

Abstract
Background and Purpose
Spinal cord injury (SCI) leads to severe motor and sensory dysfunction and significantly reduces the quality of life. The aim of the present work was to investigate the effect of administration of exogenous D‐β‐hydroxybutyrate (DBHB) on functional recovery and neuropathic pain in spinal cord‐injured mice.
Experimental Approach
Mice were given a moderate‐severe thoracic spinal contusion injury at the T9–10 level and treated with exogenous DBHB.
Key Results
Treatment of SCI mice with DBHB markedly improved locomotor function and relieved SCI‐induced hypersensitivities to mechanical and thermal stimulation. DBHB treatment partly prevented the SCI‐induced loss of motor neurons and suppressed microglial and glial activation. DBHB treatment enhanced histone acetylation and up‐regulated expression of the transcription factor FOXO3a, catalase and SOD2 in injured region of SCI mice. DBHB treatment suppressed SCI‐induced NLRP3 inflammasome activation and reduced protein expression of IL‐1β and IL‐18. In addition, DBHB treatment improved mitochondrial function and abated oxidative stress following SCI.
Conclusions and Implications
DBHB promoted functional recovery and relieved pain hypersensitivity in mice with SCI, possibly through inhibition of histone deacetylation and NLRP3 inflammasome activation and preservation of mitochondrial function. DBHB could thus be envisaged as a potential use of interventions for SCI but remains to be tested in humans.
Abbreviations
- BMS
Basso Mouse Scale
- DBHB
D‐β‐hydroxybutyrate
- GFAP
glial fibrillary acidic protein
- HDACs
histone deacetylases
- MDA
malondialdehyde
- MPO
myeloperoxidase
- SCI
spinal cord injury
Tables of Links
| LIGANDS |
|---|
| DBHB, D‐β‐hydroxybutyrate |
| H2O2 |
| IL‐1β |
| IL‐18 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a,b).
Introduction
Spinal cord injury (SCI), as a severe traumatic event that results in disturbances to normal sensory and motor function, is one of most important health problems worldwide with an annual incidence of 15 to 52.5 cases per million that appears to be rising (Singh et al., 2014). Among these patients, up to 80% also develop chronic central neuropathic pain, a complication of SCI, which contributes to unemployment and depression (Hagen and Rekand, 2015). Given the impact of SCI on these patients, effective clinical therapies that aim at improving neurologic outcomes and attenuating neuropathic pain after trauma are urgently needed.
It was reported that dietary restriction (Plunet et al., 2008), intermittent fasting (Jeong et al., 2011), ketogenic diet (Streijger et al., 2013) and exercise (Chang et al., 2014) improved functional recovery after contusion SCI. Interestingly, D‐β‐hydroxybutyrate (DBHB) levels were elevated during prolonged exercise, starvation or by the low carbohydrate ketogenic diet (Cotter et al., 2013). DBHB, a ketone body mainly produced by hepatocytes, is an alternative source of energy when glucose supply is depleted by serving as alternative source of ATP (Newman and Verdin, 2014). In vitro, DBHB prevented neuronal damage from glucose deprivation (Julio‐Amilpas et al., 2015; Camberos‐Luna et al., 2016) and protected cultured mesencephalic neurons from MPP+ toxicity and hippocampal neurons from Aβ(1–42) toxicity (Kashiwaya et al., 2000). In vivo, DBHB treatment protected neurons in models of Alzheimer's disease, Parkinson's disease and Huntington's disease (Tieu et al., 2003; Lim et al., 2011; Xie et al., 2015).
Therefore, DBHB could represent an interesting approach for management of functional impairment and neuropathic pain in spinal cord‐injured mice.
Methods
Animals
All animal care and experimental procedures complied with the Animals (Scientific Procedures) Act, 1986 of the UK Parliament, Directive 2010/63/EU of the European Parliament and the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication no. 85‐23, revised 1996) and were approved by the Animal Welfare and Ethics Review Board of the University of Bristol, UK. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). C57BL/6J (male, 12‐week‐old) mice were purchased from the Shanghai SiLaiKe Company (Shanghai, China). All the mice were housed with a 12 h light/12 h dark cycle at 22°C and free access to food and tap water.
Model of SCI and treatment
Before surgery, mice anaesthetized with ketamine and xylazine (80 and 10 mg·kg−1, respectively, i.p.) and subcutaneously injected with prophylactic antibiotics (1 mg·kg−1, Gentocin). The spinal cord was exposed by laminectomy at the T9‐T10 vertebral levels, followed by a contusion simulating moderate‐severe thoracic SCI using the Infinite Horizons Impactor (60 kdyn; Precision Systems and Instrumentation, Fairfax Station, VA, USA) (Jakeman et al., 2000). Bladders were voided manually twice, and hydration was monitored daily. Mice receiving a laminectomy at the T9‐T10 vertebral level without injury were used as sham controls.
For continuous delivery of the DBHB (Sigma‐Aldrich, MO, USA, 0.4, 0.8 and 1.6 mmol·kg−1·day−1), mini‐osmotic pumps (Model 2001, ALZET; DURECT Corp., Cupertino, CA, USA) (Tieu et al., 2003; Lim et al., 2011) were implanted subcutaneously (1 μL·h−1) 6 h after surgery. Control mice were infused with sterile PBS.
Locomotor function and sensitivity to mechanical and thermal stimulation
All experimental mice were assessed using the Basso Mouse Scale (BMS), from 0 (no ankle movement) to 9 (normal gait), for scoring hindlimb function as previously described (Basso et al., 2006). Mechanical allodynia and thermal sensitivity were recorded at times indicated post‐SCI as the methods described previously (Hargreaves et al., 1988; Martucci et al., 2008; Hoschouer et al., 2010) (these data are shown in the Supplementary data). Experiments was performed without knowledge of the treatments (blinded).
