

Abstract
Multiple sclerosis (MS) is a chronic inflammatory disease of the CNS, and one of the most common causes of disability in young adults. Over the last decade, new disease‐modifying therapies have emerged, including monoclonal antibodies (mAbs) that provide highly targeted therapies with greater efficacy than platform therapies. In particular, monoclonal antibodies directed against CD20‐positive B cells have shown remarkable results in recent clinical trials and renewed interest in the mechanism of B cell‐depleting therapies to ameliorate relapse activity and progression in MS. Here, we review the mechanisms of action and clinical evidence of approved and emerging mAbs, with a focus on B cell‐targeted therapies.
Abbreviations
- ADCC
antibody‐dependent cell‐mediated cytotoxicity
- APC
antigen‐presenting cell
- ARR
annualized relapse rate
- BAFF
B cell activating factor
- CDC
complement‐dependent cytotoxicity
- CDP
confirmed disability progression
- EAE
experimental autoimmune encephalomyelitis
- EMA
European Medicines Agency
- FDA
Food and Drug Administration
- HYP
high‐yield protein
- JCV
John Cunningham Virus
- mAb
monoclonal antibody
- MS
multiple sclerosis
- NICE
National Institute for Health and Care Excellence
- NK
natural killer
- PML
progressive multifocal leukoencephalopathy
- PPMS
primary progressive multiple sclerosis
- RRMS
relapsing–remitting multiple sclerosis
Multiple sclerosis
Multiple sclerosis (MS) can be broadly divided into two, often overlapping clinical courses: that of relapsing MS, characterized by clearly defined attacks of new or worsening neurological symptoms, or progressive MS where there is worsening neurological function independent of relapses. Clinical trials over the last 25 years have been productive in discovering an ever increasing list of medications effective in preventing relapses. However, the search for therapies to reduce or halt progression in progressive MS has remained elusive until recently, when a new anti‐CD20 monoclonal antibody (mAb), ocrelizumab, was found to significantly reduce progression in a phase III trial for primary progressive MS (Montalban et al., 2016). The emergence of mAbs in the treatment of MS, in particular those targeting B cell antigens, will be discussed in further detail.
Pathology of MS
The pathology of MS is characterized by inflammatory demyelinating lesions, which affect the grey and white matter of the CNS. It is thought that the failure of myelin repair mechanisms, oligodendrocyte loss and cumulative axonal and neuronal loss are the main drivers of disability progression in this disease (Lassmann, 2014). Although the triggers for MS remain unknown, it is widely posited that autoreactive CD4+ T helper cells, activated in the periphery by molecular mimicry, bystander activation or viral persistence, are central to the initiation of inflammatory demyelinating lesions in MS. This is based mainly on evidence derived from the most commonly used experimental model of MS, experimental autoimmune encephalomyelitis (EAE), where CNS autoimmunity is induced by inoculation with CNS antigens or by the adoptive transfer of myelin‐specific Th1 or Th17 cells (Ben‐Nun et al., 1981; Jäger et al., 2009; Robinson et al., 2014). In the EAE model, autoreactive T‐cells are necessary for blood–brain barrier breakdown and inflammatory CNS lesion development. A role for CD4+ T‐cells in MS pathogenesis is also supported by histopathological observations of these cells in MS brain lesions (Traugott et al., 1983); the increased risk of MS in association with genetic variation in the human leukocyte antigen gene region (i.e. the HLADR15 haplotype), which encodes for genes that have a critical role in antigen presentation to T‐cells (International Multiple Sclerosis Genetics Consortium, 2005); and functional studies that suggest differences in the frequency and activity of myelin reactive CD4+ T‐cells in MS cases compared with controls (Olsson et al., 1992; Zhang et al., 1994; Pender et al., 2000). However, there is now increasing evidence to support a role for B‐lymphocytes (B‐cells) as key effectors in the pathogenesis of MS, principally led by the therapeutic success of antibody‐mediated B‐cell killing.
B‐cells in MS
B‐cells develop from haematopoietic stem cells in the bone marrow, and upon entering the circulation, they mature into naïve B‐cells. Following exposure to antigen, B‐cells undergo a proliferative phase where they develop into antibody‐producing plasma blasts/plasma cells, or memory B‐cells that are primed to respond following re‐exposure to the same antigen (Claes et al., 2015; summarized in Figure 1). Terminally differentiated B‐cells can also accumulate and reside in the bone marrow as a lifetime source of stable antibody‐producing cells.
Figure 1.
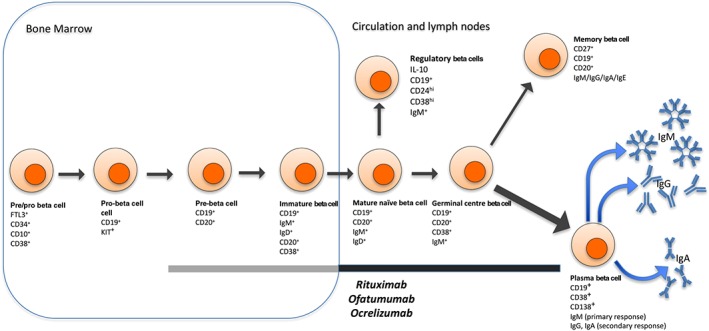
As B‐cells develop in the bone marrow, they express various combinations of surface molecules during the transition from pre/pro‐ B‐cell to immature B‐cells. After leaving the bone marrow and becoming a mature naïve B‐cells, a small subset will become IL‐10‐secreting regulatory B‐cells. Others, upon activation, develop into plasma B‐cells or memory B‐cells. Initially, plasma B‐cells secrete IgM, but after class switch recombination may secrete IgG, IgA or IgE. Rituximab, ofatumumab and ocrelizumab are mAbs that bind CD20, killing CD20+ cells through antibody‐dependent and complement‐dependent cell cytotoxicity. CD20 is expressed on pre‐B‐cells, immature B‐cells, mature naïve B‐cells, germinal centre B‐cells and memory B‐cells, rendering them vulnerable to these drugs. The black line indicates B‐cell subsets present in the periphery that are known to be affected by rituximab, ofatumumab and ocrelizumab. The grey line indicates CD20+ cells in the bone marrow – because of their location, it is unclear whether these cells are affected by the treatments.
Antibodies, visualized as intrathecal oligoclonal bands, have been used to diagnose MS for decades (Correale and de los Milagros Bassani Molinas, 2002). Despite the presence of these intrathecal oligoclonal bands in some MS patients and evidence for antibody deposits in some MS lesions (Lucchinetti et al., 2000), the role of antibodies as a component of the primary pathogenesis of MS, or alternatively, as an epiphenomenon, has remained controversial. Interestingly, elevated antibodies to myelin components including myelin basic protein, myelin proteolipid protein and myelin associated‐glycoprotein (Górny et al., 1983) as well as various other CNS and non‐CNS antigens (see Fraussen et al., 2014) have been identified in MS patients. However, proof of one or many ‘MS auto‐antigens’ is lacking.
Antibody production is the function of plasma cells, and many of these cells are long‐lived and not targeted by anti‐CD20 therapies. However, non‐plasma B‐cells also exert multiple other effector functions including cytokine production and antigen presentation, which could provide an explanation for pathogenic contribution of these cells in MS (Figure 2). Several studies, for instance, suggest that MS could be associated with abnormal B‐cell cytokine responses, including the production of abnormally high levels of TNF‐α and lymphotoxin‐α (Duddy et al., 2007; Bar‐Or et al., 2010), and an increase in the frequency of granulocyte macrophage colony‐stimulating factor (GM‐CSF)‐producing B‐cells relative to healthy controls (Li et al., 2015), following B‐cell stimulation via CD40 and the B‐cell receptor. Also, B‐cells from MS patients secrete more pro‐inflammatory IL‐6 and less regulatory IL‐10 than those from controls (Duddy et al., 2007).
Figure 2.
