

Abstract
Background and Purpose
Metformin, one of the most frequently prescribed medications for type 2 diabetes, reportedly exerts BP‐lowering effects in patients with diabetes. However, the effects and underlying mechanisms of metformin on BP in non‐diabetic conditions remain to be determined. The aim of the present study was to determine the effects of metformin on angiotensin II (Ang II) infusion‐induced hypertension in vivo.
Experimental Approach
The effects of metformin on BP were investigated in wild‐type (WT) C57BL/6J mice and in mice lacking AMP‐activated protein kinase α2 (AMPKα2) mice with or without Ang II infusion. Also, the effect of metformin on Ang II‐induced endoplasmic reticulum (ER) stress was explored in cultured human vascular smooth muscle cells (hVSMCs).
Key Results
Metformin markedly reduced BP in Ang II‐infused WT mice but not in AMPKα2‐deficient mice. In cultured hVSMCs, Ang II treatment resulted in inactivation of AMPK, as well as the subsequent induction of spliced X‐box binding protein‐1, phosphorylation of eukaryotic translation initiation factor 2α and expression of glucose‐regulated protein 78 kDa, representing three well‐characterized ER stress biomarkers. Moreover, AMPK activation by metformin ablated Ang II‐induced ER stress in hVSMCs. Mechanistically, metformin‐activated AMPKα2 suppressed ER stress by increasing phospholamban phosphorylation.
Conclusion and Implications
Metformin alleviates Ang II‐triggered hypertension in mice by activating AMPKα2, which mediates phospholamban phosphorylation and inhibits Ang II‐induced ER stress in vascular smooth muscle cells.
Abbreviations
- 4‐PBA
4‐phenylbutyric acid
- AICAR
5‐aminoimidazole‐4‐carboxamide ribonucleotide
- AMPK
AMP‐activated protein kinase
- Ang II
angiotensin II
- eIF2α
eukaryotic translation initiation factor 2α
- ER
endoplasmic reticulum
- Grp78
glucose‐regulated protein 78 kDa
- KDEL
Lys‐Asp‐Glu‐Leu
- PLB
phospholamban
- SERCA
sarcoplasmic/ER Ca2+‐ATPase
- TUDCA
tauroursodeoxycholic acid
- XBP1s
spliced X‐box binding protein‐1
Introduction
The widely used oral hypoglycaemic agent metformin (dimethylbiguanide) is a current first‐line treatment for type 2 diabetes (Pawlyk et al., 2014) and decreases hyperglycaemia (Hundal et al., 2000), body weight (Seifarth et al., 2013), hyperinsulinaemia (Kolodziejczyk et al., 2000) and cancer cell growth (Morales and Morris, 2015). Importantly, metformin was reported to markedly decrease BP in experimental rats (Verma et al., 1994a,b; Bhalla et al., 1996; Muntzel et al., 1997) and patients (Landin‐Wilhelmsen, 1992; Uehara et al., 2001). For example, metformin blunted salt‐induced hypertension (Muntzel et al., 1999) and prevented hypertension in spontaneously hypertensive rats (Tsai et al., 2014). Metformin treatment, however, failed to decrease BP in obese non‐diabetic patients with hypertension, in comparison with placebo (He et al., 2012) and in spontaneously hypertensive rats (Santure et al., 2000). In addition, metformin exerts only a minor, clinically insignificant, effect on BP in non‐diabetic hypertensives (Snorgaard et al., 1997). Thus, the anti‐hypertension effects of this hypoglycaemic agent are highly variable, depending upon the animal models or types of patients examined. The factors and/or molecular mechanisms that determine the responsiveness of blood vessels to BP‐lowering effects of metformin remain uncharacterized.
The activation of the renin–angiotensin–aldosterone system (RAAS) is established as a contributor to the pathogenesis of hypertension (Takahashi et al., 2011). Angiotensin II (Ang II) is thought to play a key role in regulating BP as well as water and sodium balance (Benigni et al., 2010). Furthermore, Ang II activates sympathetic nerve activity and arteriolar vasoconstriction, increases sodium and water retention and stimulates antidiuretic hormone secretion in the pituitary gland to increase water reabsorption in the collecting duct (Carey, 2015), ultimately resulting in elevated BP. Blockers of renin, angiotensin‐converting enzyme, Ang II AT1 receptors and aldosterone, all components of the RAAS, provide clinically useful, hypertension‐reducing treatments (Te Riet et al., 2015). Recently, AT2 receptor activation by compound 21, a highly selective non‐peptide agonist, prevented sodium retention and lowered BP in Ang II‐dependent hypertensive rats (Kemp et al., 2016). However, some discrepancies between these results and the real clinical outcomes of treatment remain, suggesting that other mechanisms by which Ang II may regulate the progression of hypertension could be important. Furthermore, endoplasmic reticulum (ER) stress in the brain subfornical organ was reported to contribute to Ang II‐dependent hypertension (Young et al., 2012). Our previous studies have shown that aberrant ER stress in vascular smooth muscle cells (VSMCs) enhances vascular contractility, resulting in high BP in mice (Liang et al., 2013). Thus, ER stress in VSMCs plays an essential role in the initiation and progression of Ang II‐induced hypertension.
AMP‐activated protein kinase (AMPK) is a central regulator of cellular metabolism and redox balance in mammalian tissues (Song and Zou, 2012). The vascular AMPK complex consists of three subunits, the α, β and γ subunits (Song and Zou, 2012). AMPKα2 deficiency causes aberrant ER stress in endothelial cells, resulting in endothelial dysfunction and accelerated atherogenesis in Western diet‐fed apolipoprotein E knockout mice (Dong et al., 2010a). AMPK activation inhibits LDL‐triggered ER stress in endothelium in vitro and in mice in vivo (Dong et al., 2010b). Moreover, AMPKα2 deletion leads to aberrant ER stress in VSMCs and subsequent high BP (Liang et al., 2013), confirming the notion that AMPKα2 and its physiological suppression of ER stress are essential for maintaining normal vascular tone.
Published work from us and others indicates that metformin exerts its therapeutic effects by activating AMPK (Zheng et al., 2013; Cho et al., 2015). Metformin represses palmitate‐induced ER stress in rat insulinoma cells (Simon‐Szabo et al., 2014), and also restores endothelial function in high‐fat diet‐induced obese mice, via blunting of ER stress (Cheang et al., 2014). AMPK inhibition results in excessive ER stress in carotid arteries of spontaneously hypertensive rats (Liu et al., 2015), whereas AMPK activation by 5‐aminoimidazole‐4‐carboxamide ribonucleotide (AICAR) paeonol, or berberine, suppresses ER stress in mouse aortas and rat carotid arteries (Liang et al., 2013; Liu et al., 2015; Choy et al., 2016). Based on our current understanding of Ang II infusion‐induced hypertension, we sought to determine whether AMPK‐suppressed ER stress is required for the BP‐lowering effects of metformin. Here, we report that Ang II‐induced aberrant ER stress in VSMCs contributes to the elevation of BP and that metformin acts via AMPKα2 activation‐induced suppression of ER stress in VSMCs to lower Ang II‐induced high BP in mice.
Methods
Animals
All animal care and experimental procedures were approved by the Institutional Animal Care and Use Committee of Georgia State University. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). Mice deficient in AMPKα1 (AMPKα1−/−) or AMPKα2 (AMPKα2−/−) were generated as described by Jorgensen et al., (2004). Wild‐type (WT) C57BL/6J, AMPKα1−/− and AMPKα2−/− mice (aged 10–12 weeks) +were anaesthetized and then implanted with osmotic minipumps containing Ang II or vehicle under the skin on the backs of the mice, as described previously (Wu et al., 2015). The mice were continuously infused with Ang II (0.8 μg·g−1·day−1, 14 days) or vehicle (saline). On the same day, half of the mice were treated with metformin (300 mg·kg−1 body weight per day in drinking water) or AICAR (500 mg·kg−1 body weight, one i.p. injection per day for 14 days, saline as vehicle). BP was measured using both the carotid catheter method and the radiotelemetry technique described previously (Liang et al., 2013). Mice were housed in temperature‐controlled cages under a 12 h light–dark cycle and given free access to water and a regular rodent diet.