Immunohistochemistry
Two weeks following surgery, mice were perfusion fixed and spinal cord sections were stained against glial fibrillary acidic protein (GFAP; 1:500, Santa Cruz Biotechnology, CA, USA), IBA1 (1:200, Abcam, Cambridge, UK) and NeuN (1:500, Cell Signaling Technology, MA, USA). Sections were digitized using a whole‐slide scanner (Leica SCN400, Leica Microsystems, Germany), and high‐magnification images were captured using the Leica Image Viewer Software (Version 4.0.4). The evaluation was performed by two observers, blinded to study design.
Western blotting analysis
Two weeks after surgery, 5 mm of tissue centred on the injury epicentre or equivalent area in sham‐operated mice was manually dissected and homogenized in RIPA buffer and the proteins were extracted. Equal amounts of proteins (15 μg) were separated by SDS‐PAGE and transferred to nitrocellulose membranes. The membranes were incubated overnight at 4°C with antibodies directed against α‐AcH3K9 (Abcam), α‐histone H3 (1:1000, Abcam), caspase‐1 (1:1000, Santa Cruz), procaspase‐1 (1:2000, Santa Cruz) and NLRP3 (1:800, Santa Cruz) and diluted in Tris‐buffered saline‐Tween 20 containing 1% non‐fat dry milk. The loading control was β‐actin (1:5000, Abcam).
Measurement of activity of histone deacetylases (HDACs)
Activities of HDAC in spinal cords were determined using the colorimetric HDACs activity assay kit (BioVision, Mountain View, CA, USA).
Quantitative RT‐PCR analysis
Two weeks after surgery, 5 mm of tissue centred on the injury epicentre or equivalent area in sham‐operated mice was manually dissected and total RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and 1 μg of total RNA was reverse transcribed with random primers by using a reverse transcription kit (QIAGEN, Shanghai, China) according to the manufacturer's protocol. Target cDNA levels were quantified by RT‐PCR using specific primers:‐ (FOXO3a), 5′‐TGCCTTGTCAAATTCTGTC‐3′ and 5′‐TGCACTAGCTGAATACAGTGAG‐3′; SOD2, 5′‐ATGTTACAACTCAGGTCGCTCTTC‐3′ and 5′‐TGATAGCCTCCAGCAACTCTCC‐3′; catalase, 5′‐AGATACTCCAAGGCGAAGGTG3′ and 5′‐AAAGCCACGAGGGTCACGAAC3′; NeuN, 5′‐GGCAATGGTGGGACTCAAAA3′ and 5′‐GGGACCCGCTCCTTCAAC3′; GFAP, 5′‐CCAGCTTCGAGCCAAGGA′ and 5′‐GAAGCTCCGCCTGGTAGACA3′; and IBA1, 5′‐GAGGAGCCATGAGCCAAAGCA‐3′ and 5′‐CGAGGAATTGCTTGTTGATCCCC‐3′;] and the 7500 Fast RT‐PCR system (Applied Biosystems, Foster City, CA, USA) containing SYBR green. Target mRNA levels were normalized to the levels of expression of the internal control β‐actin (5′‐CATTGCTGACAGGATGCAGAA‐3′, 5′‐GCTGATCCACATCTGCTGGA‐3′).
Levels of IL‐1β, IL‐18 and malondialdehyde (MDA)
Two weeks after surgery, 5 mm of tissue centred on the injury epicentre or equivalent area in sham‐operated mice was manually dissected and homogenized in RIPA buffer (50 mM Tris, 150 mM NaCl, 5 mM EDTA, 1% Triton‐X 100, 0.1% SDS and 0.5% deoxycholate). Levels of IL‐1β and IL‐18 were determined by using ELISA kits (R&D Systems, Minneapolis, MN, USA). Levels of MDA (a presumptive marker of oxidant‐mediated lipid peroxidation) were determined by using a kit (Cayman, Ann Arbor, MI, USA).
Measurement of myeloperoxidase (MPO) activity
MPO activity was an indicator of polymorphonuclear leukocyte accumulation, reflecting the number and distribution of neutrophils in the tissues. Tissues were homogenized in 50 mM potassium phosphate buffer (pH 6.0, 1:40 wt/vol) and incubated for 2 h; homogenates were then centrifuged (30 000 x g, 15 min), and supernatant was collected for measurement of MPO activity, as previously described (Mullane et al., 1985).
Measurement of mitochondrial function
Mitochondrial respiration and ATP formation were measured according to the method described previously (Patel et al., 2009; Li et al., 2015). Mitochondrial formation of H2O2 and O2 − was determined according to the method described by Elks et al., (2009).
DBHB measurement
DBHB levels in spinal cord and serum were measured by using a DBHB Assay kit (MAK041; Sigma, MO, USA).
Data and statistical analyses
The data and statistical analyses comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). The data are expressed as the mean ± SD. Differences among groups were assessed using one‐way ANOVA, followed by Newman–Keuls post hoc tests, when F achieved the necessary level of statistical significance (P < 0.05) and there was no significant variance inhomogeneity. Data were significantly different at P < 0.05 according to the post hoc ANOVA analysis.