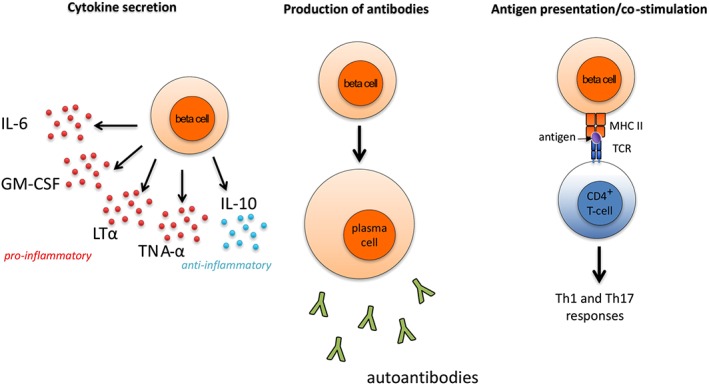
B‐cell involvement in MS. B‐cells contribute to MS pathogenesis by (i) secreting pro‐ and anti‐inflammatory cytokines; (ii) releasing antibodies including various autoantibodies; and (iii) acting as APCs that present auto‐antigens to CD4+ T‐cells, which together with cytokines promote Th1 and Th17 responses. TCR, T‐cell receptor; MHC, major histocompatibility complex.
As antigen‐presenting cells (APCs), B‐cells could play a role in the presentation of auto‐antigens to CD4+ T‐cells, hence promoting Th1 and Th17 responses. EAE models have demonstrated that, while B‐cells are not the sole APC involved, their activity increases disease severity (Parker Harp et al., 2015). Studies in humans of CD80 and CD86, co‐stimulatory molecules that help activate T‐cells, provide further evidence for a role of B‐cells as APCs. For example, the GM‐CSF‐expressing B‐cell subset described by Li et al. (2015) up‐regulates CD80 and CD86 when activated. Additionally, CD80 and CD86 expression is higher in MS patients than in healthy controls, and CD80+ lymphocyte levels increase in MS patients during exacerbations (Aung and Balashov, 2015). Therefore, B‐cells may be involved in MS not just as sources of cytokines and autoantibodies, but also as APCs that stimulate T‐cells.
Although the adaptive immune system has not been traditionally viewed as playing a role in progressive MS, descriptions of lymphoid follicle‐like structures in the meninges surrounding CNS tissue of secondary progressive MS cases suggest that B‐cells could also play a role in progressive disease (Serafini et al., 2004).
Finally, the most convincing evidence for a role of B‐cells in MS pathogenesis is derived from trials with B‐cell‐depleting therapies that have shown efficacy in both relapsing–remitting and primary progressive forms of the disease. The insights gained from MS therapeutic trials with B‐cell‐depleting mAb therapies will be discussed in more detail.
mAb therapies for relapsing MS: efficacy and mechanisms of action
Approved therapies
Natalizumab
Natalizumab is a humanized IgG4 mAb directed against the α4 subunit of the α4β1 and α4β7 integrins expressed on the surface of leukocytes (Rice et al., 2005). Binding to α4β1 integrin (also known as VLA‐4) inhibits its interaction with vascular cell adhesion molecule‐1 (VCAM‐1) expressed on cerebral vascular endothelial cells and prevents migration of leukocytes through the blood–brain barrier (Yednock et al., 1992). Natalizumab may also block binding of α4β1 integrin with other ligands such as fibronectin and osteopontin (Yaldizli and Putzki, 2009), further modulating leukocyte recruitment and activation in the CNS.
These effects on leukocyte entry, and in particular pathogenic T‐cell entry into the CNS, are thought to be the main mechanism by which natalizumab therapy reduces MS disease activity. However, it is important to consider that natalizumab therapy is also likely to inhibit B‐cell entry into the CNS, with reports of reduced numbers of B‐cells in CSF and increased numbers in the peripheral circulation of natalizumab‐treated MS patients (Stüve et al., 2006; Krumbholz et al., 2008). Furthermore, a disproportionate increase in peripheral pre B‐cells has been reported in MS patients following natalizumab therapy (Krumbholz et al., 2008; Saraste et al., 2016). Hence, it remains possible that the effects of natalizumab in MS are at least partially mediated through actions on B‐cells.
The phase III AFFIRM trial recruited 942 relapsing–remitting MS (RRMS) patients randomized 2:1 to receive monthly intravenous natalizumab 300 mg or placebo over 2 years (Polman et al., 2006). In the natalizumab group, the annualized relapse rate (ARR) was reduced by 68% over 2 years compared with placebo, and 3 month confirmed disability progression (CDP) was reduced by 42% (see Table 1 for summary of clinical trial results).
Table 1.
Clinical trials of mAbs in MS
| Active drug and MS subtype | Trial/study (phase) | Sample size | Treatment/control arms | Duration (months) | ARR (% reduction) (P‐value) | CDP % reduction (P‐value) | % reduction in the number of Gd‐enhancing lesions (P‐value) | % reduction in the number of new/enlarging T2 hyperintense lesions (P‐value) |
|---|---|---|---|---|---|---|---|---|
| Natalizumab | ||||||||
| RRMS | AFFIRM (III) | 942 |
Natalizumab i.v. Placebo |
24 |
0.23 (68) (<0.001) 0.73 |
42 (<0.001)a | 92 (<0.001) | 83 (<0.001) |
| RRMS | SENTINEL (III) | 1171 |
IFN‐β1a i.m. + natalizumab IFN‐β1a i.m. + placebo |
24 |
0.34 (55) (<0.001) 0.75 |
24 (0.02)a | 89 (<0.001) | 83 (<0.001) |
| Alemtuzumab | ||||||||
| RRMS | CARE‐MS I (III) | 581 |
Alemtuzumab i.v. IFN‐β1a s.c. |
24 |
0.18 (55) (<0.001) 0.39 |
30 (0.22)b | ReductionNS (<0.0001) | ReductionNS (0.04) |
| RRMS | CARE‐MS II (III) | 840 |
Alemtuzumab i.v. IFN‐β1a s.c. |
24 |
0.26 (49) (<0.001) 0.52 |
42 (0.008)b | ReductionNS (<0.0001) | ReductionNS (<0.0001) |
| Daclizumab | ||||||||
| RRMS | SELECT (II) | 621 |
Daclizumumab HYP s.c. 150 mg Daclizumumab HYP s.c. 300 mg Placebo |
12 |
0.21 (54) (<0.001) 0.23 (50) (<0.001) 0.46 |
57 (0.02)a
43 (0.09)a |
79 (<0.0001) 86 (<0.0001) |
70 (<0.0001) 79 (<0.0001) |
| RRMS | DECIDE (III) | 1841 |
Daclizumumab HYP s.c. 150 mg IFN‐β1a i.m. |
24 |
0.22 (45) (<0.001) 0.39 |
20 (0.16)a | 60 (<0.001) | 54 (<0.001) |
| Rituximab | ||||||||
| RRMS | HERMES (II) | 104 |
Rituximab i.v. Placebo |
12 |
0.37 (49) (0.08) 0.72 |
NS | 91 (<0.001) | NS |
| PPMS | OLYMPUS (II/III) | 439 |
Rituximab i.v. Placebo |
24 | NS | 22 (0.14)a | NS | NS |
| Ocrelizumab | ||||||||
| RRMS | OPERA I (III) | 821 |
Ocrelizumab i.v. IFN‐β1a s.c. |
24 |
0.16 (46) (<0.001) 0.29 |
(Combined OPERA I and II) | 94 (<0.001) | 77 (<0.001) |
| RRMS | OPERA II (III) | 835 |
Ocrelizumab i.v. IFN‐β1a s.c. |
24 |
0.16 (47) (<0.001) 0.29 |
40 (<0.001)a
40 (0.003)b |
95 (<0.001) | 83 (<0.001) |
| PPMS | ORATORIO (III) | 732 |
Ocrelizumab i.v. Placebo |
24 | NS |
24 (0.03)a
25 (0.04)b |
NS | NS |
| Ofatumumab | ||||||||
| RRMS | Sorensen et al. (II) | 38 |
Ofatumumab i.v. 100, 300 and 700 mg Placebo |
6 | NS | NS | >99 (<0.001) | 99 (<0.001) |
| RRMS | MIRROR (II) | 232 |
Ofatumumab s.c. 3, 30 and 60 mg Placebo |
6 | NS | NS | ≥90 (<0.001) for doses ≥30 mg | NS |
NS, not specified.
3 month.
6 month.
The SENTINEL study was another phase III trial that enrolled 1171 RRMS patients randomized equally to natalizumab combined with weekly i.m. IFN‐β1a 30 μg or IFN‐β1a alone (Rudick et al., 2006). Over 2 years, the natalizumab add‐on therapy reduced ARR by 55% compared with IFN‐β1a alone and reduced 3 month CDP by 24%.