Cell culture
Human VSMCs (hVSMCs) (ThermoFisher Scientific, Waltham, MA, USA) were cultured in M231 medium (Cascade Biologics, Portland, OR, USA) supplemented with 10% FBS, penicillin (100 U·mL−1) and streptomycin (100 μg·mL−1). All cells were incubated at 37°C in a humidified atmosphere of 5% CO2 and 95% air. Cells were grown to 70–80% confluence before being treated with the different agents.
Western blot analysis
Cell lysates were subjected to Western blot analysis as described previously (Song et al., 2009). The protein content was assayed using the BCA protein assay reagent (Pierce Chemical Co., Rockford, IL, USA). Proteins (20 μg) were separated by SDS‐PAGE and then transferred to a membrane. The membrane was incubated with a 1:1000 dilution of primary antibody, followed by a 1:5000 dilution of horseradish peroxidase‐conjugated secondary antibody. Protein bands were visualized with enhanced chemiluminescence (ECL) (Pierce Chemical Co.).
Immunohistochemistry
The mouse aorta and mesenteric artery were dissected, fixed in 4% paraformaldehyde for 24 h and embedded in paraffin. Sections were deparaffinized, rehydrated and microwaved in citrate buffer for antigen retrieval. Sections were successively incubated in endogenous peroxidase and alkaline phosphatase block buffer (Dako, Glostrup, Denmark), protein block buffer and primary antibodies, which were incubated with the sections overnight at 4°C. After rinsing in wash buffer, sections were incubated with labelled polymer–horseradish peroxidase anti‐mouse or anti‐rabbit antibodies and 3,3'‐diaminobenzidine (DAB) chromogen as described previously (Ding et al., 2016). After a final wash, the sections were counterstained with haematoxylin.
Data and statistical analysis
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Data were expressed as mean values ± SD. Distribution normality was assessed with the GraphPad Prism 5 analysis software (La Jolla, CA, USA), and all data were found to be normally distributed. Differences between groups were evaluated for significant differences using the Student's t‐test for unpaired data or the one‐way ANOVA with Bonferroni's post hoc test. All other results were analysed using a two‐tailed Student's t‐test for comparison between two groups. Values of P < 0.05 were considered significant.
Materials
Antibodies against total AMPKα, AMPKα1, AMPKα2, phospho‐AMPKα (T172; 40H9, 2535), phospho‐eukaryotic translation initiation factor 2α (eIF2α) and phospho‐phospholamban (S16/T17; 8496) were obtained from Cell Signaling Technology (Danvers, MA, USA). Antibodies against spliced X‐box binding protein‐1 (XBP1s) were obtained from Biolegend (San Diego, CA, USA). Antibodies against Lys‐Asp‐Glu‐Leu (KDEL) were obtained from Enzo Life Sciences (Farmingdale, NY). Antibodies against phospholamban (PLB) were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Osmotic minipumps were purchased from Alzet (Palo Alto, CA, USA). Tauroursodeoxycholic acid (TUDCA) and Compound C were from Calbiochem (San Diego, CA, USA). AICAR was purchased from Toronto Research Chemicals, Inc. (Toronto, Canada). All other chemicals, unless otherwise specified, were purchased from Sigma‐Aldrich (St. Louis, MO).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b).
Results
Metformin reduces Ang II‐induced hypertension in C57BL/6J mice
The model of Ang II‐induced hypertension reproduces key features of human essential hypertension (Te Riet et al., 2015). We first evaluated the effect of Ang II infusion (0.8 μg·g−1·day−1 for 14 days) on systolic (sBP) and diastolic BP (dBP) in mice. In line with previous reports (Liang et al., 2013), Ang II infusion caused a marked increase in both sBP and dBP in adult C57BL/6J wild‐type (WT) mice compared with the vehicle (saline) infusion (Figure 1A–C). Interestingly, metformin treatment did not alter the BP in vehicle‐infused mice, but did reduce sBP and dBP in Ang II‐infused mice.
Figure 1.
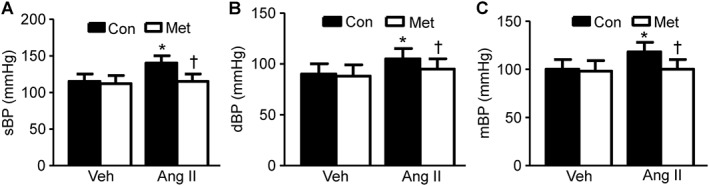
Metformin attenuates Ang II‐induced high BP in mice. (A–C) Effects of metformin (Met) on sBP (A), dBP (B) and mBP (C) in Ang II‐infused WT (C57BL/6J) mice. n = 12–16 in each group. *P < 0.05, significantly different from vehicle (Veh). †P < 0.05, significantly different from Ang II; two‐way ANOVA for repeated measures and Bonferroni post hoc test. Con, control.
Ang II induces ER stress in mice and hVSMCs
ER stress contributes to vascular smooth muscle contractility and high BP (Liang et al., 2013). Therefore, we next investigated whether Ang II infusion causes ER stress in the aorta. Immunohistochemical staining for three well‐characterized ER stress biomarkers [phosphorylated eIF2α (p‐eIF2α), XBP1s and KDEL; Zode et al., 2011] in the aortic media area from Ang II‐infused mice was stronger than that in aortas from vehicle‐treated mice (Figure 2A). Furthermore, the Immunohistochemical staining for p‐eIF2α and glucose‐regulated protein 78 kDa (Grp78) in the media area of the mesenteric artery, a resistance vessel, from Ang II‐infused mice was stronger than that from vehicle‐treated mice (Figure 2B). These results imply that ER stress in the aortic wall and resistance vessels may be associated with Ang II‐induced hypertension in vivo. We next investigated whether Ang II induces ER stress in cultured hVSMCs in vitro. As depicted in Figure 2C, the protein expression of three markers of ER stress, XBP1s, Grp78 and p‐eIF2α, was induced by Ang II in a dose‐dependent manner, peaking at 5 μM Ang II. Furthermore, the expression of ER stress biomarkers was also increased by Ang II in a time‐dependent manner, with observed increases after only 1 h Ang II exposure and a peak at 4 h treatment (Figure 2D). Intriguingly, TUDCA, a chemical chaperone that inhibits ER stress (Ben Mosbah et al., 2010), markedly decreased the Ang II‐induced increase in these three ER stress markers in hVSMCs (Figure 2E), suggesting that ER stress in VSMCs is induced by Ang II.
Figure 2.