Results
Effects of administration of exogenous DBHB on locomotor function and neuropathic pain in mice exposed to SCI
BMS scoring revealed that SCI mice showed markedly impaired motor function (Figure 1A) and decreased mechanical response threshold (Figure 1B) and thermal withdrawal latency (Figure 1C). DBHB treatment significantly promoted the recovery of motor function, and the effect was dose‐dependent (Supporting Information Table S1). Additionally, treatment of SCI mice with DBHB enhanced mechanical response threshold and thermal withdrawal latency, indicating that DBHB attenuated the hypersensitivity to mechanical and thermal stimulation in SCI mice.
Figure 1.
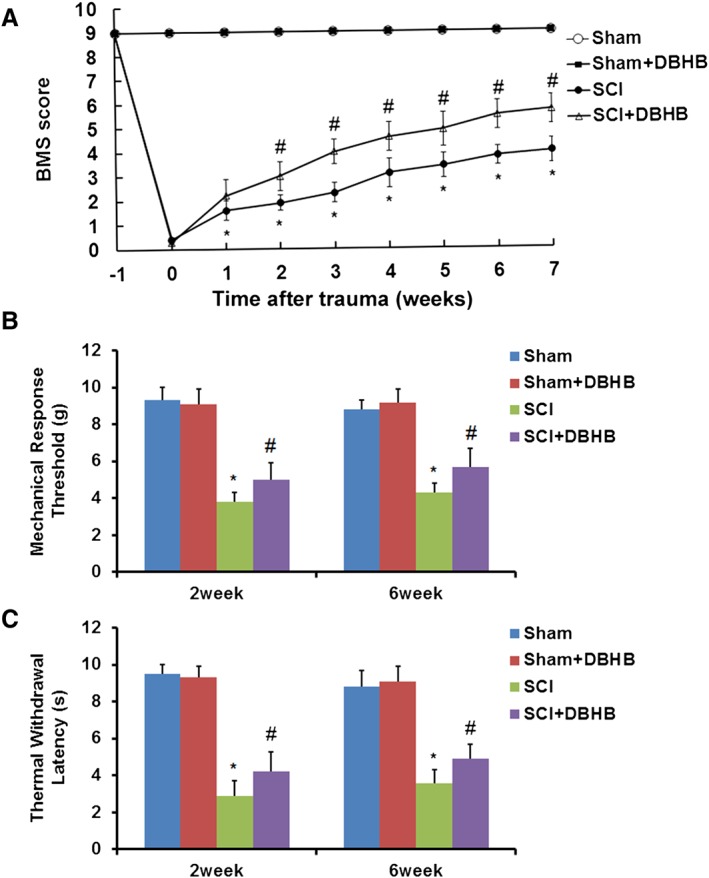
Effects of treatment with DBHB (1.6 mmol·kg−1·day−1) on locomotor function and neuropathic pain in mice exposed to SCI. The time course of locomotor recovery evaluated by the 9‐point BMS scoring (A) and sensitivities to mechanical (B) and thermal stimulation (C) after SCI are shown. Data are presented as the mean ± SD of eight mice per group. *P < 0.05, significantly different from sham‐operated mice; #P < 0.05, significantly different from SCI mice.
Effects of administration of exogenous DBHB on loss of motor neurons and microglial and glial activation in mice exposed to SCI
As shown with immunofluorescence staining, the number of NeuN‐positive cells was lower and the numbers of GFAP and IBA1‐positive cells were higher in spinal cords of SCI mice than those in sham‐operated mice (Figure 2A, B). Treatment of SCI mice with DBHB enhanced number of NeuN‐positive cells and reduced numbers of GFAP and IBA1‐positive cells, indicating that DBHB prevented the loss of motor neurons and suppressed microglial and glial activation. The mRNA levels of NeuN were lower, and mRNA levels of GFAP and IBA1 were higher in injured region of SCI mice, which was partly reversed by treatment with DBHB (Figure 2C).
Figure 2.
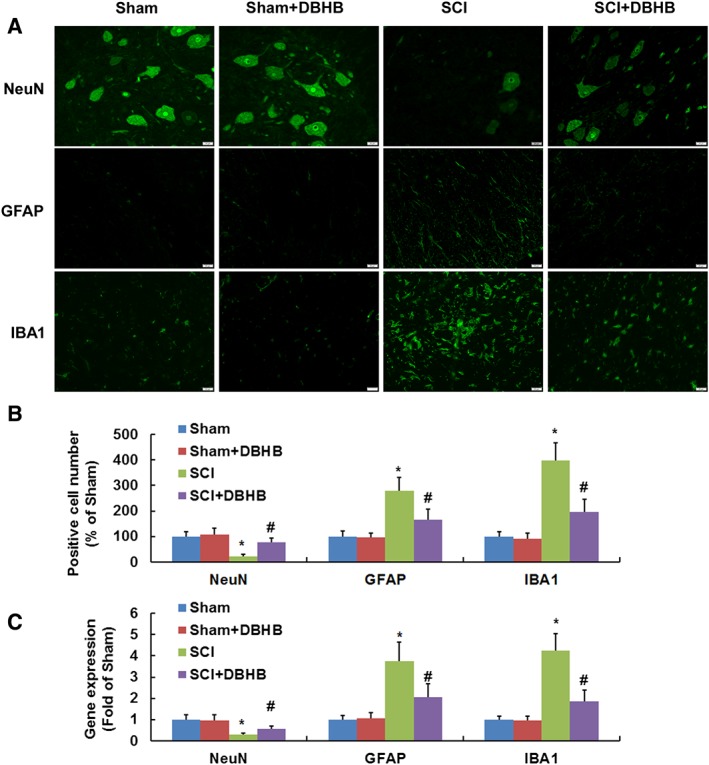
Effects of treatment with DBHB (1.6 mmol·kg−1·day−1) on loss of motor neurons and microglial and glial activation in mice exposed to SCI. Pictures (A, B) showed the sections (3 mm rostral to the epicentre, scale bar = 20 μm) immuno‐stained with antibodies recognizing NeuN, activated glia (GFAP) and microglia (IBA1). Samples of tissue (5 mm) centred on the injury epicentre or equivalent area in sham‐operated mice were manually dissected for RT‐PCR. Gene expression (C) of NeuN, GFAP and IBA1 measured by RT‐PCR are shown. Data are presented as the mean ± SD of eight mice per group. *P < 0.05, significantly different from sham‐operated mice; #P < 0.05, significantly different from SCI mice.
