

Abstract
Background and Purpose
Dysregulation of gap junction‐mediated cell coupling contributes to development of arrhythmias and myocardial damage after ischaemia/reperfusion (I/R). Connexin 43 (Cx43) is present at ventricular gap junctions and also in the mitochondria of cardiomyocytes. The dipeptide (2S, 4R)‐1‐(2‐aminoacetyl)‐4‐benzamidopyrrolidine‐2‐carboxylic acid (ZP1609) has antiarrhythmic properties and reduces infarct size when given at reperfusion. However, it is unclear, whether ZP1609 targets Cx43‐containing mitochondria and affects cardiomyocyte hypercontracture following I/R.
Experimental Approach
We studied the effects of ZP1609 on the function of murine sub‐sarcolemmal mitochondria (SSM, containing Cx43) and interfibrillar mitochondria (IFM, lacking Cx43). Murine isolated cardiomyocytes were subjected to simulated I/R without and with ZP1609 (applied during I/R or at the onset of reperfusion only), and the number of cardiomyocytes undergoing hypercontracture was quantified. Biochemical pathways targeted by ZP1609 in cardiomyocytes were analysed.
Key Results
ZP1609 inhibited ADP‐stimulated respiration and ATP production in SSM and IFM. ROS formation and calcium retention capacities in SSM and IFM were not affected by ZP1609, whereas potassium uptake was enhanced in IFM. The number of rod‐shaped cardiomyocytes was increased by ZP1609 (10 μM) when administered either during I/R or reperfusion. ZP1609 altered the phosphorylation of proteins contributing to the protection against I/R injury.
Conclusions and Implications
ZP1609 reduced mitochondrial respiration and ATP production, but enhanced potassium uptake of IFM. Additionally, ZP1609 reduced the extent of cardiomyocytes undergoing hypercontracture following I/R. The protective effect was independent of mitochondrial Cx43, as ZP1609 exerts its effects in Cx43‐containing SSM and Cx43‐lacking IFM.
Abbreviations
- Cx43
connexin 43
- GSK‐3 α/β
glycogen synthase kinase 3α/β
- HSP60
heat shock protein 60
- I/R
ischaemia/reperfusion
- IFM
interfibrillar mitochondria
- MPTP
mitochondrial permeability transition pore
- mTORC1
mechanistic target of rapamycin complex 1
- PRAS40
proline‐rich Akt substrate of 40 kDa
- SSM
subsarcolemmal mitochondria
- ZP1609
(2S, 4R)‐1‐(2‐aminoacetyl)‐4‐benzamidopyrrolidine‐2‐carboxylic acid
Introduction
In the myocardium, the coordinated contraction of cardiomyocytes is essential for the maintenance of a regular cardiac rhythm. Cardiomyocytes are coupled via gap junctions and in ventricular cardiomyocytes, connexin 43 (Cx43) is the predominant protein forming gap junctions or non‐junctional hemichannels (Schulz et al., 2015). Reductions in electrical cell coupling via decreased amounts of Cx43 or reduced Cx43 phosphorylation contribute to ischaemia‐induced arrhythmogenesis (Beardslee et al., 2000; Lerner et al., 2000). In order to maintain intercellular coupling, anti‐arrhythmic peptides were developed (De Vuyst et al., 2011). The application of the hexapeptide rotigaptide (formerly called ZP123) inhibits gap junction uncoupling by ischaemia (Xing et al., 2003; Kjolbye et al., 2007), induces gap junctional communication via Cx43‐formed channels (Clarke et al., 2006), and prevents the ischaemia‐induced dephosphorylation of myocardial Cx43 (Axelsen et al., 2006).
ZP1609, (2S, 4R)‐1‐(2‐aminoacetyl)‐4‐benzamidopyrrolidine‐2‐carboxylic acid, (also known as GAP‐134 or danegaptide) is a dipeptide analogue of rotigaptide. ZP1609 prolongs the time to conduction block in a mouse model of CaCl2‐induced arrhythmias (Butera et al., 2009) and reduces atrial fibrillation in dog hearts undergoing simultaneous atrioventricular pacing (Laurent et al., 2009) or in dogs with sterile pericarditis (Rossman et al., 2009).
Gap junctions are not only important for arrhythmias but are also involved in myocardial damage by ischaemia/reperfusion (I/R) injury since their uncoupling reduces infarct size (Rodriguez‐Sinovas et al., 2004; Shintani‐Ishida et al., 2009). Cx43‐formed hemichannels, which are predominantly closed under physiological conditions, open during ischaemia (Saez and Leybaert, 2014) and the prevention of their opening protects against I/R injury (Shintani‐Ishida et al., 2007; Wang et al., 2013). Both rotigaptide and ZP1609 decrease myocardial infarct size in dog and pig hearts when administered before reperfusion (Hennan et al., 2006; Hennan et al., 2009; Skyschally et al., 2013; Pedersen et al., 2016). In a model of renal I/R injury, however, ZP1609 did not improve renal function (Amdisen et al., 2016).
Apart from being present at the plasma membrane, Cx43 is also found in subsarcolemmal mitochondria (SSM, located directly under the sarcolemma), whereas interfibrillar mitochondria (IFM), which are present between the myofibrils, lack Cx43 (Boengler et al., 2009; Sun et al., 2015). SSM and IFM differ in their function, in that IFM consume more oxygen than SSM (Palmer et al., 1977) and take up more calcium until opening of the mitochondrial permeability transition pore (MPTP) occurs (Palmer et al., 1986). Data derived from different experimental approaches support the existence of Cx43‐formed channels within mitochondria (Miro‐Casas et al., 2009; Boengler et al., 2013; Soetkamp et al., 2014). Cx43 is involved in mitochondrial function in several aspects including respiration, potassium handling, ROS formation, MPTP opening, mitochondrial dynamics and mitophagy (Miro‐Casas et al., 2009; Tyagi et al., 2010; Boengler et al., 2012; Boengler et al., 2013; Givvimani et al., 2014; Soetkamp et al., 2014; Srisakuldee et al., 2014), parameters all contributing to myocardial I/R injury.
As the protection afforded by ZP1609 requires either predominantly open (antiarrhythmic properties) or closed (reperfusion injury) channels, it is important to investigate whether this compound directly targets Cx43. Possibly, ZP1609 might also act on a subset of mitochondria important for modulating the functional consequences of I/R injury in cardiomyocytes. To test this hypothesis, we assessed the effects of ZP1609 on mitochondrial function, especially on parameters known to be influenced by Cx43 and important for I/R injury in Cx43‐containing SSM and Cx43‐free IFM. In addition, the effects of ZP1609 on cardiomyocyte hypercontracture following I/R and biochemical pathways influenced by ZP1609 were analysed.
Methods
Animals
All animal care and experimental procedures in this study conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication No. 85–23, revised 1996) and were approved by the animal welfare office of the Justus‐Liebig‐University Giessen. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015). For the isolation of cardiomyocytes and studies of mitochondrial function 10–12 weeks old male C57Bl6J mice (25–30 g, Janvier, Le Genest‐Saint‐Isles, France) were used. These methods have been in use for several years (Schluter and Schreiber, 2005; Boengler et al., 2009). Because of the well‐established response of the mouse heart towards I/R injury, the importance of mitochondria in myocardial injury, the feasibility of characterising isolated cardiomyocytes and of studying various aspects of mitochondrial function, the mouse (heart) is appropriate for the present study. The mice (up to 5 per cage) were kept in dark/light cycles of 12 h each and had free access to standard chow and drinking water. Mice were anaesthetized with 5% isoflurane, killed by cervical dislocation, and subsequently hearts were removed.