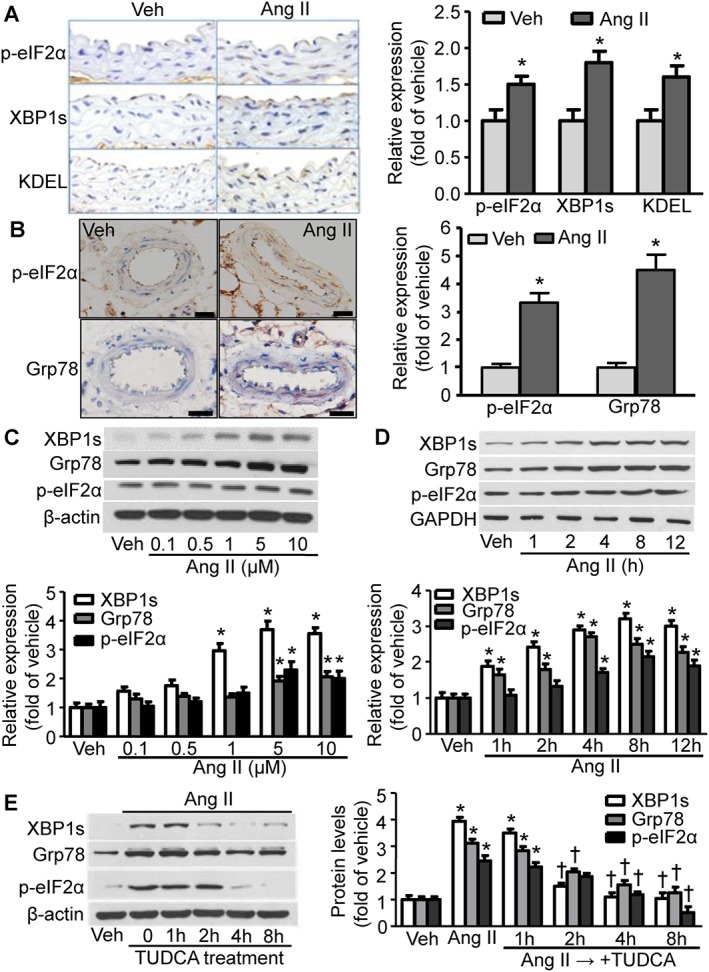
Ang II induces ER stress in mouse vessel wall and hVSMCs. (A) (Right) Representative images depict the staining of ER stress biomarkers in aorta sections isolated from mice treated with Ang II or vehicle (Veh). (Left) Quantification of the data from the right panel. n = 5, *P < 0.05, significantly different from corresponding Veh. (B) (Right) Representative images show the staining of ER stress biomarkers in sections of mesenteric artery from mice treated with Ang II or Veh. Scale bar = 20 μM. (Left) Quantification of the data from the right panel. n = 5, *P < 0.05, significantly different from corresponding Veh. (C) (Top) Representative immunoblots from four independent experiments depict the levels of ER stress biomarkers in hVSMCs treated with Ang II at different concentrations. (Bottom) Quantification of the above data. n = 5, *P < 0.05, significantly different from Veh. (D) (Top) Immunoblots showing the levels of ER stress biomarkers in hVSMCs treated with Ang II for the indicated times. (Bottom) Quantification of the above western blotting data. n = 5, *P < 0.05, significantly different from Veh. (E) (Right) Immunoblots of ER stress biomarkers in TUDCA‐treated hVSMCs also treated with Ang II. (Left) Quantification of the above immunoblot data. n = 5, *P < 0.05, significantly different from Veh. †P < 0.05, significantly different from Ang II.
Metformin eliminates Ang II‐induced ER stress in hVSMCs
We demonstrated previously that AMPK is a physiological suppressor of aberrant ER stress in endothelial cells (Dong et al., 2010a). Here, we investigated whether Ang II‐induced ER stress was dependent on AMPK inhibition in VSMCs. As shown in Figure 3A, low concentrations (0.1–1 μM) of Ang II led to minor activation of AMPK, whereas high concentrations (5 μM) of Ang II inhibited AMPKα phosphorylation in VSMCs. These data suggest that AMPK inactivation may contribute to Ang II‐induced ER stress in hVSMCs (Figure 2B). We next determined whether AMPK activation inhibits ER stress in hVSMCs. Metformin, a well‐known AMPK activator (Zou et al., 2004), dose‐dependently enhanced AMPK phosphorylation (Figure 3B), indicating AMPK activation in VSMCs (Zhang et al., 2008). Furthermore, metformin pretreatment decreased the ER stress biomarkers induced by the ER stress inducer tunicamycin (Figure 3C). As expected, treatment of hVSMCs with Ang II significantly enhanced ER stress as shown by increased levels of p‐eIF2α, KDEL and XBP1s. Importantly, metformin treatment reversed the inhibition of AMPK by Ang II and consequently ameliorated the ER stress that was induced by Ang II (Figure 3D). Furthermore, Ang II treatment significantly elevated the cytosolic Ca2+ (Figure 3E) concomitant with a reduction of Ca2+ in the ER (Figure 3F). Pretreatment with metformin substantially decreased this Ang II‐induced cytosolic Ca2+ elevation, whereas metformin pretreatment increased the ER Ca2+ levels that were reduced by Ang II (Figure 3F).
Figure 3.
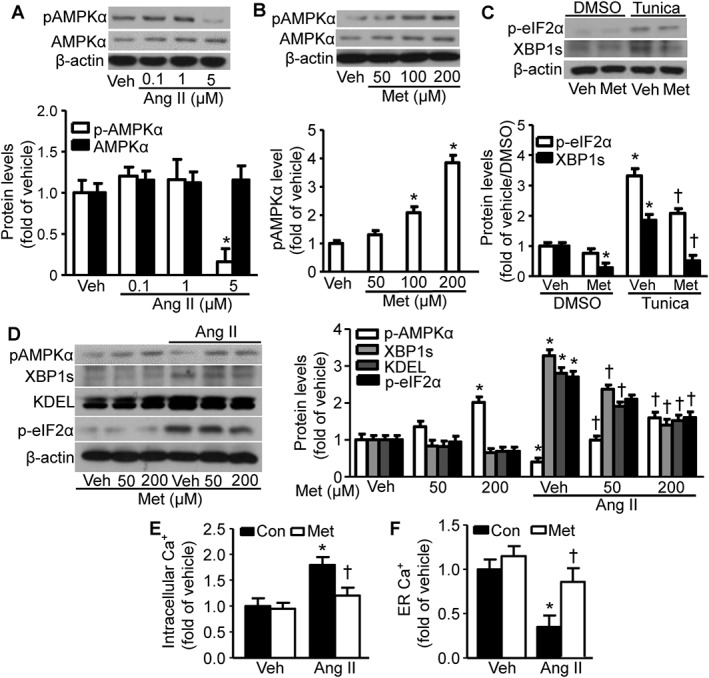
AMPKα activation by metformin (Met) reverses Ang II‐induced ER stress. (A) Effects of Ang II on AMPKα phosphorylation (pAMPKα) in hVSMCs. Representative blots from three independent experiments are shown. For each of these panels, data from these experiments were quantified. n = 5, *P < 0.05, significantly different from vehicle (Veh). (B) Effects of metformin on pAMPKα in hVSMCs. Representative blots from four independent experiments are shown. n = 5, *P < 0.05, significantly different from Veh. (C) Effects of metformin on tunicamycin (Tunica)‐induced ER stress biomarkers in hVSMCs. n = 5, *P < 0.05, significantly different from Veh/DMSO. †P < 0.05, significantly different from Tunica/Veh. (D) Effects of metformin on Ang II‐induced ER stress biomarkers in hVSMCs. Representative blots from three independent experiments are shown. n = 5, *P < 0.05, significantly different from Veh. †P < 0.05, significantly different from Veh/Ang II. (E) Effects of metformin on Ang II‐elevated intracellular Ca2+ levels in hVSMCs. n = 6,*P < 0.05, significantly different from Veh. †P < 0.05, significantly different from Ang II. (F) Effects of Met on Ang II‐reduced ER Ca2+ levels in hVSMCs. n = 6, *P < 0.05, significantly different from Veh. †P < 0.05, significantly different from Ang II. Con, control.