Effects of administration of exogenous DBHB on histone acetylation in spinal cord of mice exposed to SCI
To determine whether DBHB might have HDAC inhibitor activity, histone acetylation levels were measured by Western blotting with antibody to acetylated histone H3‐lysine 9 (AcH3K9). SCI in mice led to a reduction of histone acetylation, but DBHB treatment enhanced histone acetylation in both sham‐operated and SCI mice (Figure 3A). Furthermore, HDAC activities were higher in SCI mice than that in sham‐operated mice, and treatment with DBHB reduced HDACs activities in both sham‐operated mice and SCI mice (Figure 3B).
Figure 3.
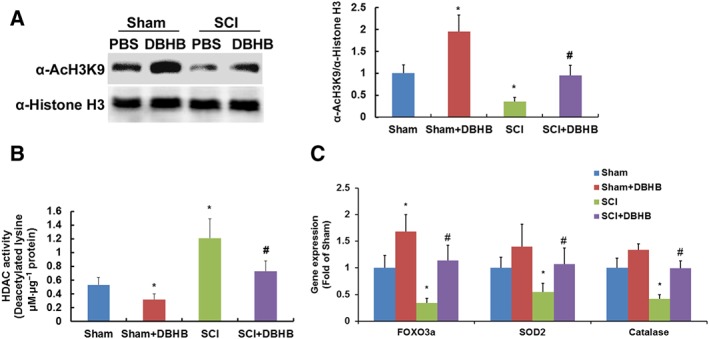
Effects of treatment with DBHB (1.6 mmol·kg−1·day−1) on histone acetylation in spinal cord of mice exposed to SCI. Samples of tissue (5 mm) centred on the injury epicentre or equivalent area in sham‐operated mice were manually dissected for measurements. Western blotting results andcorresponding quantification (A) of α‐AcH3K9 and α‐histone H3 were shown. The histone acetylation was evaluated by calculating the ratio of α‐AcH3K9/α‐histone H3. Graphs show the activities of HDACs (B) in spinal cord of mice exposed to SCI. Gene expression (C) of FOXO3a, SOD2 and catalase measured by RT‐PCR method are shown. Data are presented as the mean ± SD of eight mice per group. *P < 0.05, significantly different from sham‐operated mice; #P < 0.05, significantly different from SCI mice.
SCI led to a marked reduction of mRNA expression of FOXO3a, SOD2 and catalase (Figure 3C), which was reversed by treatment with DBHB. DBHB treatment in sham‐operated mice enhanced expression of FOXO3a significantly but expression of SOD2 and catalase was not significantly changed.
Effects of administration of exogenous DBHB on NLRP3 inflammasome activation in spinal cords of mice exposed to SCI
SCI in mice resulted in up‐regulation of NLRP3 protein expression (Figure 4A), activation of caspase‐1 (Figure 4A), up‐regulation of expression of IL‐1β (Figure 4B) and IL‐18 (Figure 4C) and activity of MPO (Figure 4D). DBHB treatment in SCI mice reduced protein expression of NLRP3, activation of caspase‐1, expression of IL‐1β and IL‐18 and activity of MPO, indicating that DBHB treatment could suppress NLRP3 inflammasome activation induced by SCI.
Figure 4.
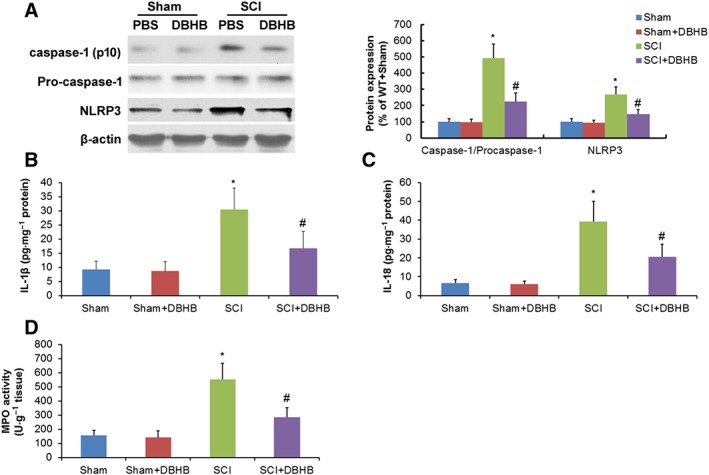
Effects of treatment with DBHB (1.6 mmol·kg−1·day−1) on NLRP3 inflammasome activation in spinal cord of mice exposed to SCI. Samples of tissue (5 mm) centred on the injury epicentre or equivalent area in sham‐operated mice were manually dissected for measurements. Western blotting results and corresponding quantification (A) of caspase‐1, pro‐caspase‐1 and NLRP3 are shown. Graphs show the levels of IL‐1β (B) and IL‐18 (C) and activity of MPO (D). Data are presented as the mean ± SD of eight mice per group. *P < 0.05, significantly different from sham‐operated mice; #P < 0.05, significantly different fromSCI mice.