In 2004, natalizumab was the first mAb to be approved by the US Food and Drug Administration (FDA) for MS. It was subsequently suspended in 2005 after two cases of progressive multifocal leukoencephalopathy (PML) were reported in the SENTINEL trial (Rudick et al., 2006). PML is a serious, often fatal demyelinating disease of the CNS, caused by infection of oligodendrocytes by the John Cunningham Virus (JCV). It has an increased incidence in those with the following three risk factors: the presence of anti‐JCV antibodies, natalizumab treatment for ≥2 years and prior use of immunosuppressive agents (Bloomgren et al., 2012). Following implementation of a risk management programme for PML, the FDA approved it again in 2006 and as did the European Union in the same year. A 2016 European Medicines Agency (EMA) label update further describes the annualized risk of PML in patients receiving natalizumab segregated by serum anti‐JCV antibody level, with patients at lower levels (<0.9) and those found to be repeatedly negative at very low risk (EMA, 2016).
The clinical efficacy of natalizumab has been highlighted recently in real‐world observational studies. Spelman et al. demonstrated superiority of natalizumab over platform therapies when used first‐line in treatment‐naïve RRMS patients, with a 68% relative reduction in ARR (Spelman et al., 2016). Other studies showed that when natalizumab was used as a second‐line agent, it was more effective than fingolimod in terms of ARR reduction and/or short‐term disability burden (Kalincik et al., 2015; Barbin et al., 2016; Lanzillo et al., 2016) and superior to alemtuzumab in facilitating disability regression, while comparable in relapse rate and disability progression outcomes in that comparison (Kalincik et al., 2017).
Alemtuzumab
Alemtuzumab is a humanized monoclonal IgG1 antibody targeting CD52, a surface antigen that is expressed at high levels on T‐ and B‐cells and to a lesser extent on monocytes and macrophages, but not on haematopoietic stem cells (Williams et al., 2013; Jones and Coles, 2014). Alemtuzumab leads to a rapid and profound depletion of CD52+ cells by three mechanisms: antibody‐dependent cell‐mediated cytotoxicity (ADCC), complement‐dependent cytotoxicity (CDC) and induction of apoptosis (Freedman et al., 2013; Ruck et al., 2015), with ADCC being the most likely predominant mechanism (Knier et al., 2014; Lycke, 2015). This is followed by repopulation of peripheral T‐ and B‐lymphocytes with an alteration in the number, proportions and functions of certain lymphocyte subsets, such as increased regulatory T‐cell subsets and memory T‐cells (Hartung et al., 2015; Milo, 2016).
Changes in the reconstituting B‐cell pool have also been reported following alemtuzumab therapy, with a predominance of immature and, later, naïve‐memory B‐cell subsets observed in association with increased serum levels of the B‐cell survival factor, B‐cell‐activating factor (BAFF) (Thompson et al., 2010). It was also noted that the recovery of memory B‐cells was incomplete in alemtuzumab‐treated patients by 12 months. It has been proposed that changes in the proportions of reconstituting immune cell subsets could be associated with long‐term efficacy of alemtuzumab in MS (Havari et al., 2014).
CARE‐MS I was a phase III trial of 581 treatment‐naive RRMS patients, randomized 2:1 to receive alemtuzumab or s.c. IFN‐β1a over 2 years (Cohen et al., 2012). Alemtuzumab was given i.v. 12 mg daily for 5 days at baseline and for 3 days at 12 months, while s.c. IFN‐β1a 44 μg was given three times a week. This study found a significant 55% reduction in ARR in the alemtuzumab group compared with the IFN‐β1a control group. There was no significant difference in 6 month CDP.
CARE‐MS II was another phase III trial of 840 RRMS patients who had relapsed on platform therapy (Coles et al., 2012). Patients were randomized 1:2:2 to s.c. IFN‐β1a 44 μg or intravenous alemtuzumab 12 or 24 mg, though the 24 mg arm was discontinued to aid recruitment. Similar to the CARE‐MS I results, the alemtuzumab arm had a significant reduction of 49% in ARR compared with IFN‐β1a. However, in contrast to CARE‐MS I, this study found a significant difference in 6 month CDP, with a 42% improvement in the alemtuzumab arm.
Infusion reactions occurred in over 90% of alemtuzumab patients. Infections were more frequent, in particular herpes infections. Secondary autoimmunity is the major long‐term adverse event, with thyroid disease occurring in a third of patients, immune thrombocytopenia in 1% and anti‐glomerular basement membrane or other nephropathies in 0.3% (Freedman et al., 2013; Lycke, 2015).
Based on the above phase III randomized control studies, the EMA‐approved alemtuzumab for the treatment of RRMS in September 2013, followed by the UK National Institute for Health and Care Excellence (NICE) in May 2014 and the US FDA in November 2014.
Daclizumab
Daclizumab is a humanized IgG1 mAb targeting the CD25 (α) subunit of high‐affinity IL‐2 receptor on activated T‐cells (Bielekova et al., 2009). Anti‐CD25 mAb decreased T‐cell proliferation and differentiation in vitro (Kircher et al., 2003) but failed to suppress T‐cell proliferation and cytokine production ex vivo (Bielekova et al., 2006). Instead, it was found to strongly induce expansion of immunoregulatory CD56 bright natural killer (NK) cells, which can utilize IL‐2 via their low‐affinity IL‐2 receptor (Knier et al., 2014). This expansion was associated with response to therapy in MS patients, presumably through NK cell‐mediated lysis of autologous activated T‐cells (Bielekova et al., 2006). More recent work suggests that daclizumab may have other effects on the innate immune system which include the inhibition of antigen‐specific T‐cell activation by dendritic cells (Wuest et al., 2011) and the inhibition of CD40 ligand expression on T‐cells (Snyder et al., 2011). Interestingly, there is also limited evidence to suggest that daclizumab treatment can inhibit intrathecal CXCL13 and IgG in MS patients, proposed to be mediated indirectly through effects on innate lymphoid cells (Perry et al., 2012).
Daclizumab was used in an early phase II MS trial (Wynn et al., 2010), while daclizumab high‐yield protein (HYP) has been used in more recent phase II and III trials (Gold et al., 2013; Kappos et al., 2015). Daclizumab HYP has the same amino acid sequence as previous versions but differs in the glycosylation profile, resulting in less immunogenicity as well as less ADCC (Gold et al., 2013).
The phase II SELECT trial was the first to use daclizumab HYP (Gold et al., 2013). A total of 621 RRMS patients were randomized 1:1:1 to monthly s.c. daclizumab HYP high‐dose 300 mg, daclizumab HYP low‐dose 150 mg or placebo and followed up for 52 weeks. There was a reduction in ARR of 54% and 50% in the low‐dose and high‐dose groups respectively compared with placebo. There was a significant 57% reduction in 3 month CDP in the low‐dose group only. In the trial, CD56bright NK cells increased in daclizumab patients from 0.6% of lymphocytes at baseline to 3.6% at the end of treatment, first becoming apparent at week 4. CD4+ and CD8+ T‐cells decreased by 7–10% at week 52.
The phase III DECIDE trial randomized 1841 RRMS patients equally to monthly s.c. daclizumab HYP 150 mg or weekly i.m. IFN‐β1a 30 μg, followed up to 144 weeks (Kappos et al., 2015). The daclizumab arm had a 45% reduction in ARR compared with IFN‐β1a. No significant change in 3 month CDP was observed.
In both the SELECT and DECIDE trials, there was an increased incidence in the daclizumab HYP groups of infections (nasopharyngitis, upper respiratory tract infections), cutaneous events (especially rash and eczema) and raised serum alanine aminotransferase or aspartate transaminase levels to more than five times the upper limit of normal (Gold et al., 2013; Kappos et al., 2015). No PML has been reported.
Based on the SELECT and DECIDE trial results, daclizumab HYP was officially approved for RRMS by the FDA in May 2016 and by the European Commission in July 2016. NICE made an initial decision in September 2016 not to recommend daclizumab be made available on the National Health Service in England and Wales, with a final statement expected in 2017.
Emerging therapies: mAbs targeting B‐cell antigens
Thus far, three anti‐CD20 mAb therapies (rituximab, ocrelizumab and ofatumumab) have been evaluated in phase II and III clinical trials and have shown high efficacy in relapsing MS. Importantly, ocrelizumab has also shown some efficacy in primary progressive MS (PPMS). The B‐lymphocyte antigen CD20 (also known as B1) is a 33–35 kD integral membrane spanning protein, belonging to the MSA4 protein family. The CD20 protein is expressed by B‐cells from the pre‐B‐cell to the mature B‐cell stage of development (Stashenko et al., 1980; Milo, 2016) and on a subset of T‐cells (Palanichamy et al., 2014). The functions of CD20 are not well characterized, but there is some evidence that it could regulate cell cycle progression (Bubien et al., 1993; Kanzaki et al., 1995) and B‐cell activation through effects on calcium mobilization and signalling (Morsy et al., 2013). As CD20 is ubiquitously expressed on the surface of B‐cell lineage cells, except stem cells and plasma cells (Figure 1), it has been demonstrated that anti‐CD20 mAb therapies can be used to rapidly deplete peripheral blood B‐cells while maintaining humoral immune responses (i.e. plasma cell‐mediated antibody production).