Isolation of mitochondria
Subarcolemmal (SSM) and IFM were isolated from C57Bl6J mice as previously described (Boengler et al., 2009). All steps were performed at 4°C. In short, hearts were washed in buffer A [100 mM KCl, 50 mM 3‐[N‐morpholino]‐propanesulfonic acid (MOPS), 5 mM MgSO4, 1 mM ATP, 1 mM EGTA, pH 7.4] and weighed. Tissue was minced in 10 mL·g−1 buffer B (buffer A + 0.04% BSA) with scissors and was then disrupted with a Potter‐Elvejhem tissue homogenizer. The homogenate was centrifuged for 10 min at 800 g. The supernatant containing the SSM was centrifuged for 10 min at 8000 g. The sedimented mitochondria were washed in buffer A and were resuspended in a small volume of buffer A. The sediment of the first centrifugation containing the IFM was resuspended in buffer B (10 mL·g−1 tissue). About 8 U·g−1 of the protease nagarse were added and incubated at 4°C for 1 min. After disruption with a Potter‐Elvejhem tissue homogenizer and centrifugation for 10 min at 800 g the supernatant was centrifuged for 10 min at 8000 g to collect the IFM. The resulting mitochondria were washed by resuspension in buffer A; they were centrifuged at 8000 g for 10 min, and finally resuspended in buffer A. These mitochondrial preparations were used to study respiration, ATP production, ROS formation, MPTP opening and potassium uptake. Animals were not randomized, but the isolated mitochondria from one animal were always subjected to all treatment groups. The order in which the samples were analysed for mitochondrial function was randomized, and the experiments and the analysis of the data were conducted by two different persons. The analysis of mitochondrial function adheres to strict rules in terms of periods of time analysed and was identical for all treatment groups. To analyse the amount of Cx43 in SSM and IFM by Western Blot, mitochondria were further purified by layering them on top of a 30% Percoll solution in isolation buffer (in mM: sucrose 250; HEPES 10; EGTA 1; 0.5% BSA; pH 7.4) and subsequent ultracentrifugation at 35 000 g for 30 min at 4°C. The mitochondrial band was collected, washed twice in isolation buffer by centrifugation at 8000 g for 5 min, and the purified mitochondria were stored at −80°C.
Mitochondrial oxygen consumption
Oxygen consumption of 100 μg·mL−1 SSM or IFM was measured with a Clark‐type oxygen electrode (Oxygen meter 782, Strathkelvin, Glasgow, UK) at 25°C in incubation buffer [containing in mM: 125 KCl, 10 Tris (titrated with MOPS), 1.2 Pi (titrated with Tris), 1.2 MgCl2, 0.02 EGTA (titrated with Tris), pH 7.4]. Complex 1‐mediated respiration was analysed in the presence of 5 mM glutamate and 2.5 mM malate, whereas complex 2‐mediated respiration was measured in the presence of 5 mM succinate and 2 μM rotenone. After recording of basal oxygen consumption, respiration was stimulated by the addition of 40 μM ADP. Oxygen consumption was analysed in nmol O2·min−1·mg protein−1). For reference see (Boengler et al., 2012). Oxygen consumption was measured from untreated SSM and IFM, or after incubation for 30 min at 4°C in isolation buffer under control conditions or with either 10, 100 nM, 1 or 10 μM ZP1609.
Mitochondrial ATP production
Mitochondrial ATP production was analysed from samples (50 μg) of SSM or IFM incubated under control conditions or after incubation for 30 min at 4°C with 1 or 10 μM ZP1609. After the incubation, mitochondria were diluted in 100 μL incubation buffer supplemented with 5 mM glutamate and 2.5 mM malate and 0.1 mM di‐adenosine‐pentaphosphate. About 100 μL of the ATP bioluminescent assay kit (1:5 diluted with incubation buffer, Sigma‐Aldrich, Munich, Germany) was added and the bioluminescence was recorded for 1 min with a Cary Eclipse spectrophotometer (Agilent Technologies, Santa Clara, CA) at room temperature to assess background values. ATP generation was initiated by the addition of 500 mM ADP and the bioluminescence was recorded for another 5 min. The mean value of the bioluminescence for 5 min after the addition of ADP was calculated, the background bioluminescence was subtracted, and the value of control SSM was set as 100%. This normalization was performed in order to eliminate variations in ATP production depending on the actual quality of the mitochondrial preparation. Three replicates were performed per group.
Calcium‐induced MPTP opening
The calcium retention capacities of 100 μg·mL−1 SSM or IFM incubated under control conditions or with ZP1609 (1, 10 μM) were analysed in incubation buffer (glutamate/malate as substrates for complex 1, no EGTA) at 25°C. Calcium Green 5 N fluorescence (0.5 μM, Invitrogen, Carlsbad, CA) was used to detect extramitochondrial calcium with a Cary Eclipse spectrophotometer at excitation and emission wavelengths of 500 and 530 nm respectively. Approximately 5 μM CaCl2 were added every third minute until a sudden increase in Calcium Green 5 N fluorescence occurred reflecting MPTP opening.
Mitochondrial ROS generation
Approximately 50 μg mitochondria (SSM or IFM) were incubated for 30 min (4°C) under control conditions, or with 1 or 10 μM ZP1609. Mitochondria were transferred to incubation buffer supplemented with 5 mM glutamate and 2.5 mM malate 2.5, 50 μM Amplex UltraRed (Invitrogen, Eugene, OR), and 0.1 U·mL−1 horseradish peroxidase. The fluorescence was measured continuously for 4 min with a Cary Eclipse spectrophotometer at the excitation/emission wavelengths of 565/581 nm respectively. As positive control, we used control mitochondria supplemented with 2 μM of the complex I inhibitor rotenone. Background fluorescence of the buffer without mitochondria was subtracted and the slope (fluorescence in arbitrary units/time) was calculated. The data on ROS formation were normalized as % of control in order to eliminate variations in ROS formation depending on the actual quality of the mitochondrial preparation.
Mitochondrial potassium influx
Freshly isolated SSM or IFM were incubated under control conditions, with 1 or 10 μM ZP1609 for 20 min at 4°C. Subsequently, mitochondria were loaded with 10 μM PBFI‐AM [acetoxymethyl ester of PBFI (potassium‐binding benzofuran isophthalate; Life Technologies, Carlsbad, CA)] diluted 2:1 with 20% pluronic F127 by additional incubation at 25°C for 10 min according to the protocol by Costa et al. (2006). Three volumes of TEA (tetraethylammonium) buffer (in mM: TEA‐Cl 120, HEPES 10, succinate 10, Na2HPO4 5, EGTA 0.1, MgCl2 0.5, rotenone 5 μM, oligomycin 0.67 μM, pH 7.2) were added, and the mitochondria were incubated for 2 min. Thereby, K+ ions are replaced within the mitochondrial matrix. Subsequently, the mitochondria were washed twice in isolation buffer and the protein concentration was measured using the Dc protein assay (BioRad, Hercules, CA). A sample of 200 μg mitochondrial proteins (SSM or IFM) was incubated for 30 min at 4°C under control conditions or with 1 or 10 μM ZP1609 respectively. Mitochondria (100 μg·mL−1) were added to isolation buffer supplemented with 1 μg·mL−1 oligomycin (inhibits ATP synthase), 50 μM glibenclamide (blocks mitochondrial ATP‐dependent potassium channels), and 1 μM cyclosporin A (CsA; inhibits opening of the MPTP). The uptake of potassium into the mitochondria was induced by adding 140 mM KCl. The PBFI fluorescence was measured in a Cary Eclipse Fluorescence Spectrophotometer at alternating excitation wavelengths of 340 (maximum potassium sensitivity of the probe) and 380 nm (isosbestic point of the probe), respectively, and an emission wavelength of 500 nm at 25°C. The maximal slope of the PBFI fluorescence after the KCl pulse – corresponding to approximately 0.03 min – was determined.
Western blot analysis
SSM and IFM purified by Percoll gradient ultracentrifugation (n = 4) were lysed in 1X NP40 buffer (25 mM Tris, 150 mM NaCl, 1 mM EDTA, 1% NP‐40, 5% glycerol, pH 7.4) supplemented with 1X PhosStop and Complete inhibitors (Roche, Basel, Switzerland) as well as 1 μM neocuproine. Protein concentration was determined using the Lowry assay. Thirty μg proteins were electrophoretically separated on 10% Bis/Tris gels and proteins were transferred to nitrocellulose membranes. After blocking, membranes were incubated with rabbit polyclonal anti‐human/rat Cx43 antibodies (Sigma) or rabbit polyclonal anti‐human manganese SOD antibodies (Merck Millipore, Darmstadt, Germany). After washing and incubation with the respective secondary antibodies, immunoreactive signals were detected by chemiluminescence (SuperSignal West Pico Chemiluminescent Substrate, ThermoFisher) and quantified using ScionImage software. The data on the predominant localization of Cx43 in SSM are confirmatory and have been published previously (Boengler et al., 2009; Soetkamp et al., 2014; Sun et al., 2015). As the general expression of Cx43 varies in every animal analysed, we set the content of Cx43 in SSM as 100% to compare with the amount of Cx43 in IFM. No statistical analysis was undertaken.
Isolation of cardiomyocytes
Mouse ventricular cardiomyocytes were isolated as described previously (Schluter and Schreiber, 2005). Briefly, hearts were digested with collagenase in the Langendorff‐mode, minced, and further digested by incubation with collagenase buffer. The resulting suspension was filtered, and the separation of cardiomyocytes from non‐myocytes was achieved by centrifugation. Finally, physiological calcium concentrations were readjusted by step‐wise increases of calcium to 1 mM concentration, and cardiomyocytes were attached on glass‐cover slips. Animals were not randomized, treatment of isolated cardiomyocytes started simultaneously in all groups.