Effects of administration of exogenous DBHB on mitochondrial dysfunction in spinal cord of mice exposed to SCI
When compared with sham‐operated mice, mitochondrial respiration rates (Figure 5A) of states III and V were lower and mitochondrial ATP formation (Figure 5B) was suppressed in injured region of SCI mice. In addition, mitochondrial formation of O2 − (Figure 5C) and H2O2 (Figure 5D) and MDA levels (Figure 5E) in injured regions were higher in SCI mice than that in sham‐operated mice. DBHB treatment in SCI mice led to enhancement of mitochondrial respiration rates and ATP formation, and reduction in mitochondrial formation of O2 − and H2O2 and MDA levels, indicating that DBHB treatment improved mitochondrial function and abated oxidative stress following SCI.
Figure 5.
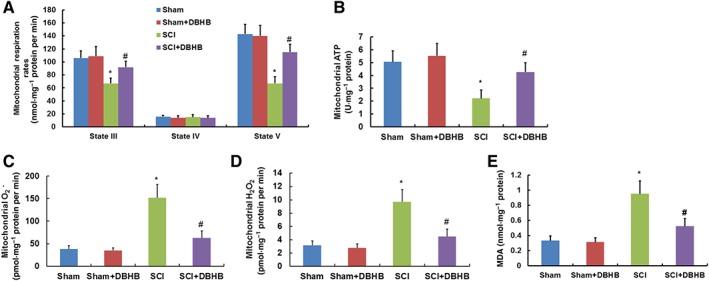
Effects of treatment with DBHB (1.6 mmol·kg−1·day−1) on mitochondrial dysfunction in spinal cord of mice exposed to SCI. Samples of tissue (5 mm) centred on the injury epicentre or equivalent area in sham‐operated mice were manually dissected for measurements. Graphs show the mitochondrial respiration rates (A) and formation of ATP (B), O2 − (C) and H2O2 (D) and MDA levels (E) in the injured site of spinal cord of mice exposed to SCI. Data are presented as the mean ± SD of eight mice per group. *P < 0.05, significantly different from sham‐operated mice; #P < 0.05, significantly different from SCI mice.
Levels of DBHB in serum and spinal cord of mice exposed to SCI and administration of exogenous DBHB
Immediately after mice were exposed to sham operation or SCI, pumps containing DBHB or PBS were implanted. Twenty‐four hours later, the blood and spinal cords were collected for determination of levels of DBHB. SCI had no significant effect on levels of DBHB in serum (Figure 6A) and spinal cord (Figure 6B). Administration of exogenous DBHB enhanced levels of DBHB in serum and spinal cord in both groups. In DBHB‐treated mice, levels of DBHB in spinal cords were lower in SCI mice than those in sham‐operated mice, indicating that the utilization of DBHB was increased when the spinal cord was exposed to trauma.
Figure 6.
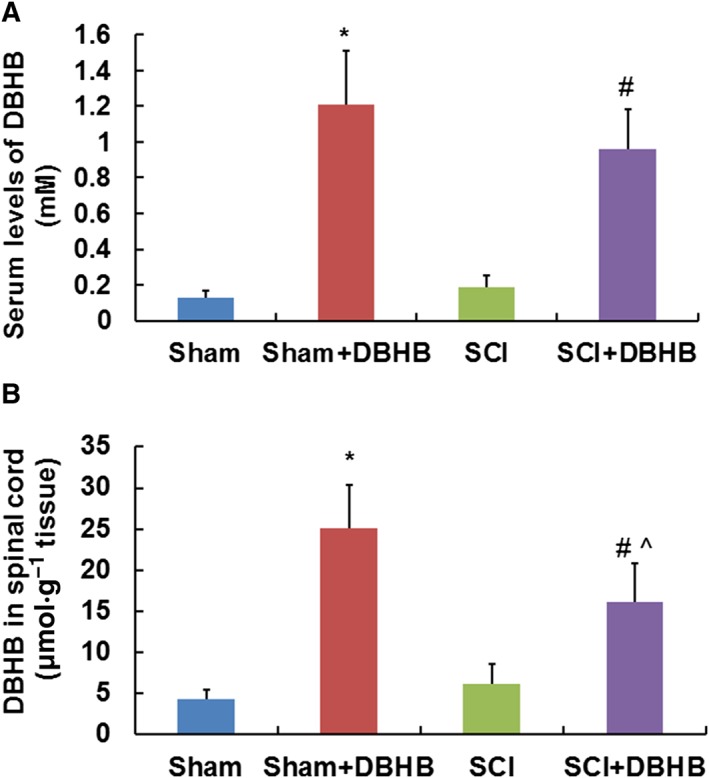
Levels of DBHB in serum and spinal cord of mice. After animals were exposed to sham‐operation or SCI, pumps containing DBHB (1.6 mmol·kg−1·day−1) or PBS were implanted. Twenty‐four hours later, the sera (A) and spinal cords (B) were collected for determination of levels of DBHB. Samples of tissue (5 mm) centred on the injury epicentre or equivalent area in sham‐operated mice were manually dissected for measurement. Data are presented as the mean ± SD of eight mice per group. *P < 0.05, significantly different from sham‐operated mice; #P < 0.05, significantly different from SCI mice; ^P < 0.05, significantly different from sham‐operated mice treated with DBHB.
Discussion
Our studies showed that infusion of the ketone body DBHB in mice conferred partial protection against SCI‐induced motor deficits and neuropathic pain, accompanied by a significant preservation of motor neurons and suppression of microglial and glial activation.