Rituximab
Rituximab is a chimeric mouse–human IgG1k mAb that binds to the CD20 cell surface epitope on circulating B‐cells (Reff et al., 1994). It lyses peripheral B‐cell populations through a threefold mechanism of action: a combination of ADCC and CDC and possibly promotion of apoptosis (Dubey et al., 2015). Rituximab is approved for patients with non‐Hodgkin's lymphoma, chronic lymphocytic leukaemia, rheumatoid arthritis, granulomatous polyangiitis and microscopic polyangiitis (Gasperi et al., 2016).
The phase II 48 week HERMES trial randomized 104 RRMS patients to receive rituximab or placebo (Hauser et al., 2008). Rituximab was given i.v. as a single course of 1 g on days 1 and 15. The primary end point was the number of gadolinium (Gd)‐enhancing lesions, a radiological marker of recent inflammatory activity, and there was a 91% reduction in the total number of Gd‐enhancing lesions up to week 24 in the rituximab group, relative to placebo. ARR in the rituximab arm was only significantly reduced at week 24, and not at week 48 compared with placebo.
Another phase II study used rituximab as an add‐on to platform therapies in 30 RRMS patients with clinical and radiological activity despite treatment with disease‐modifying therapy (Naismith et al., 2010). Rituximab was given as four weekly doses of 375 mg·m−2 i.v., and patients were followed up over a year. The rituximab arm had an 88% reduction in Gd‐enhancing lesions from pretreatment to post‐treatment scans. While the study was not powered to assess relapse rate reduction, the ARR was reduced at week 52 post treatment. Secondary end points such as the Expanded Disability Status Scale remained stable over 32 weeks.
A phase II/III placebo‐controlled trial in PPMS (OLYMPUS) was conducted on 439 patients, randomized 2:1 to receive rituximab 1 g or placebo i.v. every 24 weeks through to 96 weeks, and they were followed up to 122 weeks (Hawker et al., 2009). No significant difference was seen in the 3 month CDP. The rituximab arm had significantly less increase in T2 lesion volume, though brain volume change was similar between the groups. Interestingly, subgroup analyses showed that CDP was reduced in rituximab patients who were younger, <51 years, and had Gd‐enhancing or active inflammatory MRI lesions.
In both the HERMES and OLYMPUS trials, the incidence of adverse events and infections was similar between groups, though more infusion reactions were seen with the first rituximab dose only (Hauser et al., 2008; Hawker et al., 2009). No cases of PML have been reported in the MS trials.
While the above studies show promise for the use of rituximab in MS, no further trials are planned. This is in part due to licensing and patent issues, as well as the emergence of next‐generation anti‐CD20 antibodies, as discussed below. Although rituximab has not been approved for the treatment of MS, it can be approved for off‐label use in certain countries, and recent studies from the Swedish MS register provide insights into the real‐world experience of using off‐label rituximab. In a heterogenous cohort of 822 patients, Salzer et al. demonstrated safety and efficacy of rituximab, comparable to that reported in earlier trials (Salzer et al., 2016). Using the same registry, Alping et al. found superior efficacy and tolerability of rituximab, compared with fingolimod, in 256 stable RRMS patients who had switched from natalizumab due to JCV antibody positivity (Alping et al., 2016). The findings of these observational studies further support the utility of this anti‐CD20 mAb in MS.
Ocrelizumab
Ocrelizumab is a recombinant humanized IgG1 antibody that binds to a different but overlapping epitope compared with rituximab and is thought to bind with higher affinity to CD20 (Sorensen and Blinkenberg, 2016). As it is a humanized molecule, it is expected to be less immunogenic with repeated infusions and hence to have a more favourable benefit‐to‐risk profile (Menge et al., 2016). Compared with rituximab, ocrelizumab has increased ADCC and reduced CDC due to differences in the Fc portion of the antibody (Kappos et al., 2011). It has previously been trialled in rheumatoid arthritis, systemic lupus erythematosus and non‐Hodgkin's lymphoma.
Two identical phase III trials in RRMS patients (OPERA I and II) were recently published (Hauser et al., 2016). A total of 1656 patients were randomized 1:1 to receive ocrelizumab 600 mg i.v. every 24 weeks or IFN‐β1a 44 μg s.c. three times a week over a 96 week treatment period. In the ocrelizumab arms, the ARR at 2 years was reduced by 46% and 47% compared with the IFN‐β1a arms. A pooled analysis combining both studies showed a relative reduction of 40% in 3 month and 6 month CDP in the ocrelizumab arms.
The phase III PPMS study (ORATORIO) results were also recently published (Montalban et al., 2016). A total of 732 patients with PPMS were randomized 2:1 to receive two 300 mg infusions of ocrelizumab or placebo 14 days apart, every 24 weeks to 120 weeks. Compared with placebo, the ocrelizumab arm had a significant 24% reduction in 3 month CDP and 25% reduction in 6 month CDP.
There were more infusion‐related events in the ocrelizumab groups, highest after the first infusion and mostly mild–moderate in severity (Kappos et al., 2011; Hauser et al., 2016; Montalban et al., 2016). There were no opportunistic infections and no cases of PML. Interestingly, the clinical development of ocrelizumab in phase III trials of rheumatoid arthritis and systemic lupus erythematosus was suspended due to safety concerns surrounding the high incidence of serious and fatal opportunistic infections (Menge et al., 2016). It remains unclear why ocrelizumab was associated with a less favourable safety profile in these trials. However, it has been suggested that differences in patient population (i.e. older age and Asian ethnicity), higher dosing and use of adjunct immunosuppressive therapies may have been contributing factors (Emery et al., 2014; Sheridan, 2015; Sorensen and Blinkenberg, 2016).
The overall incidence of malignancies was higher in patients treated with ocrelizumab: 0.40 per 100 patient‐years of exposure, compared with 0.20 per 100 patient‐years of exposure in the pooled comparator groups (Hauser et al., 2016; Montalban et al., 2016). While the numbers of cancers seen after ocrelizumab treatments give some cause for concern, longer‐term studies will provide a more accurate assessment of these risks.
Based on the phase III RRMS and PPMS studies, the FDA granted ocrelizumab priority review in mid‐2016 with a decision expected in early 2017. The EMA received an application for centralized marketing authorization of ocrelizumab in November 2016.
Ofatumumab
Ofatumumab is a fully human IgG1 anti‐CD20 mAb that binds to an epitope distinct from rituximab and ocrelizumab (Knier et al., 2014; Sorensen and Blinkenberg, 2016). It is proposed to have improved efficacy over rituximab associated with a slower dissociation rate and more pronounced CDC activity with relatively less ADCC (Reagan and Castillo, 2011; Milo, 2016). In vitro studies have shown that ofatumumab depletes B‐cell lines resistant to rituximab (Wierda et al., 2011; Gupta and Jewell, 2012). As a fully human antibody, it has a very low immunogenic risk profile that could be associated with improved safety. It has demonstrated efficacy in rheumatoid arthritis and haematological malignancies and is currently approved for treatment of refractory chronic lymphocytic leukaemia (Ostergaard et al., 2010; Gupta and Jewell, 2012).
In 2014, a phase II RRMS trial randomized 38 patients to receive either placebo or two infusions of ofatumumab (100, 300 and 700 mg) 14 days apart (Sorensen et al., 2014). Another phase II RRMS placebo‐controlled study (MIRROR) was conducted using s.c. ofatumumab (Bar‐Or et al., 2014). A total of 232 patients were randomized into one of five treatment groups: placebo, ofatumumab 3 mg every 12 weeks, 30 mg every 12 weeks, 60 mg every 12 weeks or 60 mg every 4 weeks. At week 12, placebo patients received a single 3 mg ofatumumab dose. The MRI results of both studies show profound reductions on new Gd‐enhancing lesions and/or T2 hyperintense lesions (Table 1).