AMPK inactivation induces ER stress in hVSMCs
We next tested the effect of AMPK inactivation on ER stress. Compound C, a widely used AMPK inhibitor (Jin et al., 2009), clearly increased the levels of ER stress biomarkers and further enhanced the Ang II‐mediated ER stress in hVSMCs (Figure 4A, B). AMPKα1 knockdown in hVSMCs by AMPKα1‐specific siRNA led to a modest increase in ER stress. However, AMPKα2 knockdown by AMPKα2‐specific siRNA markedly induced ER stress as shown by increased expression of p‐eIF2α, protein disulfide isomerase and XBP1s, compared with levels following treatment with scrambled siRNA (Figure 4C). Furthermore, ER stress in VSMCs isolated from AMPKα2−/− mice was greater than that in VSMCs isolated from either AMPKα1−/− or WT mice (Figure 4D). These results suggest that AMPKα2 is the critical AMPKα isoform that regulates ER stress in VSMCs and that AMPKα2 plays a more important role than AMPKα1 in regulating Ang II‐induced hypertension in mice.
Figure 4.
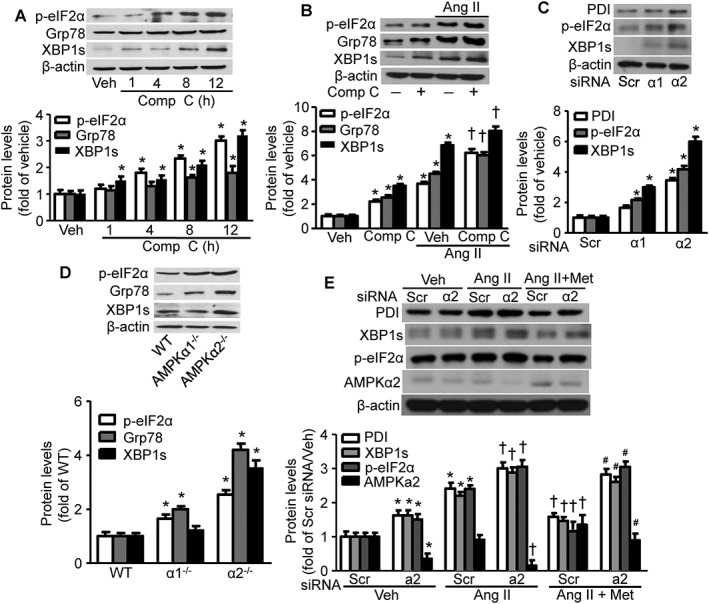
Metformin (Met) inhibition of Ang II‐induced ER stress is dependent on AMPKα2 (a2) activation. (A) Effects of Compound C (Comp C), a known AMPK inhibitor, on ER stress after incubation for the indicated times. Representative blots from four independent experiments are shown. For each panel, the quantitated data from these experiments are shown. n = 5, *P < 0.05, significantly different from vehicle (Veh). (B) Effects of Comp C on Ang II‐induced ER stress biomarkers. The blot is a representative of five blots from five individual experiments. n = 5, *P < 0.05, significantly different from Veh. †P < 0.05, significantly different from Veh/Ang II. (C) Effects of AMPKα2 knockdown on ER stress biomarkers in hVSMCs. The blot is a representative of five experiments. n = 5, *P < 0.05, significantly different from scrambled (Scr) siRNA. (D) The effects of AMPKα2 knockout in mVSMCs on ER stress biomarkers. The blot is a representative of four blots from four independent experiments. n = 5, *P < 0.05, significantly different from WT. (E) AMPKα2 is required for metformin inhibition on Ang II‐induced ER stress in hVSMCs. Representative blots from three independent experiments are shown. n = 5, *P < 0.05, significantly different from Scr siRNA. †P < 0.05, significantly different from Scr siRNA/Ang II. #P < 0.05, significantly different from Scr siRNA/Ang II + Met. PDI, protein disulfide isomerase; α1, AMPKα1.
Inhibition of Ang II‐induced ER stress by metformin is dependent on AMPKα2
We next investigated whether the protective mechanism of metformin is dependent on AMPKα2. As shown in Figure 4E, AMPKα2 siRNA, but not scrambled siRNA, resulted in ER stress in hVSMCs, in agreement with observations in AMPKα2−/− mVSMCs (Liang et al., 2013). Furthermore, metformin ameliorated the Ang II‐induced ER stress in scrambled siRNA‐transfected hVSMCs but not in AMPKα2 siRNA‐transfected hVSMCs (Figure 4E), suggesting that AMPKα2 is required for metformin alleviation of Ang II‐induced ER stress.
PLB phosphorylation is required for metformin inhibition of Ang II‐induced ER stress
Elevation of intracellular Ca2+ is a common mechanism involved in aberrant activation of ER stress and such changes of intracellular Ca2+ is regulated through the activity of sarcoplasmic/ER Ca2+‐ATPase (SERCA) (Dong et al., 2010a). PLB in its non‐phosphorylated form interacts with SERCA and inhibits its activity (Chen et al., 2007); however, upon PLB phosphorylation, SERCA functions normally and maintains ER Ca2+ levels (Koss and Kranias, 1996). Therefore, we next investigated the relevance of PLB to the inhibition by metformin of Ang II‐mediated ER stress. Ang II dose‐dependently decreased phosphorylation of PLB but did not affect total PLB protein levels (Figure 5A), while metformin restored PLB phosphorylation that had been blunted by Ang II treatment (Figure 5B). Furthermore, AMPKα2 silencing blocked the metformin‐induced inhibition of Ang II‐disrupted PLB phosphorylation (Figure 5C). Additionally, metformin suppressed Ang II‐induced ER stress in control, scrambled siRNA‐transfected hVSMCs but not in PLB siRNA‐transfected hVSMCs (Figure 5D), indicating that PLB is required for suppression of Ang II‐dependent ER stress by metformin.
Figure 5.
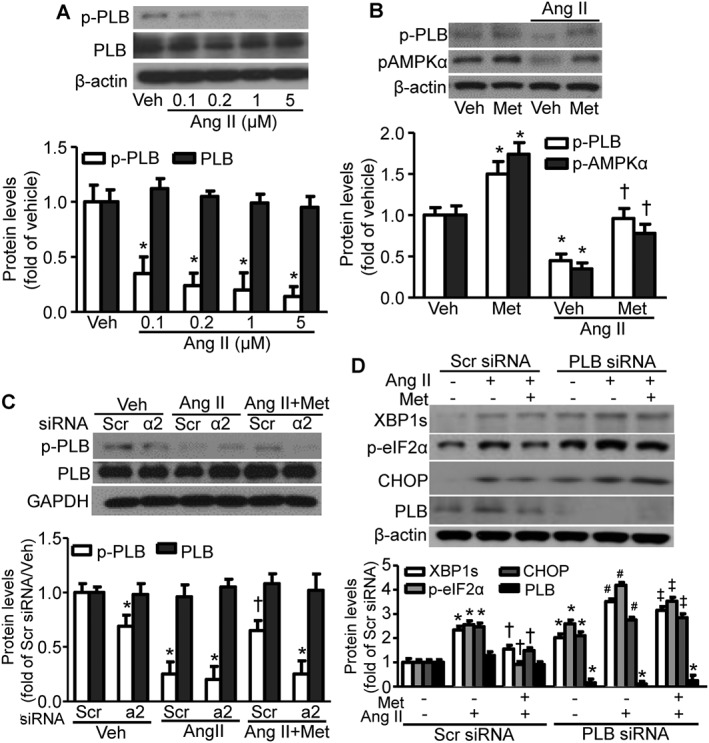
PLB phosphorylation is required for metformin (Met) inhibition of Ang II‐induced ER stress. (A) The effects of Ang II treatment of hVSMCs at the indicated concentrations on the phosphorylation of PLB. For each panel, immunoblot data from independent experiments were quantitated. n = 5, *P < 0.05, significantly different from vehicle (Veh). (B) Effects of metformin on PLB phosphorylation in the presence of Ang II treatment. n = 5, *P < 0.05, significantly different from Veh. †P < 0.05, significantly different from Veh/Ang II. (C) Effects of metformin on Ang II‐inhibited PLB phosphorylation in the presence and absence of AMPKα2. n = 4, *P < 0.05, significantly different from scrambled (Scr) siRNA/Veh. †P < 0.05 versus Scr siRNA/Ang II. (D) Effects of metformin on Ang II‐induced ER stress biomarkers in the presence and absence of PLB. The blots are representative of four blots from four independent experiments. n = 5, *P < 0.05, significantly different from Scr siRNA. †P < 0.05 versus Scr siRNA/Ang II. #P < 0.05 versus PLB siRNA. ‡P < 0.05, significantly different from Scr siRNA/Ang II + Met. p‐PLB, phosphorylated PLB. CHOP, C/EBP homologous protein.