The ketone body DBHB is usually considered to be a convenient carrier of energy from the liver to peripheral tissues during fasting, caloric restriction or exercise. However, DBHB is not only a metabolite but also exerts important cellular signalling functions. DBHB was recently reported as an endogenous inhibitor of several HDACs including classes I and IIa (Shimazu et al., 2013). HDACs remove the acetyl groups from acetylated histones, leading to a more condensed chromatin, usually associated with gene silencing. Increased mRNA and protein expression of HDAC3 has been observed in peripheral blood mononuclear cells of SCI patients (Ma et al., 2015). Furthermore, pharmacological inhibitors of HDACs have demonstrated potent therapeutic effects in animal models of SCI (Lv et al., 2011). Following spinal nerve ligation, up‐regulation of HDAC1 expression and reduction of histone H3 acetylation was associated with neuropathic pain (Cherng et al., 2014). In vivo, treatment with HDACs inhibitors promotes axon regeneration (Finelli et al., 2013). As previously reported (Lim et al., 2011; Shimazu et al., 2013), DBHB treatment promoted acetylation of histone H3 in spinal cords. Consistent with inhibition of HDACs, DBHB correlated with global changes in transcription, including induction of FOXO3, in our experiments. Treatment of HEK293 cells with DBHB increased histone acetylation at the Foxo3a promoters and caused up‐regulation of mRNA expression of FOXO3a (Shimazu et al., 2013). FOXO3a was also down‐regulated in the injured region of SCI animals (Zhang et al., 2013). Mitochondrial SOD2 and catalase are two well‐defined targets of FOXO3a (Kops et al., 2002; Nemoto and Finkel, 2002). In our experiments, DBHB treatment enhanced SOD2 and catalase gene transcription in spinal cord following SCI, which contributed to its effect on oxidative stress induced by SCI.
Apart from oxidative stress, inflammation, manifested by extensive microglial and astroglial activation and infiltration of neutrophils and macrophages, was another contributor to post‐traumatic neurodegeneration (Moghaddam et al., 2015). The NLRP3 inflammasome, as an intracellular multiprotein complex involved in the activation of caspase‐1 and the processing of the proinflammatory cytokines IL‐1β and IL‐18, is one of the most studied inflammasomes in the CNS (de Rivero Vaccari et al., 2014). The cytokine IL‐1β induced hypersensitivity to mechanical and thermal stimulation when it was injected into peripheral and central tissues (Gruber‐Schoffnegger et al., 2013), and the relationship between neuropathic pain and increased IL‐1β levels in the spinal cord in several animal models has been described (Okamoto et al., 2001). Recently, DBHB was found to reduce NLRP3 inflammasome‐mediated IL‐1β and IL‐18 production in human monocytes andto attenuate NLRP3‐mediated diseases including Muckle–Wells syndrome, familial cold autoinflammatory syndrome and urate crystal‐induced peritonitis (Youm et al., 2015). In our work, DBHB treatment suppressed SCI‐induced NLRP3 inflammasome‐mediated IL‐1β and IL‐18 production and subsequent microglial and astroglial activation and infiltration of neutrophils and macrophages, which might contribute to the protective effects of DBHB on motor function recovery and neuropathic pain.
Changes in mitochondrial morphology and function, including energy production, free radicals, calcium overload and apoptosis, also play an important role in secondary damage following SCI (McEwen et al., 2011; Jia et al., 2016). DBHB improved the mitochondrial defects by increasing mitochondrial function and ATP production in SOD1‐G93A transgenic ALS mice (Zhao et al., 2006) and in the MPTP‐induced model of Parkinson's disease (Tieu et al., 2003). Kashiwaya et al. (2000) suggested that the neuroprotective effect of DBHB might be dependent on its ability to restore the substrates for mitochondrial electron transport or its ability to decrease the production of ROS in the mitochondria. In our work, DBHB treatment of SCI mice enhanced mitochondrial respiration rates and ATP formation and reduced mitochondrial H2O2 formation and MDA levels in the injured region. However, incubation of purified brain mitochondria with DBHB did not have antioxidant effects but increased ATP production (Tieu et al., 2003), which indicated that the antioxidant property of DBHB was not dependent on its direct effect on mitochondria but might be dependent on its specific inhibition of HDACs and subsequent transcription of the genes encoding oxidative stress resistance factors as described above.
Mitochondrial changes have been associated with NLRP3 activation in disease conditions. Mitochondrial dysfunction acted upstream of NLRP3 activation by providing ROS to trigger NLRP3 oligomerization, and mitochondria acted as a platform for inflammasome assembly (Ding et al., 2016; Yu and Lee, 2016). Mitochondrial events might also lie downstream of NLRP3 activation (Zhuang et al., 2014; Yu and Lee, 2016). Therefore, the effects of DBHB on NLRP3 activation and mitochondrial function could be expressed at several levels.
The short half‐life of DBHB raises the question of whether this compound would be a feasible treatment in humans. In addition, therapeutic utility of such an approach in the clinical setting was not economical. DBHB has been given orally to two 6‐month‐old infants with persistent hyperinsulinaemic hypoglycaemia for 5 and 7 months (Plecko et al., 2002). Despite the high dosage (up to 32 g·day−1), these patients appeared to tolerate the treatment quite well, without side effects. Whether oral administration of DBHB could be an effective means of achieving therapeutic levels against SCI, requires further work in animals and patients.
In conclusion, DBHB treatment partly reversed the trauma following SCI in mice. DBHB promoted functional recovery and relieved pain hypersensitivity in mice with SCI, possibly through inhibition of histone deacetylation and NLRP3 inflammasome activation and preservation of mitochondrial function.