In these phase II trials, there were no unexpected safety signals or dose‐related safety concerns. Infusion‐related reactions were more common in the ofatumumab arm for both studies. No opportunistic infections were reported (Bar‐Or et al., 2014; Sorensen et al., 2014).
Two randomized, double‐blind, double‐dummy phase III clinical trials are currently underway to evaluate the efficacy and safety of ofatumumab compared with teriflunomide in RRMS (ASCLEPIOS I and II) (ClinicalTrials.gov identifier: NCT02792218/NCT02792231). Patients will be randomized to receive either s.c. ofatumumab every 4 weeks or teriflunomide orally once daily. Recruitment started in September 2016 and is expected to be complete by July 2019.
Immune effects of CD20 mAb therapies
Trials with CD20 mAb therapies commonly report a rapid depletion of peripheral CD19+ B‐cells, followed by a recovery phase that occurs several weeks after the final infusion. In the HERMES trial, after one course of rituximab, there was rapid depletion of CD19+ peripheral B‐cells from 2 weeks post treatment to week 24 (>95% reduction from baseline). By week 48, CD19+ cells returned to 31% of baseline (Hauser et al., 2008). The OLYMPUS trial saw a similar depletion of CD19+ B‐cells following rituximab therapy, with 35% of patients recovering peripheral B‐cell counts to within normal limits by week 122 (Hawker et al., 2009). Phase II trials of ocrelizumab and ofatumumab in RRMS suggest that these therapies could be associated with a similarly rapid, but more complete depletion of CD19+ peripheral B‐cells relative to rituximab therapy (99–100%), though head‐to‐head comparisons are lacking (Kappos et al., 2011; Sorensen et al., 2014).
Importantly, as limited changes in immunoglobulin concentrations were reported with CD20 mAb therapies in clinical trials (Hauser et al., 2008; Kappos et al., 2011; Sorensen et al., 2014), it was suggested that the unchanged immunoglobulin levels and early effects of rituximab therapy make it unlikely that the beneficial outcomes in the studies are due to depletion of pathogenic autoantibodies from the peripheral circulation. Rather, it was proposed that CD20 mAb therapies are more likely to target B‐cell cytokine production and antigen‐presenting functions as part of their mechanisms in MS.
Detailed analyses of the recovered B‐cell subsets following a single cycle of rituximab therapy in MS patients suggest that rituximab therapy could, at least in the short term, promote reduced activity in the B‐cell compartment even as B‐cell numbers recover. In their study, Palanichamy et al. (2014) demonstrated that replenishing B‐cells largely comprise the naïve (IgD+/CD27−) and transitional B‐cell subsets, possibly derived from pro‐ B‐cells that do not express CD20. The repletion of memory B‐cell subsets was more delayed, occurring from around 37–52 weeks. It remains unclear how these changes in the B‐cell compartment associate with disease activity in MS and it is also unclear if these effects are sustained over time. Nonetheless, the capacity for memory B‐cell numbers to recover over time suggests that some maintenance therapy may be required to achieve sustained therapeutic benefit with CD20 mAb therapies.
Although no significant effects on CD3+ T‐lymphocyte cells were reported in the HERMES and OLYMPUS trials for rituximab in MS, there is some evidence to suggest that rituximab therapy could deplete a small subset of CD3+ CD20dim T‐cells (<10% of total CD3+ cells) as part of its actions in MS (Palanichamy et al., 2014). Recent work suggests that although CD20+ T‐cells may show enhanced cytokine expression with stimulation in vitro (Schuh et al., 2016), it is not known if these cells contribute to MS pathogenesis or if their depletion is part of the mechanisms of rituximab therapy in MS. It is possible therefore that CD20 mAb therapies may directly target both the B‐cell and T‐cell functions as part of their mechanisms in MS.
Other B‐cell therapies in development for MS
In addition to the CD20 mAb therapies, several other biologicals targeting B‐cell surface antigens or B‐cell cytokine signalling molecules have also been trialled for MS. Importantly, the use of targeted therapeutics to modify B‐cell functions has already begun to provide novel and often unexpected insights into the functions of B‐cells in MS pathogenesis, suggesting that they are important contributors to immune regulation in MS.
CD19 mAb therapies
The CD19 antigen is expressed throughout B‐cell development and, in contrast to the CD20 antigen, is also present on plasma blasts/plasma cells (Levesque and St Clair, 2008). mAbs against CD19 may therefore show improved efficacy over CD20 mAbs for MS, as they may deplete both circulating B‐cells and pathogenic autoantibody levels (Tedder, 2009). In animal models, the anti‐CD19 mAb, MEDI‐551, is associated with prolonged depletion of most B‐cells, including pre‐B‐cells and antibody‐producing cells (Yazawa et al., 2005; Herbst et al., 2010). It remains unclear if CD19 mAb therapies will show efficacy for MS; however, a phase I randomized, blinded, placebo‐controlled dose escalation study to assess the safety and tolerability of MEDI‐551 (inebilizumab) in relapsing MS patients was recently completed (ClinicalTrials.gov identifier: NCT01585766). Further trials with these therapies are keenly anticipated as they may clarify the role of antibodies, and also CD3+ CD20dim T‐cells, which do not express CD19, in MS pathology.
Therapies targeting B‐cell cytokine signalling molecules
The survival of B‐cells is dependent on both B‐lymphocyte‐stimulating factor (BLyS/BAFF) and a proliferation‐inducing ligand (APRIL), endogenous proteins produced by activated T‐cells, monocytes and neutrophils. These molecules and their receptors represent potential therapeutic targets for autoimmune diseases with a likely B‐cell component (Hahne et al., 1998; Schneider et al., 1999).
Atacicept, a fusion protein comprising the extracellular domain of the BAFF/APRIL receptor TACI (transmembrane activator and CAML interactor and human immunoglobulin Fc domain), was developed as a biological therapy with binding activity for both BAFF and APRIL. In a phase II placebo‐controlled double‐blind trial of 255 RRMS cases, atacicept therapy was associated with reduction in circulating mature B‐cells of up to 70% and dose‐dependent reduction in serum IgM, IgG and IgA levels, suggesting an effect on antibody‐producing plasma cells (Kappos et al., 2014). Unexpectedly however, atacicept therapy was associated with an increase in the frequency of relapses, suggesting that some B‐cell or plasma cell functions are important for reducing autoimmune attacks in MS. An increase in the proportion of patients converting to clinically definite MS was also reported in trials with atacicept for optic neuritis (Sergott et al., 2015). It remains unclear why targeting of BAFF/APRIL is associated with pro‐inflammatory effects in MS and optic neuritis. However, these effects could be partially explained by the types of B‐cell subsets that are depleted in association with the neutralization of BAFF/APRIL (Lühder and Gold, 2014). Interestingly, studies in mice suggest that the survival of memory B‐cells is independent of BAFF/APRIL signalling, and hence, atacicept may not efficiently deplete B‐cells that are primed to respond to auto‐antigens (Benson et al., 2008). It was also suggested that APRIL and/or BAFF may be associated with the induction of IL‐10‐producing regulatory B‐cell types (Yang et al., 2010; Hua et al., 2016), and hence, atacicept therapy may disrupt regulatory B‐cell functions to shift immune balance towards a pro‐inflammatory state (Lühder and Gold, 2014). Finally, as atacicept reduced immunoglobulin levels, these effects on plasma cells could be, in themselves, deleterious in MS, potentially by removal of protective autoantibodies.
Future challenges for B‐cell therapies for MS
mAb therapies that are largely designed to deplete circulating lymphocytes or to prevent their migration into the CNS compartment have been successfully used to modulate the immune response in MS. Although it has long been assumed that T‐cells play an important role in the pathogenesis of this disease, targeted B‐cell‐depleting therapies have also shown surprising efficacy in clinical trials for MS, suggesting that B‐cell functions may also contribute to the pathogenesis of this disease.
Despite promising results with CD20 mAb therapies in trials for MS, it is important to note the multidimensional role of B‐cells as APCs, antibody‐secreting cells and potent sources of both pro‐inflammatory and anti‐inflammatory cytokines, suggesting that untargeted therapeutic manipulation of the B‐cell compartment in MS could be associated with long‐term deleterious effects. The most obvious concern is long‐term depletion of the slowly replenishing plasma cell compartment from the repeatedly targeted plasma cell pools, which could lead to potentially prolonged hypogammaglobulinaemia.