In vitro ischaemia and reperfusion experiments
Isolated cardiomyocytes on glass cover‐slips were introduced into a perfusion chamber and superfused with a flow rate of 1.75 mL·min−1. The buffers were transferred into the perfusion chamber through gas‐tight steel capillaries. Isolated cardiomyocytes were incubated under normoxic conditions in a HEPES buffer pH 7.4 (in mM: 118 NaCl, 4.7 KCl, 1.2 KH2PO4, 0.8 MgSO4, 2.5 CaCl2, 5 glucose, 1.9 sodium pyruvate and 10 HEPES). To simulate ischaemia, the pH of the buffer was reduced to 6.4 at 37°C, 5 mM DL‐cysteine was added, and pyruvate and glucose were omitted. The buffer was equilibrated with 100% N2 before and during experiments. Following ischaemia, the cells were reoxygenated by perfusion of a normoxic, glucose‐and pyruvate containing buffer (pH 7.4). ZP1609 was dissolved (1 or 10 μM) in the respective buffers, as indicated.
The experimental groups were as follows:
30 min normoxia
30 min normoxia with 10 μM ZP1609
15 min ischaemia and 30 min reperfusion
15 min normoxia, 15 min ischaemia and 30 min reperfusion with 1 or 10 μM ZP1609 present during the whole experiment (1 μM ZP1609‐pre, 10 μM ZP1609‐pre)
15 min ischaemia, 30 min reperfusion with 1 or 10 μM ZP1609 present during reperfusion only (1 μM ZP1609‐rep, 10 μM ZP1609‐rep)
Subsequently, cover‐slips were transferred to a microscope and five randomly selected pictures were taken for each cover‐slip. Ischaemia‐tolerant cardiomyocytes were identified by their rod‐shaped structure. Hypercontracture leading to round‐shaped cells was quantified. The total number of cells counted per experiment was 3391 ± 1385. The experiments and the evaluation of cardiomyocyte hypercontracture were performed by two different researchers.
Signal transduction
Isolated cardiomyocytes were subjected to normoxia, I/R, 10 μM ZP1609‐pre, and 10 μM ZP1609‐rep. Cells were collected using a cell scraper and stored at −80°C. Phosphorylation of 43 proteins and amounts of two related total proteins were studied using the Human Proteome Profiler Array (R&D Systems, Minneapolis, MN) according to the instructions of the manufacturer. To provide the sample size of 300 μg protein, cardiomyocytes from four mice were pooled. Immunoreactive signals were detected using SuperSignal West Femto Chemiluminescent Substrate, ThermoFisher) and were quantified using Scion Image software. Pixel intensities were corrected for the mean value of the negative controls, no statistical analysis was undertaken.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Data are presented as mean values ± SEM and group sizes are equal. Data transformation was not performed. Mitochondrial respiration rates, potassium uptake, and data on cardiomyocyte hypercontracture were analysed by one‐way ANOVA. Data on calcium‐induced MPTP opening data were compared by two‐way ANOVA. Data on mitochondrial ATP production and ROS generation were compared by Rank Sum test. A P < 0.05 indicated a significant difference. The program SigmaStat 3.5 (Systat, Software GmbH, Erkrath, Germany) was used for statistical analysis.
Materials
ZP1609 ((2S, 4R)‐1‐(2‐aminoacetyl)‐4‐benzamidopyrrolidine‐2‐carboxylic acid) was synthesized at Zealand Pharma A/S (Glostrup, Denmark) with a purity of 99.8% as determined by HPLC. Other compounds were supplied as follows: CsA, Sigma‐Aldrich; glibenclamide, Sigma‐Aldrich; oligomycin, Sigma‐Aldrich.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b,c).
Results
Mitochondria
To demonstrate the localization of Cx43 within mitochondrial subpopulations, SSM and IFM were isolated from mouse ventricles and characterized by Western blot analysis (Figure 1). The majority of Cx43 is found in SSM, whereas IFM contained negligible amounts of the protein.
Figure 1.
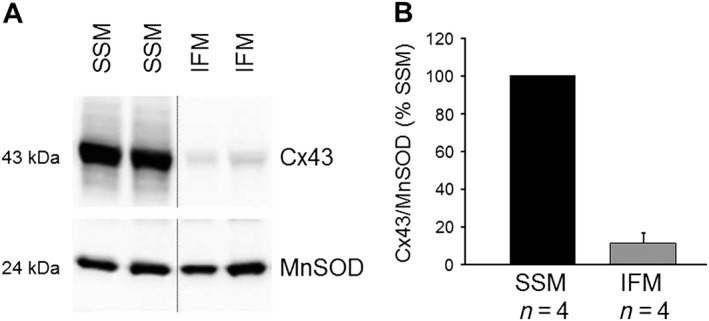
Cx43 is predominantly present in SSM. (A) Western blot analysis was performed for Cx43 and the mitochondrial marker protein manganese SOD (MnSOD) on mouse ventricular SSM and IFM mitochondria. (B) Bar graphs show the amount of Cx43 normalized to that of MnSOD in SSM and IFM.
IFM consume more oxygen than SSM (Palmer et al., 1977). In untreated mitochondria, ADP‐stimulated respiration (in nmol O2·min−1·mg protein−1) using substrates for respiratory complex 1 or complex 2 was higher in IFM (complex 1: 97.4 ± 9.7; complex 2: 175.5 ± 8.7, n = 15) than in SSM (complex 1: 67.7 ± 5.9; complex 2: 140.4 ± 7.5, n = 14, P < 0.05).
To analyse the effect of ZP1609 on mitochondrial respiration, SSM and IFM were incubated under control conditions with different concentrations of ZP1609 (10 nM to 10 μM). Subsequently, ADP‐stimulated respiration was measured using substrates for complex 1 and complex 2 and expressed as % of the untreated control value. Mitochondrial oxygen consumption was not affected by 10 nM ZP1609, but concentration‐dependently decreased in SSM and IFM when treated with 100 nM and 1 μM ZP1609. The inhibition of ADP‐stimulated respiration was comparable between 1 and 10 μM ZP1609 (Figure 2), and was more pronounced in mitochondria respiring on complex 1 than on complex 2 substrates. ZP1609 did not influence basal respiration in SSM or IFM using substrates for complex 1 and 2. The absolute values of basal and ADP‐stimulated oxygen consumption (complex 1 and 2 substrates) in SSM and IFM incubated under control conditions or with 1 and 10 μM ZP1609 are shown in Figure 3. Because 1 and 10 μM ZP1609 had the most pronounced effect on mitochondrial oxygen consumption, these concentrations were used for further experiments.
Figure 2.

ZP1609 reduces mitochondrial ADP‐stimulated respiration. ADP‐stimulated mitochondrial oxygen consumption (in % of control‐incubated mitochondria) was measured in SSM and IFM using substrates for complexes 1 or 2 in the presence of 10, 100 nM, 1 or 10 μM ZP1609. * P < 0.05, significantly different from control.
Figure 3.
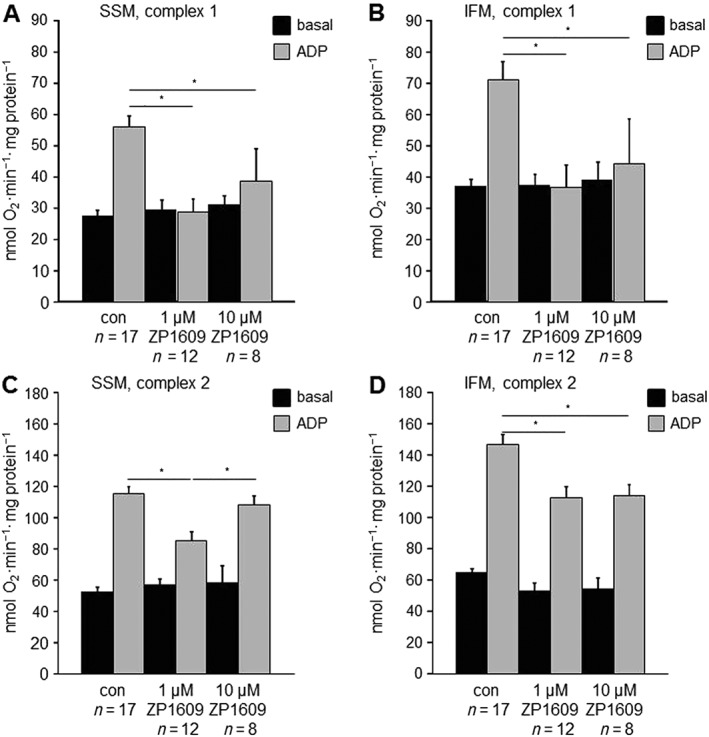
Effects of ZP1609 on mitochondrial oxygen consumption. Mouse SSM and IFM were incubated under control conditions (con) or with 1 or 10 μM ZP1609. Basal and ADP‐stimulated respiration were measured using substrates for complex 1 in SSM (A) and IFM (B), and using substrates for complex 2 in both SSM (C) and IFM (D), * P < 0.05 significantly different as indicated.