Author contributions
J.Q., W.Z. and J.Y. did the conception and design of the experiments. J.Q., W.Z., M.L., B.N. and J.Y. carried out the collection, analysis and interpretation of data. J.Q. and J.Y. performed the drafting of the article.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Table S1 Effects of treatment with DBHB (0.4, 0.8, and 1.6 mmol/kg/day) on locomotor function and neuropathic pain in mice exposed to SCI.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (81301574, 81503066) and the Shanghai Science and Technology Committee Foundation (grant nos 134119a7400 and 14ZR1408200).
Qian, J. , Zhu, W. , Lu, M. , Ni, B. , and Yang, J. (2017) D‐β‐hydroxybutyrate promotes functional recovery and relieves pain hypersensitivity in mice with spinal cord injury. British Journal of Pharmacology, 174: 1961–1971. doi: 10.1111/bph.13788.
References
- Alexander SP, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso DM, Fisher LC, Anderson AJ, Jakeman LB, McTigue DM, Popovich PG (2006). Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma 23: 635–659. [DOI] [PubMed] [Google Scholar]
- Camberos‐Luna L, Gerónimo‐Olvera C, Montiel T, Rincon‐Heredia R, Massieu L (2016). The ketone body, β‐hydroxybutyrate stimulates the autophagic flux and prevents neuronal death induced by glucose deprivation in cortical cultured neurons. Neurochem Res 41: 600–609. [DOI] [PubMed] [Google Scholar]
- Chang CK, Chou W, Lin HJ, Huang YC, Tang LY, Lin MT et al. (2014). Exercise preconditioning protects against spinal cord injury in rats by upregulating neuronal and astroglial heat shock protein 72. Int J Mol Sci 15: 19018–19036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherng CH, Lee KC, Chien CC, Chou KY, Cheng YC, Hsin ST et al. (2014). Baicalin ameliorates neuropathic pain by suppressing HDAC1 expression in the spinal cord of spinal nerve ligation rats. J Formos Med Assoc 113: 513–520. [DOI] [PubMed] [Google Scholar]
- Cotter DG, Schugar RC, Crawford PA (2013). Ketone body metabolism and cardiovascular disease. Am J Physiol Heart Circ Physiol 304: H1060–H1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Hluwalia A, Alexander SPA, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rivero Vaccari JP, Dietrich WD, Keane RW (2014). Activation and regulation of cellular inflammasomes: gaps in our knowledge for central nervous system injury. J Cereb Blood Flow Metab 34: 369–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding W, Guo H, Xu C, Wang B, Zhang M, Ding F (2016). Mitochondrial reactive oxygen species‐mediated NLRP3 inflammasome activation contributes to aldosterone‐induced renal tubular cells injury. Oncotarget 7: 17479–17491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elks CM, Mariappan N, Haque M, Guggilam A, Majid DS, Francis J (2009). Chronic NF‐κB blockade reduces cytosolic and mitochondrial oxidative stress and attenuates renal injury and hypertension in SHR. Am J Physiol Renal Physiol 296: F298–F305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finelli MJ, Wong JK, Zou H (2013). Epigenetic regulation of sensory axon regeneration after spinal cord injury. J Neurosci 33: 19664–19676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber‐Schoffnegger D, Drdla‐Schutting R, Honigsperger C, Wunderbaldinger G, Gassner M, Sandkuhler J (2013). Induction of thermal hyperalgesia and synaptic long‐term potentiation in the spinal cord lamina I by TNF‐alpha and IL‐1beta is mediated by glial cells. J Neurosci 33: 6540–6551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen EM, Rekand T (2015). Management of neuropathic pain associated with spinal cord injury. Pain Ther 4: 51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J (1988). A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 32: 77–88. [DOI] [PubMed] [Google Scholar]
- Hoschouer EL, Basso DM, Jakeman LB (2010). Aberrant sensory responses are dependent on lesion severity after spinal cord contusion injury in mice. Pain 148: 328–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakeman LB, Guan Z, Wei P, Ponnappan R, Dzwonczyk R, Popovich PG et al. (2000). Traumatic spinal cord injury produced by controlled contusion in mouse. J Neurotrauma 17: 299–319. [DOI] [PubMed] [Google Scholar]
- Jeong MA, Plunet W, Streijger F, Lee JH, Plemel JR, Park S et al. (2011). Intermittent fasting improves functional recovery after rat thoracic contusion spinal cord injury. J Neurotrauma 28: 479–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia ZQ, Li G, Zhang ZY, Li HT, Wang JQ, Fan ZK et al. (2016). Time representation of mitochondrial morphology and function after acute spinal cord injury. Neural Regen Res 11: 137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julio‐Amilpas A, Montiel T, Soto‐Tinoco E, Gerónimo‐Olvera C, Massieu L (2015). Protection of hypoglycemia‐induced neuronal death by β‐hydroxybutyrate involves the preservation of energy levels and decreased production of reactive oxygen species. J Cereb Blood Flow Metab 35: 851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwaya Y, Takeshima T, Mori N, Nakashima K, Clarke K, Veech RL (2000). D‐beta‐hydroxybutyrate protects neurons in models of Alzheimer's and Parkinson's disease. Proc Natl Acad Sci U S A 97: 5440–5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). NC3Rs reporting guidelines working group. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ et al. (2002). Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 419: 316–321. [DOI] [PubMed] [Google Scholar]
