized Ca(2+) signals. The Journal of physiology. 2016;594:19–30. doi: 10.1113/jphysiol.2014.275107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jin X, Shah S, Liu Y, Zhang H, Lees M, Fu Z, Lippiat JD, Beech DJ, Sivaprasadarao A, Baldwin SA, Zhang H, Gamper N. Activation of the Cl− channel ANO1 by localized calcium signals in nociceptive sensory neurons requires coupling with the IP3 receptor. Science signaling. 2013;6:ra73. doi: 10.1126/scisignal.2004184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kunzelmann K, Schreiber R, Kmit A, Jantarajit W, Martins JR, Faria D, Kongsuphol P, Ousingsawat J, Tian Y. Expression and function of epithelial anoctamins. Experimental physiology. 2012;97:184–192. doi: 10.1113/expphysiol.2011.058206. [DOI] [PubMed] [Google Scholar]
- 82.McCarl CA, Picard C, Khalil S, Kawasaki T, Rother J, Papolos A, Kutok J, Hivroz C, Ledeist F, Plogmann K, Ehl S, Notheis G, Albert MH, Belohradsky BH, Kirschner J, Rao A, Fischer A, Feske S. ORAI1 deficiency and lack of store-operated Ca2+ entry cause immunodeficiency, myopathy, and ectodermal dysplasia. The Journal of allergy and clinical immunology. 2009;124:1311–1318 e1317. doi: 10.1016/j.jaci.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fuchs S, Rensing-Ehl A, Speckmann C, Bengsch B, Schmitt-Graeff A, Bondzio I, Maul-Pavicic A, Bass T, Vraetz T, Strahm B, Ankermann T, Benson M, Caliebe A, Folster-Holst R, Kaiser P, Thimme R, Schamel WW, Schwarz K, Feske S, Ehl S. Antiviral and regulatory T cell immunity in a patient with stromal interaction molecule 1 deficiency. Journal of immunology. 2012;188:1523–1533. doi: 10.4049/jimmunol.1102507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schaballie H, Rodriguez R, Martin E, Moens L, Frans G, Lenoir C, Dutre J, Canioni D, Bossuyt X, Fischer A, Latour S, Meyts I, Picard C. A novel hypomorphic mutation in STIM1 results in a late-onset immunodeficiency. The Journal of allergy and clinical immunology. 2015;136:816–819. e814. doi: 10.1016/j.jaci.2015.03.009. [DOI] [PubMed] [Google Scholar]
- 85.Parry DA, Holmes TD, Gamper N, El-Sayed W, Hettiarachchi NT, Ahmed M, Cook GP, Logan CV, Johnson CA, Joss S, Peers C, Prescott K, Savic S, Inglehearn CF, Mighell AJ. A homozygous STIM1 mutation impairs store-operated calcium entry and natural killer cell effector function without clinical immunodeficiency. The Journal of allergy and clinical immunology. 2016;137:955–957. e958. doi: 10.1016/j.jaci.2015.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Courtois G, Smahi A. NF-kappaB-related genetic diseases. Cell death and differentiation. 2006;13:843–851. doi: 10.1038/sj.cdd.4401841. [DOI] [PubMed] [Google Scholar]
- 87.Sadier A, Viriot L, Pantalacci S, Laudet V. The ectodysplasin pathway: from diseases to adaptations. Trends in genetics: TIG. 2014;30:24–31. doi: 10.1016/j.tig.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 88.Doffinger R, Smahi A, Bessia C, Geissmann F, Feinberg J, Durandy A, Bodemer C, Kenwrick S, Dupuis-Girod S, Blanche S, Wood P, Rabia SH, Headon DJ, Overbeek PA, Le Deist F, Holland SM, Belani K, Kumararatne DS, Fischer A, Shapiro R, Conley ME, Reimund E, Kalhoff H, Abinun M, Munnich A, Israel A, Courtois G, Casanova JL. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nature genetics. 2001;27:277–285. doi: 10.1038/85837. [DOI] [PubMed] [Google Scholar]
- 89.Fusco F, Pescatore A, Conte MI, Mirabelli P, Paciolla M, Esposito E, Lioi MB, Ursini MV. EDA-ID and IP, two faces of the same coin: how the same IKBKG/NEMO mutation affecting the NF-kappaB pathway can cause immunodeficiency and/or inflammation. International reviews of immunology. 2015;34:445–459. doi: 10.3109/08830185.2015.1055331. [DOI] [PubMed] [Google Scholar]
- 90.Jain A, Ma CA, Liu S, Brown M, Cohen J, Strober W. Specific missense mutations in NEMO result in hyper-IgM syndrome with hypohydrotic ectodermal dysplasia. Nature immunology. 2001;2:223–228. doi: 10.1038/85277. [DOI] [PubMed] [Google Scholar]