Mitochondrial ATP‐production was measured in SSM and IFM without or with 1 and 10 μM ZP1609 in the presence of complex 1 substrates. In accordance to the effect of ZP1609 on respiration, both SSM and IFM demonstrated less ATP generation in mitochondria treated with 10 μM ZP1609 (Figure 4).
Figure 4.
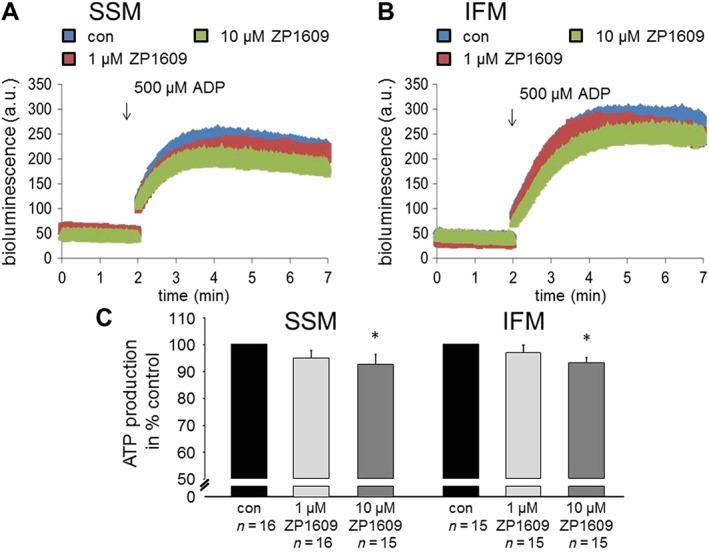
ZP1609 decreases mitochondrial ATP production. Original traces show the bioluminescence indicating ATP production from mouse SSM (A) and IFM (B) incubated under control conditions (con) or with 1 or 10 μM ZP1609. Background bioluminescence was recorded for 2 min, then 500 μM ADP was added to stimulate ATP generation, and the bioluminescence was monitored for another 5 min. (C) Quantification of ATP generation from control and ZP1609‐treated SSM and IFM. Control‐incubated SSM or IFM were set as 100%, * P < 0.05, significantly different from control (con).
Calcium‐induced MPTP opening was analysed in SSM and IFM, without or with ZP1609 (Figure 5). MPTP opening was calculated as the amount of CaCl2 added to the mitochondrial preparations until a sudden increase in Calcium Green fluorescence appeared (Figure 5A).
Figure 5.
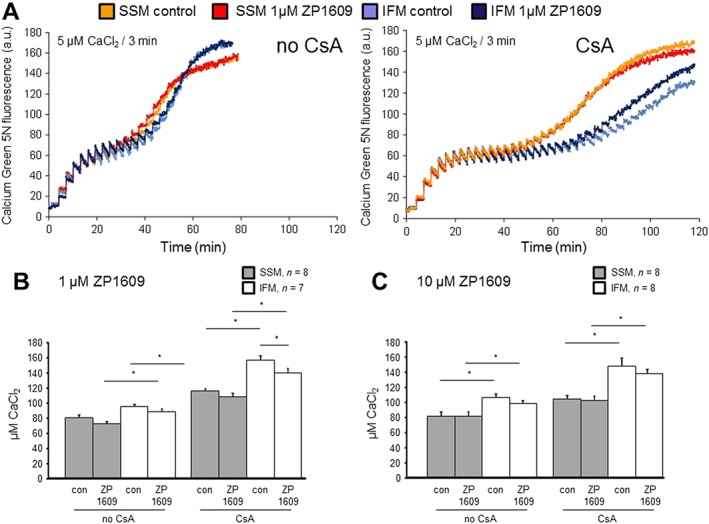
ZP1609 does not affect mitochondrial calcium retention capacity. (A) Original traces showing Calcium Green 5 N fluorescence of control mitochondria (SSM and IFM) and mitochondria treated with 1 μM ZP1609 in the absence (left panel) or presence (right panel) of cyclosporin A (CsA). CaCl2 (5 μM) was added every third minute until a sudden increase in Calcium Green 5 N fluorescence occurred reflecting MPTP opening. (B, C) The concentration of CaCl2 at which MPTP opening takes place was calculated in SSM and IFM in the absence or presence of CsA in mitochondria incubated under control conditions (con) or with 1 μM ZP1609 (ZP, B) or 10 μM ZP1609 (C), respectively, * P < 0.05, significantly different as indicated.
In SSM and in IFM, the incubation with 1 (Figure 5B) and 10 μM of ZP1609 (Figure 5C) did not alter the calcium concentration required to induce MPTP opening. As expected, in the presence of the MPTP inhibitor CsA SSM and IFM retained more calcium until MPTP opening occurred.
As enhanced amounts of ROS facilitate MPTP opening, ROS formation was measured in SSM and IFM treated with ZP1609. However, neither 1 nor 10 μM ZP1609 influenced ROS formation in SSM or IFM (Figure 6).
Figure 6.

ZP1609 does not affect mitochondrial ROS formation. ROS formation was measured using Amplex UltraRed fluorescence and the slope of control‐incubated mitochondria was set as 100%. Incubation of both SSM and IFM with 1 and 10 μM ZP1609 had no effect on ROS formation, n = 8.
The influence of ZP1609 on mitochondrial potassium uptake was measured as the increase in PBFI fluorescence after the addition of KCl in SSM and IFM incubated under control conditions, with 1 or 10 μM ZP1609 respectively. The excitation at 380 nm (isosbestic point) resulted in a small increase in the PBFI fluorescence, which was similar between groups (Figure 7). However, the excitation at 340 nm demonstrated a rapid increase in the PBFI fluorescence until a plateau was reached. The maximal slope of the increase in the PBFI fluorescence over time was calculated. In SSM, the incubation with ZP1609 (either 1 or 10 μM) did not affect the potassium uptake. However, incubation of IFM with 10 μM ZP1609 significantly increased the maximal slope of the PBFI fluorescence, indicating enhanced mitochondrial potassium influx.
Figure 7.
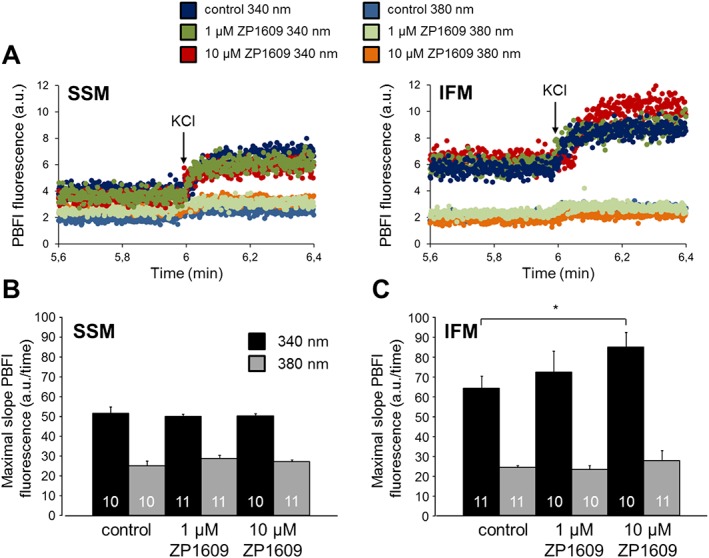
ZP1609 enhances the uptake of K+ in IFM, but not in SSM. (A) Original traces showing PBFI fluorescence in arbitrary units (a.u.) in SSM (left panel) and IFM (right panel) incubated under control conditions or with 1 or 10 μM ZP1609 respectively. After 6 min, a bolus of 140 mM KCl was added. PBFI fluorescence is shown for the excitation wavelengths 340 nm (probe is sensitive for potassium) and 380 nm (isosbestic point of the probe). Bar graphs show the maximal slope of the PBFI fluorescence/time for the excitation wavelengths 340 and 380 nm after the addition of KCl to SSM (B) or IFM (C).
