

SUMMARY
Histone citrullination regulates diverse cellular processes. Here, we report that SMARCAD1 preferentially associates with H3 Arginine 26 citrullination (H3R26Cit) peptides present on arrays composed of 384 histone peptides harboring distinct post-transcriptional modifications. Among 10 histone modifications assayed by ChIP-seq, H3R26Cit exhibited the most extensive genomewide co-localization with SMARCAD1 binding. Increased Smarcad1 expression correlated with naïve pluripotency in pre-implantation embryos. In the presence of LIF, Smarcad1 knockdown (KD) embryonic stem cells lost naïve state phenotypes but remained pluripotent, as suggested by morphology, gene expression, histone modifications, alkaline phosphatase activity, energy metabolism, embryoid bodies, teratoma, and chimeras. The majority of H3R26Cit ChIP-seq peaks occupied by SMARCAD1 were associated with increased levels of H3K9me3 in Smarcad1 KD cells. Inhibition of H3Cit induced H3K9me3 at the overlapping regions of H3R26Cit peaks and SMARCAD1 peaks. These data suggest a model in which SMARCAD1 regulates naïve pluripotency by interacting with H3R26Cit and suppressing heterochromatin formation.
Keywords: SMARCAD1, histone modification, citrullination, stem cells, naïve state, pluripotency, ChIP-seq, protein array
eTOC blurb
Xiao et al. identify SMARCAD1 as a candidate reader protein of the H3R26Cit histone modification. Knockdown of Smarcad1 causes mouse ES cells to exit the naïve pluripotent state and leads to increased H3K9me3 at SMARCAD1 binding regions. This suggests a model where SMARCAD1 guards naïve pluripotency by interacting with H3R26Cit and suppressing H3K9me3.
INTRODUCTION
Histone citrullination, also called deimination, is a post-translational conversion of arginine into the amino acid citrulline (Klose and Zhang, 2007). The functional importance of H3 citrullination (H3Cit) includes regulation of naïve pluripotency (Christophorou et al., 2014), gene regulation in cancer (Zhang et al., 2012), immune response (Neeli et al., 2008) and autoimmune disease (Sharma et al., 2012). However, the mechanisms underlying histone citrullination mediated regulation of cell physiology remain elusive. The available information includes that citrullination is antagonistic to arginine methylation (Cuthbert et al., 2004), negatively regulates chromatin compaction (Christophorou et al., 2014), partially by weakening H3K9me3 mediated heterochromatin formation (Sharma et al., 2012). It is unknown whether any protein could read histone citrullination and relay its regulatory signal.
To search for proteins that may interact with citrullinated histones, we leveraged the discovery that H3Cit is associated with the establishment of naïve pluripotency during embryonic development and cellular reprogramming (Christophorou et al., 2014). We searched for proteins that can be attached to chromatin and are correlated with the establishments of inner cell mass (ICM) and naïve-state pluripotent stem cells. This search identified two genes with temporal expression patterns that peak at blastocyst stage and are elevated in the ICM that share sequence similarities to chromatin modification protein genes. These genes are Smarca4 (a.k.a Brg1) and Smarcad1. Because the role of Smarca4 on ES cell regulation has been characterized and the published data do not involve H3Cit (Kidder et al., 2009), we focused on the less studied Smarcad1.
Smarcad1, a.k.a. Etl1 or Hel1, is one of the mammalian SNF2 family genes (Soininen et al., 1992). It contains DEAD/H ATP-binding domains and a bipartite nuclear localization signal (Schoor et al., 1993, Adra et al., 2000). It is expressed in all assayed embryonic and adult tissues (Adra et al., 2000), but its expression levels are exceptionally high in embryonal, mammary, and lymphoid tumors (Okazaki et al., 2008). SMARCAD1 is attached to chromatin in both cancer and normal cells (Okazaki et al., 2008, Rowbotham et al., 2011).