- Li L, Guo JD, Wang HD, Shi YM, Yuan YL, Hou SX (2015). Prohibitin 1 gene delivery promotes functional recovery in rats with spinal cord injury. Neuroscience 286: 27–36. [DOI] [PubMed] [Google Scholar]
- Lim S, Chesser AS, Grima JC, Rappold PM, Blum D, Przedborski S et al. (2011). D‐β‐hydroxybutyrate is protective in mouse models of Huntington's disease. PLoS One 6: e24620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv L, Sun Y, Han X, Xu CC, Tang YP, Dong Q (2011). Valproic acid improves outcome after rodent spinal cord injury: potential roles of histone deacetylase inhibition. Brain Res 1396: 60–68. [DOI] [PubMed] [Google Scholar]
- Ma YD, Fang J, Liu H, Zhou L (2015). Increased HDAC3 and decreased miRNA‐130a expression in PBMCs through recruitment HDAC3 in patients with spinal cord injuries. Int J Clin Exp Pathol 8: 1682–1689. [PMC free article] [PubMed] [Google Scholar]
- Martucci C, Trovato AE, Costa B, Borsani E, Franchi S, Magnaghi V et al. (2008). The purinergic antagonist PPADS reduces pain related behaviours and interleukin‐1 beta, interleukin‐6, iNOS and nNOS overproduction in central and peripheral nervous system after peripheral neuropathy in mice. Pain 137: 81–95. [DOI] [PubMed] [Google Scholar]
- McEwen ML, Sullivan PG, Rabchevsky AG, Springer JE (2011). Targeting mitochondrial function for the treatment of acute spinal cord injury. Neurotherapeutics 8: 168–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghaddam A, Child C, Bruckner T, Gerner HJ, Daniel V, Biglari B (2015). Posttraumatic inflammation as a key to neuroregeneration after traumatic spinal cord injury. Int J Mol Sci 16: 7900–7916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullane KM, Kraemer R, Smith B (1985). Myeloperoxidase activity as a quantitative assessment of neutrophil infiltration into ischemic myocardium. J Pharmacol Methods 14: 157–167. [DOI] [PubMed] [Google Scholar]
- Nemoto S, Finkel T (2002). Redox regulation of forkhead proteins through a p66shc‐dependent signaling pathway. Science 295: 2450–2452. [DOI] [PubMed] [Google Scholar]
- Newman JC, Verdin E (2014). Ketone bodies as signaling metabolites. Trends Endocrinol Metab 25: 42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Martin DP, Schmelzer JD, Mitsui Y, Low PA (2001). Pro‐ and anti‐inflammatory cytokine gene expression in rat sciatic nerve chronic constriction injury model of neuropathic pain. Exp Neurol 169: 386–391. [DOI] [PubMed] [Google Scholar]
- Patel SP, Sullivan PG, Pandya JD, Rabchevsky AG (2009). Differential effects of the mitochondrial uncoupling agent, 2,4‐dinitrophenol, or the nitroxide antioxidant, tempol, on synaptic or nonsynaptic mitochondria after spinal cord injury. J Neurosci Res 87: 130–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plecko B, Stoeckler‐Ipsiroglu S, Schober E, Harrer G, Mlynarik V, Gruber S et al. (2002). Oral beta‐hydroxybutyrate supplementation in two patients with hyperinsulinemic hypoglycemia: monitoring of beta‐hydroxybutyrate levels in blood and cerebrospinal fluid, and in the brain by in vivo magnetic resonance spectroscopy. Pediatr Res 52: 301–306. [DOI] [PubMed] [Google Scholar]
- Plunet WT, Streijger F, Lam CK, Lee JH, Liu J, Tetzlaff W (2008). Dietary restriction started after spinal cord injury improves functional recovery. Exp Neurol 213: 28–35. [DOI] [PubMed] [Google Scholar]
- Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N et al. (2013). Suppression of oxidative stress by β‐hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 339: 211–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Tetreault L, Kalsi‐Ryan S, Nouri A, Fehlings MG (2014). Global prevalence and incidence of traumatic spinal cord injury. Clin Epidemiol 6: 309–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streijger F, Plunet WT, Lee JH, Liu J, Lam CK, Park S et al. (2013). Ketogenic diet improves forelimb motor function after spinal cord injury in rodents. PLoS One 8: e78765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieu K, Perier C, Caspersen C, Teismann P, Wu DC, Yan SD et al. (2003). D‐beta‐hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J Clin Invest 112: 892–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie G, Tian W, Wei T, Liu F (2015). The neuroprotective effects of β‐hydroxybutyrate on Aβ‐injected rat hippocampus in vivo and in Aβ‐treated PC‐12 cells in vitro. Free Radic Res 49: 139–150. [DOI] [PubMed] [Google Scholar]
- Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D et al. (2015). The ketone metabolite β‐hydroxybutyrate blocks NLRP3 inflammasome‐mediated inflammatory disease. Nat Med 21: 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JW, Lee MS (2016). Mitochondria and the NLRP3 inflammasome: physiological and pathological relevance. Arch Pharm Res 39: 1503–1518. [DOI] [PubMed] [Google Scholar]
- Zhang S, Huan W, Wei H, Shi J, Fan J, Zhao J et al. (2013). FOXO3a/p27kip1 expression and essential role after acute spinal cord injury in adult rat. J Cell Biochem 114: 354–365. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Lange DJ, Voustianiouk A, MacGrogan D, Ho L, Suh J et al. (2006). A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci 7: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang Y, Ding G, Zhao M, Bai M, Yang L, Ni J et al. (2014). NLRP3 inflammasome mediates albumin‐induced renal tubular injury through impaired mitochondrial function. J Biol Chem 289: 25101–25111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Effects of treatment with DBHB (0.4, 0.8, and 1.6 mmol/kg/day) on locomotor function and neuropathic pain in mice exposed to SCI.