- 91.Zonana J, Elder ME, Schneider LC, Orlow SJ, Moss C, Golabi M, Shapira SK, Farndon PA, Wara DW, Emmal SA, Ferguson BM. A novel X-linked disorder of immune deficiency and hypohidrotic ectodermal dysplasia is allelic to incontinentia pigmenti and due to mutations in IKK-gamma (NEMO) American journal of human genetics. 2000;67:1555–1562. doi: 10.1086/316914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ambudkar IS. Calcium signalling in salivary gland physiology and dysfunction. The Journal of physiology. 2016;594:2813–2824. doi: 10.1113/JP271143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Davis FM, Janoshazi A, Janardhan KS, Steinckwich N, D’Agostin DM, Petranka JG, Desai PN, Roberts-Thomson SJ, Bird GS, Tucker DK, Fenton SE, Feske S, Monteith GR, Putney JW., Jr Essential role of Orai1 store-operated calcium channels in lactation. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:5827–5832. doi: 10.1073/pnas.1502264112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xing J, Petranka JG, Davis FM, Desai PN, Putney JW, Bird GS. Role of Orai1 and store-operated calcium entry in mouse lacrimal gland signalling and function. The Journal of physiology. 2014;592:927–939. doi: 10.1113/jphysiol.2013.267740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lur G, Sherwood MW, Ebisui E, Haynes L, Feske S, Sutton R, Burgoyne RD, Mikoshiba K, Petersen OH, Tepikin AV. InsP(3)receptors and Orai channels in pancreatic acinar cells: co-localization and its consequences. The Biochemical journal. 2011;436:231–239. doi: 10.1042/BJ20110083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Son A, Park S, Shin DM, Muallem S. Orai1 and STIM1 in ER/PM junctions: roles in pancreatic cell function and dysfunction. American journal of physiology Cell physiology. 2016;310:C414–422. doi: 10.1152/ajpcell.00349.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cheng KT, Liu X, Ong HL, Ambudkar IS. Functional requirement for Orai1 in store-operated TRPC1-STIM1 channels. The Journal of biological chemistry. 2008;283:12935–12940. doi: 10.1074/jbc.C800008200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cheng KT, Liu X, Ong HL, Swaim W, Ambudkar IS. Local Ca(2)+ entry via Orai1 regulates plasma membrane recruitment of TRPC1 and controls cytosolic Ca(2)+ signals required for specific cell functions. PLoS Biol. 2011;9:e1001025. doi: 10.1371/journal.pbio.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ong HL, Cheng KT, Liu X, Bandyopadhyay BC, Paria BC, Soboloff J, Pani B, Gwack Y, Srikanth S, Singh BB, Gill DL, Ambudkar IS. Dynamic assembly of TRPC1-STIM1-Orai1 ternary complex is involved in store-operated calcium influx. Evidence for similarities in store-operated and calcium release-activated calcium channel components. The Journal of biological chemistry. 2007;282:9105–9116. doi: 10.1074/jbc.M608942200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu X, Cheng KT, Bandyopadhyay BC, Pani B, Dietrich A, Paria BC, Swaim WD, Beech D, Yildrim E, Singh BB, Birnbaumer L, Ambudkar IS. Attenuation of store-operated Ca2+ current impairs salivary gland fluid secretion in TRPC1(−/−) mice. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:17542–17547. doi: 10.1073/pnas.0701254104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hong JH, Li Q, Kim MS, Shin DM, Feske S, Birnbaumer L, Cheng KT, Ambudkar IS, Muallem S. Polarized but differential localization and recruitment of STIM1, Orai1 and TRPC channels in secretory cells. Traffic. 2011;12:232–245. doi: 10.1111/j.1600-0854.2010.01138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gerasimenko JV, Gryshchenko O, Ferdek PE, Stapleton E, Hebert TO, Bychkova S, Peng S, Begg M, Gerasimenko OV, Petersen OH. Ca2+ release-activated Ca2+ channel blockade as a potential tool in antipancreatitis therapy. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:13186–13191. doi: 10.1073/pnas.1300910110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wen L, Voronina S, Javed MA, Awais M, Szatmary P, Latawiec D, Chvanov M, Collier D, Huang W, Barrett J, Begg M, Stauderman K, Roos J, Grigoryev S, Ramos S, Rogers E, Whitten J, Velicelebi G, Dunn M, Tepikin AV, Criddle DN, Sutton R. Inhibitors of ORAI1 Prevent Cytosolic Calcium-Associated Injury of Human Pancreatic Acinar Cells and Acute Pancreatitis in 3 Mouse Models. Gastroenterology. 2015;149:481–492. e487. doi: 10.1053/j.gastro.2015.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Saint-Criq V, Gray MA. Role of CFTR in epithelial physiology. Cellular and molecular life sciences: CMLS. 2016 doi: 10.1007/s00018-016-2391-y. [DOI] [PMC free article] [PubMed] [Google Scholar]