Animal studies have highlighted the ability of particular B‐cell subsets to negatively regulate immune responses through the production of anti‐inflammatory cytokines, which modulate pathogenic CD4+ T‐cell subtypes and innate immune cell responses, as well as via interactions with regulatory T‐cells. Reduced regulatory B‐cell outputs (i.e. IL‐10) in MS patients and the severe exacerbation of disease in patients treated with atacicept provide some perspective on the consequences of targeting the majority of B‐cell populations as a therapeutic strategy. Future studies will require a focus on the specific consequences of the various B‐cell therapies, with the ultimate goal being a balance between removal of the drivers of pathogenic immune responses and maintenance of the beneficial immune regulatory functions of B‐cell subsets in MS.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a, b).
Conflict of interest
A.N. received consulting fees from EMD Serono and grants from Novartis and Merck Serono. M.G. received speaker honoraria and travel expenses from Biogen. H.B. reports consulting fees from Novartis, Biogen, TEVA, Merck and Oxford Pharmagenesis.
Acknowledgements
M.G. is supported by a Multiple Sclerosis Research Australia and Charityworks for MS postdoctoral fellowship.
Nguyen, A.‐L. , Gresle, M. , Marshall, T. , Butzkueven, H. , and Field, J. (2017) Monoclonal antibodies in the treatment of multiple sclerosis: emergence of B‐cell‐targeted therapies. British Journal of Pharmacology, 174: 1895–1907. doi: 10.1111/bph.13780.
References
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alping P, Frisell T, Novakova L, Islam‐Jakobsson P, Salzer J, Bjorck A et al. (2016). Rituximab versus fingolimod after natalizumab in multiple sclerosis patients. Ann Neurol 79: 950–958. [DOI] [PubMed] [Google Scholar]
- Aung LL, Balashov KE (2015). Decreased Dicer expression is linked to increased expression of co‐stimulatory molecule CD80 on B cells in multiple sclerosis. Mult Scler 21: 1131–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbin L, Rousseau C, Jousset N, Casey R, Debouverie M, Vukusic S et al. (2016). Comparative efficacy of fingolimod vs natalizumab: a French multicenter observational study. Neurology 86: 771–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar‐Or A, Grove R, Austin D, Tolson J, Vanmeter S, Lewis E et al. (2014). The MIRROR study: a randomized, double‐blind, placebo‐controlled, parallel‐group, dose‐ranging study to investigate the safety and mri efficacy of subcutaneous ofatumumab in subjects with relapsing–remitting multiple sclerosis (RRMS). Neurology 82 (Suppl. 10): I7–I1. [Google Scholar]
- Bar‐Or A, Fawaz L, Fan B, Darlington PJ, Rieger A, Ghorayeb C et al. (2010). Abnormal B‐cell cytokine responses a trigger of T‐cell‐mediated disease in MS? Ann Neurol 67: 452–461. [DOI] [PubMed] [Google Scholar]
- Ben‐Nun A, Wekerle H, Cohen IR (1981). The rapid isolation of clonable antigen‐specific T lymphocyte lines capable of mediating autoimmune encephalomyelitis. Eur J Immunol 11: 195–199. [DOI] [PubMed] [Google Scholar]
- Benson MJ, Dillon SR, Castigli E, Geha RS, Xu S, Lam K‐P et al. (2008). Cutting edge: the dependence of plasma cells and independence of memory B cells on BAFF and APRIL. J Immunol 180: 3655–3659. [DOI] [PubMed] [Google Scholar]
- Bielekova B, Catalfamo M, Reichert‐Scrivner S, Packer A, Cerna M, Waldmann TA et al. (2006). Regulatory CD56(bright) natural killer cells mediate immunomodulatory effects of IL‐2Ralpha‐targeted therapy (daclizumab) in multiple sclerosis. Proc Natl Acad Sci U S A 103: 5941–5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielekova B, Howard T, Packer AN, Richert N, Blevins G, Ohayon J et al. (2009). Effect of anti‐CD25 antibody daclizumab in the inhibition of inflammation and stabilization of disease progression in multiple sclerosis. Arch Neurol 66: 483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloomgren G, Richman S, Hotermans C, Subramanyam M, Goelz S, Natarajan A et al. (2012). Risk of natalizumab‐associated progressive multifocal leukoencephalopathy. N Engl J Med 366: 1870–1880. [DOI] [PubMed] [Google Scholar]
- Bubien JK, Zhou LJ, Bell PD, Frizzell RA, Tedder TF (1993). Transfection of the CD20 cell surface molecule into ectopic cell types generates a Ca2+ conductance found constitutively in B lymphocytes. J Cell Biol 121: 1121–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claes N, Fraussen J, Stinissen P, Hupperts R, Somers V (2015). B cells are multifunctional players in multiple sclerosis pathogenesis: insights from therapeutic interventions. Front Immunol 6: 642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JA, Coles AJ, Arnold DL, Confavreux C, Fox EJ, Hartung HP et al. (2012). Alemtuzumab versus interferon beta 1a as first‐line treatment for patients with relapsing–remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet 380: 1819–1828. [DOI] [PubMed] [Google Scholar]
- Coles AJ, Twyman CL, Arnold DL, Cohen JA, Confavreux C, Fox EJ et al. (2012). Alemtuzumab for patients with relapsing multiple sclerosis after disease‐modifying therapy: a randomised controlled phase 3 trial. Lancet 380: 1829–1839. [DOI] [PubMed] [Google Scholar]
- Correale J, de los Milagros Bassani Molinas M (2002). Oligoclonal bands and antibody responses in multiple sclerosis. J Neurol 249: 375–389. [DOI] [PubMed] [Google Scholar]
- Dubey D, Kieseier BC, Hartung HP, Hemmer B, Miller‐Little WA, Stuve O (2015). Clinical management of multiple sclerosis and neuromyelitis optica with therapeutic monoclonal antibodies: approved therapies and emerging candidates. Expert Rev Clin Immunol 11: 93–108. [DOI] [PubMed] [Google Scholar]
- Duddy M, Niino M, Adatia F, Hebert S, Freedman M, Atkins H et al. (2007). Distinct effector cytokine profiles of memory and naive human B cell subsets and implication in multiple sclerosis. J Immunol 178: 6092–6099. [DOI] [PubMed] [Google Scholar]
- EMA (2016). EMA confirms recommendations to minimise risk of brain infection PML with Tysabri: European Medicines Agency.