Cardiomyocytes
The effect of ZP1609 on hypercontracture was measured in freshly isolated mouse cardiomyocytes as the percentage of rod‐shaped cardiomyocytes after incubation under normoxic conditions or following I/R. The percentage of rod‐shaped cardiomyocytes following 30 min incubation under normoxic conditions was similar between control cells (77.3 ± 3.3%) and cells treated with 10 μM ZP1609 (79.1 ± 1.4%, n = 4 independent isolations).
Approximately 15 min ischaemia and 30 min reperfusion decreased the percentage of rod‐shaped cardiomyocytes compared to normoxic conditions (Figure 8). The lower concentration of ZP1609 (1 μM ) had no effect on cardiomyocyte hypercontracture when present during 15 min normoxia followed by I/R, where 18.8 ± 8.7% (n = 3) of the cardiomyocytes had a rod‐shaped phenotype or when added at the onset of reperfusion (23.4 ± 1.8%, n = 3) compared to I/R alone (19.9 ± 8.8%, n = 3). In contrast, the higher concentration of ZP1609 (10 μM) enhanced the number of rod‐shaped cardiomyocytes, regardless of the time at which ZP1609 was applied (10 μM ZP1609‐pre and 10 μM ZP1609‐rep, Figure 8).
Figure 8.
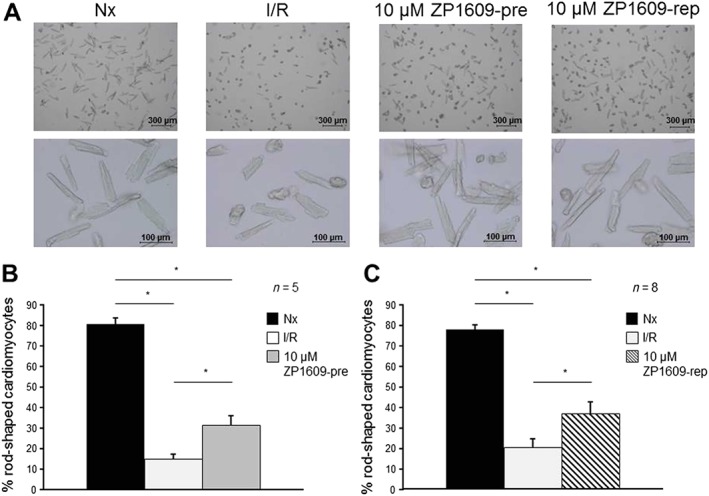
ZP1609 reduces cardiomyocyte hypercontracture following I/R. (A) Representative images of cardiomyocytes subjected to normoxia (Nx), I/R in the absence or presence of 10 μM ZP1609 when applied during I/R (10 μM ZP1609‐pre) or at the onset of reperfusion (10 μM ZP1609‐rep). Cardiomyocytes are shown in two magnifications. Bar graphs show the percentage of rod‐shaped cardiomyocytes following Nx, I/R in the absence or in the presence of 10 μM ZP1609 when applied during I/R (B) or at the onset of reperfusion (C).
To study biochemical pathways influenced by ZP1609 (apart from Cx43), the phosphorylation of several protein kinases was measured in cardiomyocytes undergoing normoxia, I/R, 10 μM ZP1609‐pre, and 10 μM ZP1609‐rep (Figure 9). Whereas I/R increased the phosphorylation of PRAS40 (proline‐rich Akt substrate of 40 kDa), compared with that under normoxic conditions, this phosphorylation was decreased in the groups treated with 10 μM ZP1609‐pre or 10 μM ZP1609‐rep. The phosphorylation of glycogen synthase kinase 3α/β (GSK‐3 α/β) was reduced by I/R compared to normoxia but enhanced by ZP1609 (both 10 μM ZP1609‐pre and 10 μM ZP1609‐rep). In contrast to PRAS40 and GSK‐3 α/β, which were similarly influenced by ZP1609, an increase in the level of HSP60 (heat shock protein 60) was found specifically in the group 10 μM ZP1609‐rep. The exact pixel intensities are shown in Table S1.
Figure 9.
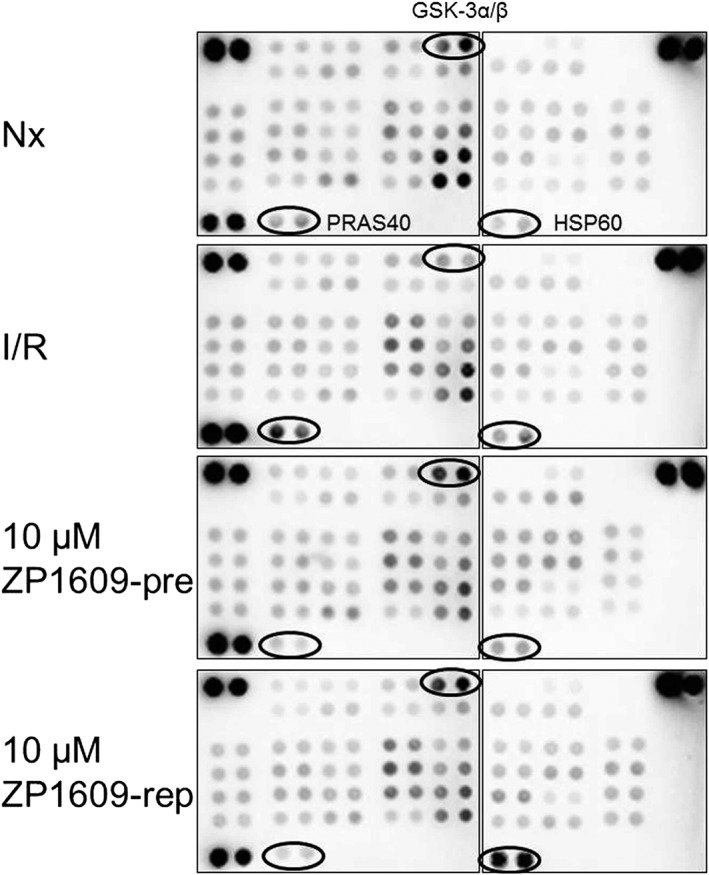
ZP1609 alters the phosphorylation or amounts of proteins in cardiomyocytes undergoing I/R. The phospho‐kinase array detects the phosphorylation status of several protein kinases in extracts of cardiomyocytes under normoxic conditions (Nx), I/R without or with 10 μM ZP1609 when applied during I/R (10 μM ZP1609‐pre) or at the onset of reperfusion (10 μM ZP1609‐rep). The proteins with the most obvious differences between groups are indicated. For the analysis, cardiomyocytes from four mice were pooled.
Discussion and conclusions
The compound ZP1609 was developed as a dipeptide with similar characteristics to those of rotigaptide, that is, enhancing gap junction conductance in order to decrease ischaemia‐induced arrhythmias. Whereas the exact molecular target of ZP1609 is unclear up to now, the anti‐arrhythmic properties of the peptide suggested Cx43 was a potential target. As Cx43 is not only localized at the plasma membrane but also within cardiomyocyte SSM, we tested putative effects of ZP1609 on mitochondrial function. The fact that IFM did not contain Cx43 allowed these mitochondria to be used as an internal negative control.
Previous studies showed that mitochondria of conditional Cx43‐knockout mice or mitochondria in which Cx43 was inhibited by the mimetic peptide Gap27 demonstrated decreased complex 1‐mediated respiration, whereas complex 2‐mediated respiration was unaffected. This effect was specific for Cx43‐containing SSM (Boengler et al., 2012). In the present study, ZP1609 decreased complex 1‐mediated respiration and, to a lower extent, complex 2‐mediated respiration in both SSM and IFM. In accordance with the data on mitochondrial oxygen consumption, ZP1609 inhibited ATP production in SSM and IFM. The magnitude of the reduction of ATP generation was comparable to that observed in a previous study (Boengler et al., 2012). ATP production was more effectively inhibited by 10 μM ZP1609 compared to 1 μM ZP1609, whereas the decrease in complex 1‐mediated respiration was similar between the two concentrations of ZP1609. This effect may be due to partial uncoupling of oxidative phosphorylation by the higher dose of ZP1609. The finding that ZP1609 affected the Cx43‐lacking IFM suggests that ZP1609 may have other molecular targets, apart from Cx43, within these mitochondria.
The opening of the MPTP contributes to reperfusion injury in animal studies and in humans (Piot et al., 2008; Hausenloy et al., 2009). Previous data showed that treatment of cardiac mitochondria with Gap27 reduced mitochondrial calcium retention capacity specifically in SSM (Srisakuldee et al., 2014). In the present study, in all control groups, IFM took up more calcium until MPTP opening occurred, than the SSM – a phenomenon already described before (Palmer et al., 1986) – thereby indicating accurate separation of the two mitochondrial populations. The incubation of SSM and IFM with 1 or 10 μM ZP1609 did not change MPTP opening in both mitochondrial populations, again suggesting that ZP1609 acted through targets other than Cx43 – at least under basal conditions.