AMPKα2 is required for metformin alleviation of Ang II‐induced hypertension in mice
In line with previous reports (Wang et al., 2011; Liang et al., 2013), the sBP, dBP and mean BP (mBP) in AMPKα2−/− mice were higher than those in WT mice under basal conditions (Figure 6A–C). Metformin treatment alone did not alter the BP in either WT or AMPKα2−/− mice under normal conditions (Figure 6A–C). Ang II infusion significantly increased BP (sBP, dBP and mBP) in both WT and AMPKα2−/− mice compared with vehicle treatment (Figure 6A–C). Importantly, metformin treatment alleviated Ang II‐induced increases in sBP, dBP and mBP in WT mice but not in AMPK α2−/− mice (Figure 6A–C), indicating that AMPKα2 is indeed essential for metformin alleviation of Ang II‐induced high BP. Interestingly, AICAR, a non‐selective AMPK activator (Zhang et al., 2009a), significantly reduced Ang II‐induced high sBP in both WT and AMPKα2−/− mice, although AICAR did not change the sBP in both WT and AMPKα2−/− mice under normal condition (Figure 6D).
Figure 6.
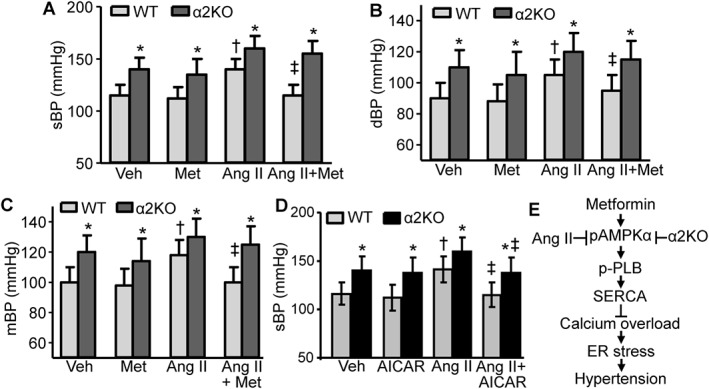
AMPKα2 is responsible for metformin (Met)‐induced reduction of Ang II‐induced high BP in vivo. (A–C) Effects of Met on sBP, dBP and mBP in Ang II‐infused AMPKα2 knockout mice (α2KO) and WT mice. n = 6–10 animals per group. *P < 0.05, significantly different from corresponding WT. †P < 0.05, significantly different from WT/vehicle (Veh). ‡P < 0.05, significantly different from WT/Ang II; two‐way ANOVA for repeated measures and Bonferroni post hoc test. (D) AICAR ameliorates Ang II‐induced sBP in both WT and α2KO mice. n = 5–8 mice per group. *P < 0.05, significantly different from corresponding WT. †P < 0.05 versus WT/Veh. ‡P < 0.05, significantly different from WT/Ang II or α2KO/Ang II respectively; two‐way ANOVA for repeated measures and Bonferroni post hoc test. (E) Proposed mechanism underlying the metformin‐induced reversal of Ang II‐induced abnormal calcium homeostasis, ER stress and hypertension. pAMPKα, phosphorylated AMPKα.
Discussion
In the present study, we demonstrated that Ang II‐induced aberrant ER stress in VSMCs contributes to the elevation of BP and that metformin lowers this Ang II‐induced high BP via AMPKα2 activation‐induced suppression of ER stress in VSMCs in mice. Mechanistically, metformin‐mediated AMPK activation promotes the phosphorylation of PLB, which ultimately restores cellular calcium homeostasis mediated by SERCA activation, inhibits ER stress and alleviates Ang II‐induced hypertension in mice (Figure 6E).
Elevated ER stress plays critical roles in the development and progression of cardiovascular diseases, including atherosclerotic plaque rupture (Saksi et al., 2014), coronary artery disease and diabetic cardiomyopathy (Yang et al., 2015b). Emerging evidence from humans (Du et al., 2017) as well as animal models indicates that enhanced ER stress is an important contributor to the development of hypertension (Hasty and Harrison, 2012; Liang et al., 2013). For example, prolonged ER stress in the rostral ventrolateral medulla contributes to oxidative stress‐associated neurogenic hypertension in spontaneously hypertensive rats (Chao et al., 2013), and accordingly, the ER stress inhibitor TUDCA decreases sBP in the spontaneously hypertensive rats (Choi et al., 2016). 4‐Phenylbutyric acid (4‐PBA), which is an ER stress inhibitor that is structurally unrelated to TUDCA, also lowers monocrotaline‐induced pulmonary artery pressure in male Wistar rats (Wu et al., 2016). Furthermore, ER stress may be responsible for obesity‐induced hypertension. Importantly, the main finding of the present work is that ER stress, mediated by AMPK inhibition, in VSMCs is a major cause underlying Ang II‐dependent hypertension. We also demonstrated that the effect of metformin on suppression of ER stress in VSMCs is responsible for its BP‐lowering effect. Thus, metformin alleviates Ang II‐dependent hypertension via reduction of ER stress mediated by activation of AMPK. In agreement with these conclusions, we have demonstrated that AMPKα2 deletion promoted ER stress in the mouse aorta (Liang et al., 2013). As Ang II‐inhibited AMPK activation (Figure 3A) and AMPK activation by metformin (Figure 3B) decreased Ang II‐mediated ER stress in VSMCs (Figure 3D), Ang II‐triggered ER stress may occur via AMPK inhibition. This finding is consistent with our previous study which demonstrated that AMPK acted as a physiological suppressor of ER stress by maintaining SERCA activity and intracellular Ca2+ homeostasis in endothelial cells (Dong et al., 2010a). In addition, AMPK activation by irisin, a polypeptide containing 112 amino acids, secreted mainly by skeletal muscle cells during exercise, has been shown to lower BP in spontaneously hypertensive rats by improving NO bioactivity and endothelial cell function (Fu et al., 2016a). This has been mechanistically attributed to the higher NO release by enhancing the phosphorylation and activation of endothelial NOS (eNOS) at Ser1177 and Ser633 (Chen et al., 2009; Zhang et al., 2009b) and by blocking NO inactivation by reactive oxygen species (Deng et al., 2010). Indeed, a recent study demonstrates that the endothelium‐specific AMPKα2 knockout mice have normotensive phenotype and endothelium‐specific AMPKα1 knockout mice are hypertensive (Enkhjargal et al., 2014), although the level of ER stress of these mice was not investigated. Taken together, ER stress‐mediated endothelial dysfunction associated with eNOS uncoupling or endothelial oxidative stress might also be a critical trigger for the development of high BP (Cheang et al., 2014; Galan et al., 2014). Further study on a causative effect of AMPK deletion‐induced ER stress in VSMCs and BP elevation is needed.