- Emery P, Rigby W, Tak PP, Dorner T, Olech E, Martin C et al. (2014). Safety with ocrelizumab in rheumatoid arthritis: results from the ocrelizumab phase III program. PLoS One 9: e87379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraussen J, Claes N, de Bock L, Somers V (2014). Targets of the humoral autoimmune response in multiple sclerosis. Autoimmun Rev 13: 1126–1137. [DOI] [PubMed] [Google Scholar]
- Freedman MS, Kaplan JM, Markovic‐Plese S (2013). Insights into the mechanisms of the therapeutic efficacy of alemtuzumab in multiple sclerosis. J Clin Cell Immunol 4: 1000152. [PMC free article] [PubMed] [Google Scholar]
- Gasperi C, Stuve O, Hemmer B (2016). B cell‐directed therapies in multiple sclerosis. Neurodegener Dis Manag 6: 37–47. [DOI] [PubMed] [Google Scholar]
- Gold R, Giovannoni G, Selmaj K, Havrdova E, Montalban X, Radue EW et al. (2013). Daclizumab high‐yield process in relapsing–remitting multiple sclerosis (SELECT): a randomised, double‐blind, placebo‐controlled trial. Lancet 381: 2167–2175. [DOI] [PubMed] [Google Scholar]
- Górny MK, Wróblewska Z, Pleasure D, Miller SL, Wajgt A, Koprowski H (1983). CSF antibodies to myelin basic protein and oligodendrocytes in multiple sclerosis and other neurological diseases. Acta Neurol Scand 67: 338–347. [DOI] [PubMed] [Google Scholar]
- Gupta IV, Jewell RC (2012). Ofatumumab, the first human anti‐CD20 monoclonal antibody for the treatment of B cell hematologic malignancies. Ann N Y Acad Sci 1263: 43–56. [DOI] [PubMed] [Google Scholar]
- Hahne M, Kataoka T, Schröter M, Hofmann K, Irmler M, Bodmer J‐L et al. (1998). APRIL, a new ligand of the tumor necrosis factor family, stimulates tumor cell growth. J Exp Med 188: 1185–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartung HP, Aktas O, Boyko AN (2015). Alemtuzumab: a new therapy for active relapsing–remitting multiple sclerosis. Mult Scler 21: 22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser SL, Bar‐Or A, Comi G, Giovannoni G, Hartung HP, Hemmer B et al. (2016). Ocrelizumab versus interferon Beta‐1a in relapsing multiple sclerosis. N Engl J Med https://doi.org/10.1056/NEJMoa1601277. [DOI] [PubMed] [Google Scholar]
- Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ et al. (2008). B‐cell depletion with rituximab in relapsing–remitting multiple sclerosis. N Engl J Med 358: 676–688. [DOI] [PubMed] [Google Scholar]
- Havari E, Turner MJ, Campos‐Rivera J, Shankara S, Nguyen TH, Roberts B et al. (2014). Impact of alemtuzumab treatmet on the survival and function of human refulatory T cells in vitro. Immunology 141: 123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawker K, O'Connor P, Freedman MS, Calabresi PA, Antel J, Simon J et al. (2009). Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double‐blind placebo‐controlled multicenter trial. Ann Neurol 66: 460–471. [DOI] [PubMed] [Google Scholar]
- Herbst R, Wang Y, Gallagher S, Mittereder N, Kuta E, Damschroder M et al. (2010). B‐cell depletion in vitro and in vivo with an afucosylated anti‐CD19 antibody. J Pharmacol Exp Ther 335: 213–222. [DOI] [PubMed] [Google Scholar]
- Hua C, Audo R, Yeremenko N, Baeten D, Hahne M, Combe B et al. (2016). A proliferation inducing ligand (APRIL) promotes IL‐10 production and regulatory functions of human B cells. J Autoimmun 73: 64–72. [DOI] [PubMed] [Google Scholar]
- International Multiple Sclerosis Genetics Consortium (2005). A high density screen for linkage in multiple sclerosis. Am J Hum Genet 77: 454–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäger A, Dardalhon V, Sobel RA, Bettelli E, Kuchroo VK (2009). Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J Immunol 183: 7169–7177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JL, Coles AJ (2014). Mode of action and clinical studies with alemtuzumab. Exp Neurol 262 (Pt A): 37–43. [DOI] [PubMed] [Google Scholar]
- Kalincik T, Brown JW, Robertson N, Willis M, Scolding N, Rice CM et al. (2017). Treatment effectiveness of alemtuzumab compared with natalizumab, fingolimod, and interferon beta in relapsing–remitting multiple sclerosis: a cohort study. Lancet Neurol https://doi.org/10.1016/S1474‐4422(17)30007‐8. [DOI] [PubMed] [Google Scholar]
- Kalincik T, Horakova D, Spelman T, Jokubaitis V, Trojano M, Lugaresi A et al. (2015). Switch to natalizumab versus fingolimod in active relapsing–remitting multiple sclerosis. Ann Neurol 77: 425–435. [DOI] [PubMed] [Google Scholar]
- Kanzaki M, Shibata H, Mogami H, Kojima I (1995). Expression of calcium‐permeable cation channel CD20 accelerates progression through the G1 phase in Balb/c 3T3 cells. J Biol Chem 270: 13099–13104. [DOI] [PubMed] [Google Scholar]
- Kappos L, Hartung H‐P, Freedman MS, Boyko A, Radü EW, Mikol DD et al., for the ATAMS Study Group (2014). Atacicept in multiple sclerosis (ATAMS): a randomised, placebo‐controlled, double‐blind, phase 2 trial. Lancet Neurol 13: 353–363. [DOI] [PubMed] [Google Scholar]
- Kappos L, Li D, Calabresi PA, O'Connor P, Bar‐Or A, Barkhof F et al. (2011). Ocrelizumab in relapsing–remitting multiple sclerosis: a phase 2, randomised, placebo‐controlled, multicentre trial. Lancet 378: 1779–1787. [DOI] [PubMed] [Google Scholar]
- Kappos L, Wiendl H, Selmaj K, Arnold DL, Havrdova E, Boyko A et al. (2015). Daclizumab HYP versus interferon beta‐1a in relapsing multiple sclerosis. N Engl J Med 373: 1418–1428. [DOI] [PubMed] [Google Scholar]
- Kircher B, Latzer K, Gastl G, Nachbaur D (2003). Comparative in vitro study of the immunomodulatory activity of humanized and chimeric anti‐CD25 monoclonal antibodies. Clin Exp Immunol 134: 426–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knier B, Hemmer B, Korn T (2014). Novel monoclonal antibodies for therapy of multiple sclerosis. Expert Opin Biol Ther 14: 503–513. [DOI] [PubMed] [Google Scholar]
- Krumbholz M, Meinl I, Kumpfel T, Hohlfeld R, Meinl E (2008). Natalizumab disproportionately increased circulating pre‐B and B cells in multiple sclerosis. Neurology 71: 1350–1354. [DOI] [PubMed] [Google Scholar]
- Lanzillo R, Carotenuto A, Moccia M, Sacca F, Russo CV, Massarelli M et al. (2016). A longitudinal real‐life comparison study of natalizumab and fingolimod. Acta Neurol Scand https://doi.org/10.1111/ane.12718. [DOI] [PubMed] [Google Scholar]
- Lassmann H (2014). Multiple sclerosis: lessons from molecular neuropathology. Exp Neurol 262 (Part A): 2–7. [DOI] [PubMed] [Google Scholar]
- Levesque MC, St Clair EW (2008). B cell‐directed therapies for autoimmune disease and correlates of disease response and relapse. J Allergy Clin Immunol 121: 13–21. 2–3. [DOI] [PubMed] [Google Scholar]
- Li R, Rezk A, Miyazaki Y, Hilgenberg E, Touil H, Shen P et al., Canadian B cells in MS Team (2015). Proinflammatory GM‐CSF‐producing B cells in multiple sclerosis and B cell depletion therapy. Sci Transl Med 7: 310. [DOI] [PubMed] [Google Scholar]
- Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H (2000). Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol 47: 707–717. [DOI] [PubMed] [Google Scholar]
- Lühder F, Gold R (2014). Trial and error in clinical studies: lessons from ATAMS. Lancet Neurol 13: 340–341. [DOI] [PubMed] [Google Scholar]
- Lycke J (2015). Monoclonal antibody therapies or the treatment of relapsing‐remitting multiple sclerosis: differentiating mechanisms and clinical outcomes. Ther Adv Neurol Disord 8: 274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menge T, Dubey D, Warnke C, Hartung HP, Stuve O (2016). Ocrelizumab for the treatment of relapsing–remitting multiple sclerosis. Expert Rev Neurother 16: 1131–1139. [DOI] [PubMed] [Google Scholar]
- Milo R (2016). Therapeutic strategies targeting B‐cells in multiple sclerosis. Autoimmun Rev 15: 714–718. [DOI] [PubMed] [Google Scholar]
- Montalban X, Hauser SL, Kappos L, Arnold DL, Bar‐Or A, Comi G et al. (2016). Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med https://doi.org/10.1056/NEJMoa1606468. [DOI] [PubMed] [Google Scholar]
- Morsy DE, Sanyal R, Zaiss AK, Deo R, Muruve DA, Deans JP (2013). Reduced T‐dependent humoral immunity in CD20‐deficient mice. J Immunol 191: 3112–3118. [DOI] [PubMed] [Google Scholar]
- Naismith RT, Piccio L, Lyons JA, Lauber J, Tutlam NT, Parks BJ et al. (2010). Rituximab add‐on therapy for breakthrough relapsing multiple sclerosis: a 52‐week phase II trial. Neurology 74: 1860–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson T, Sun J, Hillert J, Höjeberg B, Ekre H‐P, Andersson G et al. (1992). Increased numbers of T cells recognizing multiple myelin basic protein epitopes in multiple sclerosis. Eur J Immunol 22: 1083–1087. [DOI] [PubMed] [Google Scholar]