Mitochondrial ROS are formed by the respiratory chain, but many other mitochondrial proteins contribute as well (Di Lisa et al., 2017). As ZP1609 reduced mitochondrial respiration, ROS formation was measured in the presence or absence of ZP1609. Although ZP1609 reduced respiration it did not affect ROS formation. One explanation for this finding relates to the small amount of ROS produced by the respiratory chain; only 0.1% of the total oxygen consumption is used to form ROS (St‐Pierre et al., 2002) and any minor change in the production rate might fall below the detection limit of the technique used for ROS determination. As ROS facilitate MPTP opening (Di Lisa et al., 2011), the lack of any effect of ZP1609 on ROS formation is, however, in accordance with the lack of effect of ZP1609 on calcium‐induced MPTP‐opening.
Under physiological conditions, the open probability of mitochondrial potassium channels is suggested to be low, whereas an opening of these channels prior to sustained I/R confers cardioprotection (Schulz and Di Lisa, 2016). Because pharmacological inhibition or genetic ablation of Cx43 reduces mitochondrial potassium influx (Miro‐Casas et al., 2009; Boengler et al., 2013), we assessed the effects of ZP1609 on mitochondrial potassium uptake. Whereas 1 μM ZP1609 did not alter potassium influx, the higher concentration (10 μM ZP1609) increased the entry of potassium into IFM, whereas it had no effect on SSM. Therefore, regarding all mitochondrial parameters analysed in the present study, potassium entry is the only factor where differences between SSM and IFM after treatment with ZP1609 were observed. The finding that the Cx43‐free IFM are targeted by ZP1609 again strengthens the hypothesis that ZP1609 acts through factors other than Cx43.
Taken together, our data on the effect of ZP1609 on mitochondrial function demonstrated no SSM‐dependent influence on respiration, ATP‐production, ROS‐generation, MPTP‐opening, or potassium uptake. Therefore, it is likely that, within mitochondria under basal conditions, Cx43 is not a direct target of ZP1609.
Gap junctions are also involved in myocardial damage by I/R injury and their uncoupling reduces infarct size (Rodriguez‐Sinovas et al., 2004; Shintani‐Ishida et al., 2009). Cx43‐formed hemichannels, which are predominantly closed under physiological conditions, open during ischaemia (Saez and Leybaert, 2014) and the prevention of their opening protects against I/R injury (Shintani‐Ishida et al., 2007; Wang et al., 2013). Therefore, gap junctional uncoupling or the prevention of hemichannel opening mediate protection, presumably by limiting the passage of a so‐called ‘death factor’. The passage of Na+‐ions through gap junctions from hypercontracting cardiomyocytes with a damaged sarcolemma to adjacent cells and secondary entry of Ca2+‐ions via reverse Na+/Ca2+‐exchange contributes to the propagation of cell damage (Ruiz‐Meana et al., 1999). On the other hand, a down‐regulation of Cx43 increases apoptosis (Yasui et al., 2000), showing that preserved cell–cell interaction may increase the transfer of a ‘survival factor’. Also, Cx43‐formed hemichannels participate in the transduction of cytoprotective factors and this may involve the activation of protein kinases (Plotkin et al., 2002).
With reperfusion following periods of prolonged ischaemia, restoration of mitochondrial ATP synthesis in the presence of still elevated intracellular calcium concentrations caused hypercontracture of cardiomyocytes leading to cell death (Garcia‐Dorado et al., 2012). As ZP1609 reduced mitochondrial ATP production and presumably targets sarcolemmal (hemi)‐channels, we assessed the influence of the drug on the hypercontracture of isolated cardiomyocytes following I/R and were able to demonstrate a reduced amount of hypercontracting cardiomyocytes when ZP1609 was applied prior to ischaemia or prior to reperfusion only. Blocking hypercontracture by 2,3‐butanedione monoxime or maintaining low pH during the early phase of reperfusion not only attenuated hypercontracture of cardiomyocytes but ultimately reduced infarct size (Garcia‐Dorado et al., 1992; Cohen et al., 2008). Similarly, ZP1609 decreased myocardial infarction when administered at reperfusion in vivo (Hennan et al., 2009; Skyschally et al., 2013).
In glioma cells, ZP1609 (Butera et al., 2009) reduced dye‐uptake via Cx43‐formed hemichannels, thereby suggesting hemichannel closure by ZP1609. Because opening of Cx43‐formed hemichannels in cardiomyocytes contributes to loss of ionic gradients, cellular volume overload and finally cell damage (Schulz et al., 2015), inhibition of hemichannel opening is cardioprotective (Wang et al., 2013). However, as opening of Cx43‐formed hemichannels is supposed to predominantly occur during ischaemia, the similar magnitude of protection by ZP1609 when applied before ischaemia or at reperfusion cannot be explained by hemichannel blockade.
Although there is no evidence from the data obtained in the present study that ZP1609 acts on Cx43 in cardiomyocytes, the potential for a direct or indirect interaction of ZP1609 with Cx43 remains. Indirect interaction through modulation of kinase activity has already been demonstrated for the anti‐arrhythmic peptide AAP10 which modulated the phosphorylation status of Cx43 (Jozwiak and Dhein, 2008). Also, rotigaptide prevented Cx43 de‐phosphorylation induced by ischaemia (Kjolbye et al., 2007). Specifically, the ischaemia‐induced de‐phosphorylation of Cx43 at Ser297 and Ser368 – which are targeted by PKC – was reduced following treatment with rotigaptide (Axelsen et al., 2006). A putative effect of ZP1609 on Cx43 phosphorylation would presumably be restricted to sarcolemmal Cx43, as this compound exerts no specific effects on Cx43‐containing SSM. Within mitochondria, ZP1609 probably acts independently of Cx43.
In order to identify target proteins of ZP1609 (other than Cx43), the influence of ZP1609 on different biochemical pathways was studied. The analysis of the phosphorylation status of several proteins revealed that the drug prevented the I/R‐induced de‐phosphorylation of GSK3 α/β. The de‐phosphorylation of the protein by I/R has already been described before and corresponds to its activation, whereas the phosphorylation of GSK3 inactivates the protein (Miura and Tanno, 2010). Inhibition of GSK3 exerts cardioprotective effects via the inhibition of MPTP opening at reperfusion (Juhaszova et al., 2004), which also prevents cardiomyocyte hypercontracture (Ruiz‐Meana et al., 2007). Whereas in the present study no effects of ZP1609 on MPTP opening under basal conditions were observed, such effects may still occur after I/R.
I/R induced the phosphorylation of PRAS40 and ZP1609 prevented such phosphorylation. PRAS40 was originally identified as a substrate of Akt and is known to inhibit the mTOR complex 1 (mTORC1) (Sancak et al., 2007). An increase in the phosphorylation of PRAS40 is known to prevent the inhibition of mTORC1, and the inhibition of mTORC1 with PRAS40 decreases tissue damage after myocardial infarction (Volkers et al., 2013). Therefore, the decreased phosphorylation of PRAS40 by ZP1609 may exert protection via decreased activity of mTORC1.
Whereas the effects of ZP1609 on the phosphorylation of GSK3 α/β or PRAS40 were not dependent on the time when ZP1609 was administered, differences occurred in the levels of HSP60, which was specifically increased by ZP1609‐rep and not by ZP1609‐pre. Together with HSP10, HSP60 acts as a mitochondrial chaperone and both factors are important for the folding of mitochondrial proteins. The overexpression of HSP60 in cardiomyocytes protects against I/R injury by maintaining mitochondrial integrity (Lin et al., 2001) and ZP1609 may contribute to the activation of such cardioprotective program. Taken together, the analysis of biochemical pathways influenced by ZP1609 results in the identification of putative target proteins, which may be involved in the protection of cardiomyocytes from I/R injury and which may function independently of Cx43.
ZP1609 is known to exert pharmacological effects in animal models of ventricular and atrial arrhythmias and its oral bioavailability presents the possibility for the prevention of cardiac arrhythmias in man. In addition, ZP1609 protects against myocardial I/R injury in animals. However, although published data suggest that ZP1609 acts on gap junctions via the modulation of Cx43, the present study indicates that ZP1609 can act independently of a direct interference with Cx43, at least at the level of mitochondria. In addition, new putative targets of ZP1609 were identified in the present study. This putative interference of ZP1609 with other factors has to be taken into account, when ZP1609 is further tested in clinical trials, and the identification of the exact protein(s) targeted by ZP1609 requires further studies.