In addition, the ER stress suppressors, 4‐phenylbutyric acid (4‐PBA) (Tabas, 2010) or TUDCA, lower Ang II‐elevated BP (Young et al., 2012; Liang et al., 2013). Although metformin alone did not alter the BP in either WT or AMPKα2−/− mice under normal conditions, metformin effectively normalized Ang II‐elevated BP in WT mice but not in AMPKα2−/− mice (Figure 6A–C). However, the non‐FDA‐approved drug AICAR ameliorates Ang II‐increased BP in AMPKα2−/− mice (Figure 6D), which may be due to the ER stress reduction and recovery of endothelial function (Dong et al., 2010b; Li et al., 2015). These data suggest that the AMPKα2 isoform is required for maintaining normal BP and is involved in metformin alleviation of Ang II‐induced hypertension. These results are in line with the notion that AMPKα2 is esssential for the BP‐lowering effects of resveratrol in deoxycorticosterone acetate–salt hypertensive mice (Sun et al., 2015). Together, these results offer a possible explanation for the lack of effects of metformin on BP reduction in obese hypertensive patients (He et al., 2012). Indeed, obesity is known to decrease or inhibit AMPKα in animals (Decleves et al., 2011; Davies et al., 2014; Fu et al., 2016b) and patients (Xu et al., 2012). In addition, metformin may have no effect on high BP in the absence of ER stress or in cases of hypertension with normal AMPK activity. Thus, metformin only exerts its role in lowering BP under conditions where AMPKα2 is present, AMPK activity is inhibited and ER stress is elevated. These requirements offer a clear explanation of the paradoxical effects of metformin on hypertension in the clinic.
PLB is the principal physiological inhibitor of SERCA (Koss and Kranias, 1996), and phosphorylation of PLB at Ser16 or Thr17 enhances SERCA function to maintain ER calcium levels by decreasing the efficacy of PLB‐mediated inhibition (Traister et al., 2014). Recently, metformin was reported to enhance PLB degradation via autophagy in cardiomyocytes, exerting a protective function in hearts (Teng et al., 2015). Furthermore, AMPK activation by A769662 increases PLB phosphorylation at Thr17 in pooled femoral arteries (Schneider et al., 2015). In agreement with these earlier data, we demonstrated that treatment of hVSMCs with Ang II reduced the level of phosphorylated PLB. The AMPKα2 isoform in VSMCs is required for metformin to block the inhibition, by Ang II, of PLB phosphorylation. Whether AMPKα2 directly or indirectly phosphorylates PLB, however, warrants further investigation.
It remains to be determined whether other mechanisms are involved in the effects of metformin on lowering BP in humans. For example, metformin prevents hypertension in spontaneously hypertensive rats by reducing levels of asymmetric dimethylarginine (Tsai et al., 2014). Metformin decreases NAD(P)H oxidase activity in mouse podocytes, leading to reduction of oxidative stress (Piwkowska et al., 2010). Metformin restores endothelial function inhibited by changes in glucose levels, via AMPK‐dependent eNOS recoupling and reduction of p47‐phox, a subunit of NADPH oxidase (An et al., 2016). Such reduction of oxidative stress and restoration of endothelial function contribute to the reduced BP in streptozotocin‐induced diabetic rats (Majithiya and Balaraman, 2006). In addition, metformin decreases Ang II‐elevated BP in mice, which may occur via induction of urinary sodium excretion (Deji et al., 2012). Alternatively, metformin may exert these effects via induction of ER stress in prostate cancer cells (Yang et al., 2015a). Therefore, further investigation of these mechanisms is warranted in light of our findings.
In summary, our results indicate that aberrant ER stress accompanied the development of Ang II‐mediated, high BP. Metformin inhibited Ang II‐induced ER stress via an AMPKα2–PLB–SERCA pathway, and this pathway may provide a novel therapeutic target for treating hypertension.
Author contributions
Q.D., P.S. and M.‐H.Z. designed the experiments and performed data analysis. Q.D. and Y.D. conducted the experiments. P.S. and M.‐H.Z. wrote and revised the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This study was supported by grants from the National Institutes of Health (HL079584, HL080499, HL089220, HL110488, HL128014, HL132500, AG047776 and CA213022). This work was, in part, supported by the Georgia Research Alliance. Dr Zou is a Georgia Research Alliance Eminent Scholar in Molecular Medicine.
Duan, Q. , Song, P. , Ding, Y. , and Zou, M.‐H. (2017) Activation of AMP‐activated protein kinase by metformin ablates angiotensin II‐induced endoplasmic reticulum stress and hypertension in mice in vivo . British Journal of Pharmacology, 174: 2140–2151. doi: 10.1111/bph.13833.
References
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An H, Wei R, Ke J, Yang J, Liu Y, Wang X et al. (2016). Metformin attenuates fluctuating glucose‐induced endothelial dysfunction through enhancing GTPCH1‐mediated eNOS recoupling and inhibiting NADPH oxidase. J Diabetes Complications 30: 1017–1024. [DOI] [PubMed] [Google Scholar]
- Ben Mosbah I, Alfany‐Fernandez I, Martel C, Zaouali MA, Bintanel‐Morcillo M, Rimola A et al. (2010). Endoplasmic reticulum stress inhibition protects steatotic and non‐steatotic livers in partial hepatectomy under ischemia–reperfusion. Cell Death Dis 1: e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benigni A, Cassis P, Remuzzi G (2010). Angiotensin II revisited: new roles in inflammation, immunology and aging. EMBO Mol Med 2: 247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla RC, Toth KF, Tan E, Bhatty RA, Mathias E, Sharma RV (1996). Vascular effects of metformin. Possible mechanisms for its antihypertensive action in the spontaneously hypertensive rat. Am J Hypertens 9: 570–576. [DOI] [PubMed] [Google Scholar]
- Carey RM (2015). The intrarenal renin–angiotensin system in hypertension. Adv Chronic Kidney Dis 22: 204–210. [DOI] [PubMed] [Google Scholar]
- Chao YM, Lai MD, Chan JY (2013). Redox‐sensitive endoplasmic reticulum stress and autophagy at rostral ventrolateral medulla contribute to hypertension in spontaneously hypertensive rats. Hypertension 61: 1270–1280. [DOI] [PubMed] [Google Scholar]
- Cheang WS, Tian XY, Wong WT, Lau CW, Lee SS, Chen ZY et al. (2014). Metformin protects endothelial function in diet‐induced obese mice by inhibition of endoplasmic reticulum stress through 5′ adenosine monophosphate‐activated protein kinase–peroxisome proliferator‐activated receptor delta pathway. Arterioscler Thromb Vasc Biol 34: 830–836. [DOI] [PubMed] [Google Scholar]
- Chen Z, Akin BL, Jones LR (2007). Mechanism of reversal of phospholamban inhibition of the cardiac Ca2+‐ATPase by protein kinase A and by anti‐phospholamban monoclonal antibody 2D12. J Biol Chem 282: 20968–20976. [DOI] [PubMed] [Google Scholar]