- Ostergaard M, Baslund B, Rigby W, Rojkovich B, Jorgensen C, Dawes PT et al. (2010). Ofatumumab, a human anti‐CD20 monoclonal antibody, for treatment of rheumatoid arthritis with an inadequate response to one or more disease‐modifying antirheumatic drugs: results of a randomized, double‐blind, placebo‐controlled, phase I/II study. Arthritis Rheum 62: 2227–2238. [DOI] [PubMed] [Google Scholar]
- Palanichamy A, Jahn S, Nickles D, Derstine M, Abounasr A, Hauser SL et al. (2014). Rituximab efficiently depleted increased CD20‐expressing T‐cells in multiple sclerosis patients. J Immunol 193: 580–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker Harp CR, Archambault AS, Sim J, Ferris ST, Mikesell RJ, Koni PA et al. (2015). B cell antigen presentation is sufficient to drive neuroinflammation in an animal model of multiple sclerosis. J Immunol 194: 5077–5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pender MP, Csurhes PA, Greer JM, Mowat PD, Henderson RD, Cameron KD et al. (2000). Surges of increased T cell reactivity to an encephalitogenic region of myelin proteolipid protein occur more often in patients with multiple sclerosis than in healthy subjects. J Immunol 165: 5322–5331. [DOI] [PubMed] [Google Scholar]
- Perry JS, Han S, Xu Q, Herman ML, Kennedy LB, Csako G et al. (2012). Inhibition of LTi cell development by CD25 blockade is associated with decreased intrathecal inflammation in multiple sclerosis. Sci Transl Med 4 https://doi.org/10.1126/scitranslmed.3004140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polman CH, O'Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH et al. (2006). A randomized, placebo‐controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med 354: 899–910. [DOI] [PubMed] [Google Scholar]
- Reagan JL, Castillo JJ (2011). Ofatumumab for newly diagnosed and relapsed/refractory chronic lymphocytic leukemia. Expert Rev Anticancer Ther 11: 151–160. [DOI] [PubMed] [Google Scholar]
- Reff ME, Carner K, Chambers KS, Chinn PC, Leonard JE, Raab R et al. (1994). Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood 83: 435–445. [PubMed] [Google Scholar]
- Rice GP, Hartung HP, Calabresi PA (2005). Anti‐alpha4 integrin therapy for multiple sclerosis: mechanisms and rationale. Neurology 64: 1336–1342. [DOI] [PubMed] [Google Scholar]
- Robinson AP, Harp CT, Noronha A, Miller SD (2014). The experimental autoimmune encephalomyelitis (EAE) model of MS: utility for understanding disease pathophysiology and treatment. Handb Clin Neurol 122: 173–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruck T, Bittner S, Wiendl H, Meuth SG (2015). Alemtuzumab in multiple sclerosis: mechanism of action and beyond. Int J Mol Sci 16: 16414–16439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudick RA, Stuart WH, Calabresi PA, Confavreux C, Galetta SL, Radue EW et al. (2006). Natalizumab plus interferon beta‐1a for relapsing multiple sclerosis. N Engl J Med 354: 911–923. [DOI] [PubMed] [Google Scholar]
- Salzer J, Svenningsson R, Alping P, Novakova L, Bjorck A, Fink K et al. (2016). Rituximab in multiple sclerosis: a retrospective observational study on safety and efficacy. Neurology 87: 2074–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraste M, Penttilä T‐L, Airas L (2016). Natalizumab treatment leads to an increase in circulating CXCR3‐expressing B cells. Neurol Neuroimmunol Neuroinflamm 3: e292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider P, Mackay F, Steiner V, Hofmann K, Bodmer J‐L, Holler N et al. (1999). BAFF, a novel ligand of the tumor necrosis factor family, stimulates B cell growth. J Exp Med 189: 1747–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuh E, Berer K, Mulazzani M, Feil K, Meinl I, Lahm H et al. (2016). Features of human CD3+CD20+ T cells. J Immunol 197: 1111–1117. [DOI] [PubMed] [Google Scholar]
- Serafini B, Rosicarelli B, Magliozzi R, Stiglian E, Aloisi F (2004). Detection of ectopic B‐cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol 14: 164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergott RC, Bennett JL, Rieckmann P, Montalban X, Mikol D, Freudensprung U et al., ATON Trial Group (2015). ATON: results from a phase II randomized trial of the B‐cell‐targeting agent atacicept in patients with optic neuritis. J Neurol Sci 351: 174–178. [DOI] [PubMed] [Google Scholar]
- Sheridan C (2015). Anti‐CD20 antibody wows in multiple sclerosis. Nat Biotechnol 33: 1215–1216. [DOI] [PubMed] [Google Scholar]
- Snyder JT, Shen J, Azmi H, Hou J, Fowler DH, Ragheb JA (2011). Direct inhibition of CD40L expression can contribute to the clinical efficacy of daclizumab independently of its effects on cell division and Th1/Th2 cytokine production. Blood 109: 5399–5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen PS, Blinkenberg M (2016). The potential role for ocrelizumab in the treatment of multiple sclerosis: current evidence and future prospects. Ther Adv Neurol Disord 9: 44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen PS, Lisby S, Grove R, Derosier F, Shackelford S, Havrdova E et al. (2014). Safety and efficacy of ofatumumab in relapsing–remitting multiple sclerosis: a phase 2 study. Neurology 82: 573–581. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spelman T, Kalincik T, Jokubaitis V, Zhang A, Pellegrini F, Wiendl H et al. (2016). Comparative efficacy of first‐line natalizumab vs IFN‐beta or glatiramer acetate in relapsing MS. Neurol Clin Pract 6: 102–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stashenko P, Nadler LM, Hardy R, Schlossman SF (1980). Characterization of a human B lymphocyte‐specific antigen. J Immunol 125: 1678–1685. [PubMed] [Google Scholar]
- Stüve O, Marra CM, Jerome KR, Cook L, Cravens PD, Cepok S et al. (2006). Immune surveillance in multiple sclerosis patients treated with natalizumab. Ann Neurol 59: 743–747. [DOI] [PubMed] [Google Scholar]
- Tedder TF (2009). CD19: a promising B cell target for rheumatoid arthritis. Nat Rev Rheumatol 5: 572–577. [DOI] [PubMed] [Google Scholar]
- Thompson SA, Jones JL, Cox AL, Compton DAS, Coles AJ (2010). B‐cell reconsitution and BAFF after alemtuzumab (Campath‐1H) treatment of multiple sclerosis. J Clin Immunol 30: 99–105. [DOI] [PubMed] [Google Scholar]
- Traugott U, Reinherz EL, Raine CS (1983). Multiple sclerosis. Distribution of T cells, T cell subsets and Ia‐positive macrophages in lesions of different ages. J Neuroimmunol 4: 201–221. [DOI] [PubMed] [Google Scholar]
- Wierda WG, Padmanabhan S, Chan GW, Gupta IV, Lisby S, Osterborg A (2011). Ofatumumab is active in patients with fludarabine‐refractory CLL irrespective of prior rituximab: results from the phase 2 international study. Blood 118: 5126–5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams T, Coles A, Azzopardi L (2013). The outlook for alemtuzumab in multiple sclerosis. BioDrugs 27: 181–189. [DOI] [PubMed] [Google Scholar]
- Wuest SC, Edwan J, Martin JF, Han S, Perry JA, Cartagena CM et al. (2011). A vital role for IL‐2 trans‐presentation in DC‐mediated T cell activation in humans as revealed by daclizumab therapy. Nat Med 17: 604–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn D, Kaufman M, Montalban X, Vollmer T, Simon J, Elkins J et al. (2010). Daclizumab in active relapsing multiple sclerosis (CHOICE study): a phase 2, randomised, double‐blind, placebo‐controlled, add‐on trial with interferon beta. Lancet Neurol 9: 381–390. [DOI] [PubMed] [Google Scholar]
- Yaldizli O, Putzki N (2009). Natalizumab in the treatment of multiple sclerosis. Ther Adv Neurol Disord 2: 115–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Sun L, Wang S, Ko KH, Xu H, Zheng BJ et al. (2010). Novel function of B cell‐activating factor in the induction of IL‐10‐producing regulatory B cells. J Immunol 184: 3321–3325. [DOI] [PubMed] [Google Scholar]
- Yazawa N, Hamaguchi Y, Poe JC, Tedder TF (2005). Immunotherapy using unconjugated CD19 monoclonal antibodies in animal models for B lymphocyte malignancies and autoimmune disease. Proc Natl Acad Sci U S A 102: 15178–15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yednock TA, Cannon C, Fritz LC, Sanchez‐Madrid F, Steinman L, Karin N (1992). Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha 4 beta 1 integrin. Nature 356: 63–66. [DOI] [PubMed] [Google Scholar]
- Zhang J, Markovic‐Plese S, Lacet B, Raus J, Weiner HL, Hafler DA (1994). Increased frequency of interleukin 2‐responsive T cells specific for myelin basic protein and proteolipid protein in peripheral blood and cerebrospinal fluid of patients with multiple sclerosis. J Exp Med 179: 973–984. [DOI] [PMC free article] [PubMed] [Google Scholar]