Taken together, our data show that ZP1609 might exert cardioprotection by reducing the proportion of hypercontracting cardiomyocytes following I/R. It is possible that this protection is achieved through a reduction in mitochondrial respiration and ATP production rate. However, the protective effect of ZP1609 was independent of mitochondrial Cx43, as no difference in mitochondrial function was observed between Cx43‐containing SSM and Cx43‐free IFM.
Author contributions
K.B. performed the research on isolated mitochondria and wrote the paper. M.B. and R.S. performed the research on isolated cardiomyocytes. K.‐D.S. and R.S. designed the research study and helped in data analyses, interpretation and preparation of the manuscript.
Conflict of interest
Rainer Schulz has received honoraria for lectures provided to AstraZeneca, Recordati and Sanofi. The study was in part funded by a grant obtained from Zealand Pharma A/S.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Table S1 ZP1609 alters the phosphorylation and amounts of proteins in cardiomyocytes undergoing ischemia/reperfusion. Protein phosphorylation was studied using the human proteome profiler array (R&D Systems). The Table indicates the proteins identified, as well as the phosphorylation site(s) analyzed. The mean value of the measured pixel intensities (in arbitrary units, a.u.) of the negative controls were subtracted. Pixel intensities are shown for cardiomyocytes undergoing normoxia (Nx), ischemia/reperfusion in the absence (I/R) or presence of 10 μM ZP1609 when applied during ischemia/reperfusion (10 μM ZP1609‐pre) or at the onset of reperfusion (10 μM ZP1609‐rep).
Acknowledgements
We thank Elvira Ungefug, Sabrina Böhme, Anna Reis and Nadine Woitasky for their helpful technical assistance.
Boengler, K. , Bulic, M. , Schreckenberg, R. , Schlüter, K.‐D. , and Schulz, R. (2017) The gap junction modifier ZP1609 decreases cardiomyocyte hypercontracture following ischaemia/reperfusion independent from mitochondrial connexin 43. British Journal of Pharmacology, 174: 2060–2073. doi: 10.1111/bph.13804.
References
- Alexander SP, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015a). The concise guide to PHARMACOLOGY 2015/16: Other ion channels. Br J Pharmacol 172: 5942–5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5734–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amdisen C, Keller AK, Hansen RS, Norregaard R, Krag SP, Moldrup U et al. (2016). Testing Danegaptide effects on kidney function after ischemia/reperfusion injury in a new porcine two week model. PLoS One 11: e0164109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelsen LN, Stahlhut M, Mohammed S, Larsen BD, Nielsen MS, Holstein‐Rathlou NH et al. (2006). Identification of ischemia‐regulated phosphorylation sites in connexin43: a possible target for the antiarrhythmic peptide analogue rotigaptide (ZP123). J Mol Cell Cardiol 40: 790–798. [DOI] [PubMed] [Google Scholar]
- Beardslee MA, Lerner DL, Tadros PN, Laing JG, Beyer EC, Yamada KA et al. (2000). Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ Res 87: 656–662. [DOI] [PubMed] [Google Scholar]
- Boengler K, Ruiz‐Meana M, Gent S, Ungefug E, Soetkamp D, Miro‐Casas E et al. (2012). Mitochondrial connexin 43 impacts on respiratory complex I activity and mitochondrial oxygen consumption. J Cell Mol Med 16: 1649–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boengler K, Stahlhofen S, Van De Sand A, Gres P, Ruiz‐Meana M, Garcia‐Dorado D et al. (2009). Presence of connexin 43 in subsarcolemmal, but not in interfibrillar cardiomyocyte mitochondria. Basic Res Cardiol 104: 141–147. [DOI] [PubMed] [Google Scholar]
- Boengler K, Ungefug E, Heusch G, Leybaert L, Schulz R (2013). Connexin 43 impacts on mitochondrial potassium uptake. Front Pharmacol 4: 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butera JA, Larsen BD, Hennan JK, Kerns E, Di L, Alimardanov A et al. (2009). Discovery of (2S,4R)‐1‐(2‐aminoacetyl)‐4‐benzamidopyrrolidine‐2‐carboxylic acid hydrochloride (GAP‐134)13, an orally active small molecule gap‐junction modifier for the treatment of atrial fibrillation. J Med Chem 52: 908–911. [DOI] [PubMed] [Google Scholar]
- Clarke TC, Thomas D, Petersen JS, Evans WH, Martin PE (2006). The antiarrhythmic peptide rotigaptide (ZP123) increases gap junction intercellular communication in cardiac myocytes and HeLa cells expressing connexin 43. Br J Pharmacol 147: 486–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MV, Yang XM, Downey JM (2008). Acidosis, oxygen, and interference with mitochondrial permeability transition pore formation in the early minutes of reperfusion are critical to postconditioning's success. Basic Res Cardiol 103: 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa AD, Jakob R, Costa CL, Andrukhiv K, West IC, Garlid KD (2006). The mechanism by which the mitochondrial ATP‐sensitive K+ channel opening and H2O2 inhibit the mitochondrial permeability transition. J Biol Chem 281: 20801–20808. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vuyst E, Boengler K, Antoons G, Sipido KR, Schulz R, Leybaert L (2011). Pharmacological modulation of connexin‐formed channels in cardiac pathophysiology. Br J Pharmacol 163: 469–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lisa F, Carpi A, Giorgio V, Bernardi P (2011). The mitochondrial permeability transition pore and cyclophilin D in cardioprotection. Biochim Biophys Acta 1813: 1316–1322. [DOI] [PubMed] [Google Scholar]
- Di Lisa F, Giorgio M, Ferdinandy P, Schulz R (2017). New aspects of p66Shc in ischaemia reperfusion injury and cardiovascular diseases. Br J Pharmacol 174: 1690–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Dorado D, Ruiz‐Meana M, Inserte J, Rodriguez‐Sinovas A, Piper HM (2012). Calcium‐mediated cell death during myocardial reperfusion. Cardiovasc Res 94: 168–180. [DOI] [PubMed] [Google Scholar]
- Garcia‐Dorado D, Theroux P, Duran JM, Solares J, Alonso J, Sanz E et al. (1992). Selective inhibition of the contractile apparatus. A new approach to modification of infarct size, infarct composition, and infarct geometry during coronary artery occlusion and reperfusion. Circulation 85: 1160–1174. [DOI] [PubMed] [Google Scholar]
- Givvimani S, Pushpakumar S, Veeranki S, Tyagi SC (2014). Dysregulation of Mfn2 and Drp‐1 proteins in heart failure. Can J Physiol Pharmacol 92: 583–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausenloy DJ, Ong SB, Yellon DM (2009). The mitochondrial permeability transition pore as a target for preconditioning and postconditioning. Basic Res Cardiol 104: 189–202. [DOI] [PubMed] [Google Scholar]
- Hennan JK, Swillo RE, Morgan GA, Keith JC Jr, Schaub RG, Smith RP et al. (2006). Rotigaptide (ZP123) prevents spontaneous ventricular arrhythmias and reduces infarct size during myocardial ischemia/reperfusion injury in open‐chest dogs. J Pharmacol Exp Ther 317: 236–243. [DOI] [PubMed] [Google Scholar]
- Hennan JK, Swillo RE, Morgan GA, Rossman EI, Kantrowitz J, Butera J et al. (2009). GAP‐134 ([2S,4R]‐1‐[2‐aminoacetyl]4‐benzamidopyrrolidine‐2‐carboxylic acid) prevents spontaneous ventricular arrhythmias and reduces infarct size during myocardial ischemia/reperfusion injury in open‐chest dogs. J Cardiovasc Pharmacol Ther 14: 207–214. [DOI] [PubMed] [Google Scholar]
- Jozwiak J, Dhein S (2008). Local effects and mechanisms of antiarrhythmic peptide AAP10 in acute regional myocardial ischemia: electrophysiological and molecular findings. Naunyn Schmiedebergs Arch Pharmacol 378: 459–470. [DOI] [PubMed] [Google Scholar]
- Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW et al. (2004). Glycogen synthase kinase‐3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest 113: 1535–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjolbye AL, Haugan K, Hennan JK, Petersen JS (2007). Pharmacological modulation of gap junction function with the novel compound rotigaptide: a promising new principle for prevention of arrhythmias. Basic Clin Pharmacol Toxicol 101: 215–230. [DOI] [PubMed] [Google Scholar]