- Chen Z, Peng IC, Sun W, Su MI, Hsu PH, Fu Y et al. (2009). AMP‐activated protein kinase functionally phosphorylates endothelial nitric oxide synthase Ser633. Circ Res 104: 496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho K, Chung JY, Cho SK, Shin H‐W, Jang I‐J, Park J‐W et al. (2015). Antihyperglycemic mechanism of metformin occurs via the AMPK/LXRα/POMC pathway. Sci Rep 5: 8145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SK, Lim M, Byeon SH, Lee YH (2016). Inhibition of endoplasmic reticulum stress improves coronary artery function in the spontaneously hypertensive rats. Sci Rep 6: 31925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy KW, Mustafa MR, Lau YS, Liu J, Murugan D, Lau CW et al. (2016). Paeonol protects against endoplasmic reticulum stress‐induced endothelial dysfunction via AMPK/PPARdelta signaling pathway. Biochem Pharmacol 116: 51–62. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies M, Fraser SA, Galic S, Choy SW, Katerelos M, Gleich K et al. (2014). Novel mechanisms of Na+ retention in obesity: phosphorylation of NKCC2 and regulation of SPAK/OSR1 by AMPK. Am J Physiol Renal Physiol 307: F96–F106. [DOI] [PubMed] [Google Scholar]
- Decleves AE, Mathew AV, Cunard R, Sharma K (2011). AMPK mediates the initiation of kidney disease induced by a high‐fat diet. J Am Soc Nephrol 22: 1846–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deji N, Kume S, Araki S, Isshiki K, Araki H, Chin‐Kanasaki M et al. (2012). Role of angiotensin II‐mediated AMPK inactivation on obesity‐related salt‐sensitive hypertension. Biochem Biophys Res Commun 418: 559–564. [DOI] [PubMed] [Google Scholar]
- Deng G, Long Y, Yu YR, Li MR (2010). Adiponectin directly improves endothelial dysfunction in obese rats through the AMPK–eNOS Pathway. Int J Obes (Lond) 34: 165–171. [DOI] [PubMed] [Google Scholar]
- Ding Y, Chen J, Okon IS, Zou MH, Song P (2016). Absence of AMPKalpha2 accelerates cellular senescence via p16 induction in mouse embryonic fibroblasts. Int J Biochem Cell Biol 71: 72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Zhang M, Liang B, Xie Z, Zhao Z, Asfa S et al. (2010a). Reduction of AMP‐activated protein kinase {alpha}2 increases endoplasmic reticulum stress and atherosclerosis in vivo . Circulation 121: 792–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Zhang M, Wang S, Liang B, Zhao Z, Liu C et al. (2010b). Activation of AMP‐activated protein kinase inhibits oxidized LDL‐triggered endoplasmic reticulum stress in vivo . Diabetes 59: 1386–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L, He F, Kuang L, Tang W, Li Y, Chen D (2017). eNOS/iNOS and endoplasmic reticulum stress‐induced apoptosis in the placentas of patients with preeclampsia. J Hum Hypertens 31: 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enkhjargal B, Godo S, Sawada A, Suvd N, Saito H, Noda K et al. (2014). Endothelial AMP‐activated protein kinase regulates blood pressure and coronary flow responses through hyperpolarization mechanism in mice. Arterioscler Thromb Vasc Biol 34: 1505–1513. [DOI] [PubMed] [Google Scholar]
- Fu J, Han Y, Wang J, Liu Y, Zheng S, Zhou L et al. (2016a). Irisin lowers blood pressure by improvement of endothelial dysfunction via AMPK–Akt–eNOS–NO pathway in the spontaneously hypertensive rat. J Am Heart Assoc 5: e003433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X, Zhu M, Zhang S, Foretz M, Viollet B, Du M (2016b). Obesity impairs skeletal muscle regeneration through inhibition of AMPK. Diabetes 65: 188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan M, Kassan M, Kadowitz PJ, Trebak M, Belmadani S, Matrougui K (2014). Mechanism of endoplasmic reticulum stress‐induced vascular endothelial dysfunction. Biochim Biophys Acta 1843: 1063–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasty AH, Harrison DG (2012). Endoplasmic reticulum stress and hypertension – a new paradigm? J Clin Invest 122: 3859–3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H, Zhao Z, Chen J, Ni Y, Zhong J, Yan Z et al. (2012). Metformin‐based treatment for obesity‐related hypertension: a randomized, double‐blind, placebo‐controlled trial. J Hypertens 30: 1430–1439. [DOI] [PubMed] [Google Scholar]
- Hundal RS, Krssak M, Dufour S, Laurent D, Lebon V, Chandramouli V et al. (2000). Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes 49: 2063–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Mullen TD, Hou Q, Bielawski J, Bielawska A, Zhang X et al. (2009). AMPK inhibitor compound C stimulates ceramide production and promotes Bax redistribution and apoptosis in MCF7 breast carcinoma cells. J Lipid Res 50: 2389–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen SB, Viollet B, Andreelli F, Frosig C, Birk JB, Schjerling P et al. (2004). Knockout of the alpha2 but not alpha1 5′‐AMP‐activated protein kinase isoform abolishes 5‐aminoimidazole‐4‐carboxamide‐1‐beta‐4‐ribofuranosidebut not contraction‐induced glucose uptake in skeletal muscle. J Biol Chem 279: 1070–1079. [DOI] [PubMed] [Google Scholar]
- Kemp BA, Howell NL, Keller SR, Gildea JJ, Padia SH, Carey RM (2016). AT2 receptor activation prevents sodium retention and reduces blood pressure in angiotensin II‐dependent hypertension. Circ Res 119: 532–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodziejczyk B, Duleba AJ, Spaczynski RZ, Pawelczyk L (2000). Metformin therapy decreases hyperandrogenism and hyperinsulinemia in women with polycystic ovary syndrome. Fertil Steril 73: 1149–1154. [DOI] [PubMed] [Google Scholar]
- Koss KL, Kranias EG (1996). Phospholamban: a prominent regulator of myocardial contractility. Circ Res 79: 1059–1063. [DOI] [PubMed] [Google Scholar]
- Landin‐Wilhelmsen K (1992). Metformin and blood pressure. J Clin Pharm Ther 17: 75–79. [DOI] [PubMed] [Google Scholar]
- Li J, Wang Y, Wang Y, Wen X, Ma XN, Chen W et al. (2015). Pharmacological activation of AMPK prevents Drp1‐mediated mitochondrial fission and alleviates endoplasmic reticulum stress‐associated endothelial dysfunction. J Mol Cell Cardiol 86: 62–74. [DOI] [PubMed] [Google Scholar]
- Liang B, Wang S, Wang Q, Zhang W, Viollet B, Zhu Y et al. (2013). Aberrant endoplasmic reticulum stress in vascular smooth muscle increases vascular contractility and blood pressure in mice deficient of AMP‐activated protein kinase‐alpha2 in vivo . Arterioscler Thromb Vasc Biol 33: 595–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Liu J, Huang Z, Yu X, Zhang X, Dou D et al. (2015). Berberine improves endothelial function by inhibiting endoplasmic reticulum stress in the carotid arteries of spontaneously hypertensive rats. Biochem Biophys Res Commun 458: 796–801. [DOI] [PubMed] [Google Scholar]
- Majithiya JB, Balaraman R (2006). Metformin reduces blood pressure and restores endothelial function in aorta of streptozotocin‐induced diabetic rats. Life Sci 78: 2615–2624. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales DR, Morris AD (2015). Metformin in cancer treatment and prevention. Annu Rev Med 66: 17–29. [DOI] [PubMed] [Google Scholar]
- Muntzel MS, Abe A, Petersen JS (1997). Effects of adrenergic, cholinergic and ganglionic blockade on acute depressor responses to metformin in spontaneously hypertensive rats. J Pharmacol Exp Ther 281: 618–623. [PubMed] [Google Scholar]
- Muntzel MS, Hamidou I, Barrett S (1999). Metformin attenuates salt‐induced hypertension in spontaneously hypertensive rats. Hypertension 33: 1135–1140. [DOI] [PubMed] [Google Scholar]
- Pawlyk AC, Giacomini KM, McKeon C, Shuldiner AR, Florez JC (2014). Metformin pharmacogenomics: current status and future directions. Diabetes 63: 2590–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piwkowska A, Rogacka D, Jankowski M, Dominiczak MH, Stepinski JK, Angielski S (2010). Metformin induces suppression of NAD(P)H oxidase activity in podocytes. Biochem Biophys Res Commun 393: 268–273. [DOI] [PubMed] [Google Scholar]
- Saksi J, Ijas P, Mayranpaa MI, Nuotio K, Isoviita PM, Tuimala J et al. (2014). Low‐expression variant of fatty acid‐binding protein 4 favors reduced manifestations of atherosclerotic disease and increased plaque stability. Circ Cardiovasc Genet 7: 588–598. [DOI] [PubMed] [Google Scholar]
- Santure M, Pitre M, Gaudreault N, Marette A, Nadeau A, Bachelard H (2000). Effect of metformin on the vascular and glucose metabolic actions of insulin in hypertensive rats. Am J Physiol Gastrointest Liver Physiol 278: G682–G692. [DOI] [PubMed] [Google Scholar]
- Schneider H, Schubert KM, Blodow S, Kreutz CP, Erdogmus S, Wiedenmann M et al. (2015). AMPK dilates resistance arteries via activation of SERCA and BKCa channels in smooth muscle. Hypertension 66: 108–116. [DOI] [PubMed] [Google Scholar]
- Seifarth C, Schehler B, Schneider HJ (2013). Effectiveness of metformin on weight loss in non‐diabetic individuals with obesity. Exp Clin Endocrinol Diabetes 121: 27–31. [DOI] [PubMed] [Google Scholar]
- Simon‐Szabo L, Kokas M, Mandl J, Keri G, Csala M (2014). Metformin attenuates palmitate‐induced endoplasmic reticulum stress, serine phosphorylation of IRS‐1 and apoptosis in rat insulinoma cells. PLoS One 9: e97868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snorgaard O, Kober L, Carlsen J (1997). The effect of metformin on blood pressure and metabolism in nondiabetic hypertensive patients. J Intern Med 242: 407–412. [DOI] [PubMed] [Google Scholar]
- Song P, Zhang M, Wang S, Xu J, Choi HC, Zou MH (2009). Thromboxane A2 receptor activates a Rho‐associated kinase/LKB1/PTEN pathway to attenuate endothelium insulin signaling. J Biol Chem 284: 17120–17128. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Song P, Zou MH (2012). Regulation of NAD(P)H oxidases by AMPK in cardiovascular systems. Free Radic Biol Med 52: 1607–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun GQ, Li YB, Du B, Meng Y (2015). Resveratrol via activation of AMPK lowers blood pressure in DOCA‐salt hypertensive mice. Clin Exp Hypertens 37: 616–621. [DOI] [PubMed] [Google Scholar]
- Tabas I (2010). The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ Res 107: 839–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Yoshika M, Komiyama Y, Nishimura M (2011). The central mechanism underlying hypertension: a review of the roles of sodium ions, epithelial sodium channels, the renin–angiotensin–aldosterone system, oxidative stress and endogenous digitalis in the brain. Hypertens Res 34: 1147–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Te Riet L, van Esch JH, Roks AJ, van den Meiracker AH, Danser AH (2015). Hypertension: renin–angiotensin–aldosterone system alterations. Circ Res 116: 960–975. [DOI] [PubMed] [Google Scholar]
- Teng AC, Miyake T, Yokoe S, Zhang L, Rezende LM Jr, Sharma P et al. (2015). Metformin increases degradation of phospholamban via autophagy in cardiomyocytes. Proc Natl Acad Sci U S A 112: 7165–7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traister A, Li M, Aafaqi S, Lu M, Arab S, Radisic M et al. (2014). Integrin‐linked kinase mediates force transduction in cardiomyocytes by modulating SERCA2a/PLN function. Nat Commun 5: 4533. [DOI] [PubMed] [Google Scholar]
- Tsai CM, Kuo HC, Hsu CN, Huang LT, Tain YL (2014). Metformin reduces asymmetric dimethylarginine and prevents hypertension in spontaneously hypertensive rats. Transl Res 164: 452–459. [DOI] [PubMed] [Google Scholar]
- Uehara MH, Kohlmann NE, Zanella MT, Ferreira SR (2001). Metabolic and haemodynamic effects of metformin in patients with type 2 diabetes mellitus and hypertension. Diabetes Obes Metab 3: 319–325. [DOI] [PubMed] [Google Scholar]
- Verma S, Bhanot S, McNeill JH (1994a). Antihypertensive effects of metformin in fructose‐fed hyperinsulinemic, hypertensive rats. J Pharmacol Exp Ther 271: 1334–1337. [PubMed] [Google Scholar]
- Verma S, Bhanot S, McNeill JH (1994b). Metformin decreases plasma insulin levels and systolic blood pressure in spontaneously hypertensive rats. Am J Physiol 267: H1250–H1253. [DOI] [PubMed] [Google Scholar]
- Wang S, Liang B, Viollet B, Zou MH (2011). Inhibition of the AMP‐activated protein kinase‐alpha2 accentuates agonist‐induced vascular smooth muscle contraction and high blood pressure in mice. Hypertension 57: 1010–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Adi D, Long M, Wang J, Liu F, Gai MT et al. (2016). 4‐Phenylbutyric acid induces protection against pulmonary arterial hypertension in rats. PLoS One 11: e0157538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Song P, Zhang W, Liu J, Dai X, Liu Z et al. (2015). Activation of AMPKalpha2 in adipocytes is essential for nicotine‐induced insulin resistance in vivo . Nat Med 21: 373–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XJ, Gauthier MS, Hess DT, Apovian CM, Cacicedo JM, Gokce N et al. (2012). Insulin sensitive and resistant obesity in humans: AMPK activity, oxidative stress, and depot‐specific changes in gene expression in adipose tissue. J Lipid Res 53: 792–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Wei J, Wu Y, Wang Z, Guo Y, Lee P et al. (2015a). Metformin induces ER stress‐dependent apoptosis through miR‐708‐5p/NNAT pathway in prostate cancer. Oncogene 4: e158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Zhao D, Ren J, Yang J (2015b). Endoplasmic reticulum stress and protein quality control in diabetic cardiomyopathy. Biochim Biophys Acta 1852: 209–218. [DOI] [PubMed] [Google Scholar]
- Young CN, Cao X, Guruju MR, Pierce JP, Morgan DA, Wang G et al. (2012). ER stress in the brain subfornical organ mediates angiotensin‐dependent hypertension. J Clin Invest 122: 3960–3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang BB, Zhou G, Li C (2009a). AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab 9: 407–416. [DOI] [PubMed] [Google Scholar]
- Zhang M, Dong Y, Xu J, Xie Z, Wu Y, Song P et al. (2008). Thromboxane receptor activates the AMP‐activated protein kinase in vascular smooth muscle cells via hydrogen peroxide. Circ Res 102: 328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang QJ, McMillin SL, Tanner JM, Palionyte M, Abel ED, Symons JD (2009b). Endothelial nitric oxide synthase phosphorylation in treadmill‐running mice: role of vascular signalling kinases. J Physiol 587: 3911–3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Yang W, Wu F, Wang C, Yu L, Tang L et al. (2013). Prognostic significance of AMPK activation and therapeutic effects of metformin in hepatocellular carcinoma. Clin Cancer Res 19: 5372–5380. [DOI] [PubMed] [Google Scholar]
- Zode GS, Kuehn MH, Nishimura DY, Searby CC, Mohan K, Grozdanic SD et al. (2011). Reduction of ER stress via a chemical chaperone prevents disease phenotypes in a mouse model of primary open angle glaucoma. J Clin Invest 121: 3542–3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou MH, Kirkpatrick SS, Davis BJ, Nelson JS, Wiles WG, Schlattner U et al. (2004). Activation of the AMP‐activated protein kinase by the anti‐diabetic drug metformin in vivo. Role of mitochondrial reactive nitrogen species. J Biol Chem 279: 43940–43951. [DOI] [PubMed] [Google Scholar]