- Laurent G, Leong‐Poi H, Mangat I, Moe GW, Hu X, So PP et al. (2009). Effects of chronic gap junction conduction‐enhancing antiarrhythmic peptide GAP‐134 administration on experimental atrial fibrillation in dogs. Circ Arrhythm Electrophysiol 2: 171–178. [DOI] [PubMed] [Google Scholar]
- Lerner DL, Yamada KA, Schuessler RB, Saffitz JE (2000). Accelerated onset and increased incidence of ventricular arrhythmias induced by ischemia in Cx43‐deficient mice. Circulation 101: 547–552. [DOI] [PubMed] [Google Scholar]
- Lin KM, Lin B, Lian IY, Mestril R, Scheffler IE, Dillmann WH (2001). Combined and individual mitochondrial HSP60 and HSP10 expression in cardiac myocytes protects mitochondrial function and prevents apoptotic cell deaths induced by simulated ischemia‐reoxygenation. Circulation 103: 1787–1792. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–318m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miro‐Casas E, Ruiz‐Meana M, Agullo E, Stahlhofen S, Rodriguez‐Sinovas A, Cabestrero A et al. (2009). Connexin43 in cardiomyocyte mitochondria contributes to mitochondrial potassium uptake. Cardiovasc Res 83: 747–756. [DOI] [PubMed] [Google Scholar]
- Miura T, Tanno M (2010). Mitochondria and GSK‐3beta in cardioprotection against ischemia/reperfusion injury. Cardiovasc Drugs Ther 24: 255–263. [DOI] [PubMed] [Google Scholar]
- Palmer JW, Tandler B, Hoppel CL (1977). Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J Biol Chem 252: 8731–8739. [PubMed] [Google Scholar]
- Palmer JW, Tandler B, Hoppel CL (1986). Heterogeneous response of subsarcolemmal heart mitochondria to calcium. Am J Physiol 250: H741–H748. [DOI] [PubMed] [Google Scholar]
- Pedersen CM, Venkatasubramanian S, Vase H, Hyldebrandt JA, Contractor H, Schmidt MR et al. (2016). Rotigaptide protects the myocardium and arterial vasculature from ischaemia reperfusion injury. Br J Clin Pharmacol 81: 1037–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piot C, Croisille P, Staat P, Thibault H, Rioufol G, Mewton N et al. (2008). Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med 359: 473–481. [DOI] [PubMed] [Google Scholar]
- Plotkin LI, Manolagas SC, Bellido T (2002). Transduction of cell survival signals by connexin‐43 hemichannels. J Biol Chem 277: 8648–8657. [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Sinovas A, Garcia‐Dorado D, Ruiz‐Meana M, Soler‐Soler J (2004). Enhanced effect of gap junction uncouplers on macroscopic electrical properties of reperfused myocardium. J Physiol 559: 245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman EI, Liu K, Morgan GA, Swillo RE, Krueger JA, Gardell SJ et al. (2009). The gap junction modifier, GAP‐134 [(2S,4R)‐1‐(2‐aminoacetyl)‐4‐benzamido‐pyrrolidine‐2‐carboxylic acid], improves conduction and reduces atrial fibrillation/flutter in the canine sterile pericarditis model. J Pharmacol Exp Ther 329: 1127–1133. [DOI] [PubMed] [Google Scholar]
- Ruiz‐Meana M, Abellan A, Miro‐Casas E, Garcia‐Dorado D (2007). Opening of mitochondrial permeability transition pore induces hypercontracture in Ca2+ overloaded cardiac myocytes. Basic Res Cardiol 102: 542–552. [DOI] [PubMed] [Google Scholar]
- Ruiz‐Meana M, Garcia‐Dorado D, Hofstaetter B, Piper HM, Soler‐Soler J (1999). Propagation of cardiomyocyte hypercontracture by passage of Na(+) through gap junctions. Circ Res 85: 280–287. [DOI] [PubMed] [Google Scholar]
- Saez JC, Leybaert L (2014). Hunting for connexin hemichannels. FEBS Lett 588: 1205–1211. [DOI] [PubMed] [Google Scholar]
- Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E et al. (2007). PRAS40 is an insulin‐regulated inhibitor of the mTORC1 protein kinase. Mol Cell 25: 903–915. [DOI] [PubMed] [Google Scholar]
- Schluter KD, Schreiber D (2005). Adult ventricular cardiomyocytes: isolation and culture. Methods Mol Biol 290: 305–314. [DOI] [PubMed] [Google Scholar]
- Schulz R, Di Lisa F (2016). Mitochondrial potassium homeostasis: a central player in cardioprotection. Cardiovasc Res 110: 4–5. [DOI] [PubMed] [Google Scholar]
- Schulz R, Gorge PM, Gorbe A, Ferdinandy P, Lampe PD, Leybaert L (2015). Connexin 43 is an emerging therapeutic target in ischemia/reperfusion injury, cardioprotection and neuroprotection. Pharmacol Ther 153: 90–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani‐Ishida K, Uemura K, Yoshida K (2007). Hemichannels in cardiomyocytes open transiently during ischemia and contribute to reperfusion injury following brief ischemia. Am J Physiol Heart Circ Physiol 293: H1714–H1720. [DOI] [PubMed] [Google Scholar]
- Shintani‐Ishida K, Unuma K, Yoshida K (2009). Ischemia enhances translocation of connexin43 and gap junction intercellular communication, thereby propagating contraction band necrosis after reperfusion. Circ J 73: 1661–1668. [DOI] [PubMed] [Google Scholar]
- Skyschally A, Walter B, Schultz Hansen R, Heusch G (2013). The antiarrhythmic dipeptide ZP1609 (danegaptide) when given at reperfusion reduces myocardial infarct size in pigs. Naunyn Schmiedebergs Arch Pharmacol 386: 383–391. [DOI] [PubMed] [Google Scholar]
- Soetkamp D, Nguyen TT, Menazza S, Hirschhauser C, Hendgen‐Cotta UB, Rassaf T et al. (2014). S‐nitrosation of mitochondrial connexin 43 regulates mitochondrial function. Basic Res Cardiol 109: 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srisakuldee W, Makazan Z, Nickel BE, Zhang F, Thliveris JA, Pasumarthi KB et al. (2014). The FGF‐2‐triggered protection of cardiac subsarcolemmal mitochondria from calcium overload is mitochondrial connexin 43‐dependent. Cardiovasc Res 103: 72–80. [DOI] [PubMed] [Google Scholar]
- St‐Pierre J, Buckingham JA, Roebuck SJ, Brand MD (2002). Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem 277: 44784–44790. [DOI] [PubMed] [Google Scholar]
- Sun J, Nguyen T, Aponte AM, Menazza S, Kohr MJ, Roth DM et al. (2015). Ischaemic preconditioning preferentially increases protein S‐nitrosylation in subsarcolemmal mitochondria. Cardiovasc Res 106: 227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi N, Vacek JC, Givvimani S, Sen U, Tyagi SC (2010). Cardiac specific deletion of N‐methyl‐d‐aspartate receptor 1 ameliorates mtMMP‐9 mediated autophagy/mitophagy in hyperhomocysteinemia. J Recept Signal Transduct Res 30: 78–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkers M, Konstandin MH, Doroudgar S, Toko H, Quijada P, Din S et al. (2013). Mechanistic target of rapamycin complex 2 protects the heart from ischemic damage. Circulation 128: 2132–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, De Vuyst E, Ponsaerts R, Boengler K, Palacios‐Prado N, Wauman J et al. (2013). Selective inhibition of Cx43 hemichannels by Gap19 and its impact on myocardial ischemia/reperfusion injury. Basic Res Cardiol 108: 309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing D, Kjolbye AL, Nielsen MS, Petersen JS, Harlow KW, Holstein‐Rathlou NH et al. (2003). ZP123 increases gap junctional conductance and prevents reentrant ventricular tachycardia during myocardial ischemia in open chest dogs. J Cardiovasc Electrophysiol 14: 510–520. [DOI] [PubMed] [Google Scholar]
- Yasui K, Kada K, Hojo M, Lee JK, Kamiya K, Toyama J et al. (2000). Cell‐to‐cell interaction prevents cell death in cultured neonatal rat ventricular myocytes. Cardiovasc Res 48: 68–76. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 ZP1609 alters the phosphorylation and amounts of proteins in cardiomyocytes undergoing ischemia/reperfusion. Protein phosphorylation was studied using the human proteome profiler array (R&D Systems). The Table indicates the proteins identified, as well as the phosphorylation site(s) analyzed. The mean value of the measured pixel intensities (in arbitrary units, a.u.) of the negative controls were subtracted. Pixel intensities are shown for cardiomyocytes undergoing normoxia (Nx), ischemia/reperfusion in the absence (I/R) or presence of 10 μM ZP1609 when applied during ischemia/reperfusion (10 μM ZP1609‐pre) or at the onset of reperfusion (10 μM ZP1609‐rep).