RESULTS
Increased SMARCAD1 expression is associated with naïve pluripotency in developing embryos and in cell culture
We analyzed the association between Smarcad1 expression levels and pluripotent states in mouse and human preimplantation embryos. We generated RNA sequencing (RNA-seq) data from the ICM and the whole blastocyst of preimplantation mouse embryos. Smarcad1 was expressed 2.7 times higher in ICM (RPKM=132.4) than in the whole blastocyst (RPKM=49.4) (Figure 1A). Consistently, in a published RNA-seq dataset (Tang et al., 2010), Smarcad1 exhibited 1.5 times higher expression in the ICM than in trophectoderm. Next, we re-analyzed our previously generated gene expression data from mouse preimplantation embryos at 7 developmental stages with 3 biological replicates at each stage (Xie et al., 2010). Smarcad1 mRNA was strongly and reproducibly induced at the 8-cell stage (Figure 1B), approximately 2 days prior to implantation. At the blastocyst stage, mouse SMARCAD1 protein expression is restricted to the ICM (see Figure 4 of (Schoor et al., 1993)). In a single-cell RNA-seq dataset from human preimplantation embryos (GEO: GSE36552, (Yan et al., 2013)), SMARCAD1 expression increased from 2-cell to blastocyst stage, peaking in some single cells in morula in ICM (Figure 1C). All except one human mural trophectoderm cells had low SMARCAD1 expression (Figure 1C). Human 32-cell morula is formed approximately 2 days prior to implantation (Cockburn and Rossant, 2010), therefore the SMARCAD1 mRNA peaked at approximately the same amount of time prior for implantation in mice and in humans.
Figure 1. Smarcad1 expression patterns in early embryonic development and in pluripotent stem cells.

(A) RNA-seq derived Smarcad1 mRNA levels in ICM, whole blastocyst, trophectoderm, and epiblast. dpc: days post conception. (B) Expression heatmap of genes related to DNA methylation, H3K9 methylation, and histone citrullination. Three biological replicates (a, b, c) were analyzed in each of 7 stages in mouse preimplantation development. L-Morula: late morula stage. Gene expression levels were normalized across samples, clustered (dendrogram), and visualized (yellow: high expression, blue: low expression). (C) SMARCAD1 expression (y axis) in single cells of human preimplantation embryos (GEO: GSE36552). Each column represents a single cell, and the cells were grouped by developmental stage (x axis). TE: trophectoderm. (D–E) Western blots of SMARCAD1 and ACTIN in mouse ES cells and EpiSCs (D), and in pig naïve and primed pluripotent cells (E). (F) Microarray derived Smarcad1 expression levels in mouse ES cells, EpiSC, and epiblast. P4, P24: denotes passages 4 and 24.
Furthermore, we tested the association between Smarcad1 expression and pluripotent states using various cell lines. SMARCAD1 protein levels were significantly higher in mouse embryonic stem (ES) cells (naïve state) than in epiblast derived stem cells (EpiSCs, primed state) (Figure 1D). In humans, SMARCAD1 mRNA was more abundant in naïve cells as compared to primed ES cells (RNA-seq data from ArrayExpress: E-MTAB-2857 (Takashima et al., 2014), p-value = 0.012, two-tailed T test). In addition, our western blots suggest that SMARCAD1 proteins were more abundant in pig naïve ES cells (Telugu et al., 2011) than in the primed pig iPS cells (Ezashi et al., 2009) (Figure 1E). We do not have data to directly compare Smarcad1 expression levels in preimplantation ICM and postimplantation epiblasts. However, re-analysis of a published microarray dataset (Tesar et al., 2007) suggested higher Smarcad1 mRNA expression in ES cells than in postimplantation epiblasts (p-value=0.00039) or in EpiSCs (p-value=0.00015) (Figure 1F). Taken together, higher Smarcad1 mRNA and protein levels are associated with naïve pluripotent stem cells in developing embryos and in cell culture.
SMARCAD1 possesses binding specificity to histone modification but not to DNA sequence
SMARCAD1 binds to chromatin (Okazaki et al., 2008, Rowbotham et al., 2011). To determine whether the specificity of SMARCAD1-chromatin interaction is achieved by SMARCAD1’s recognition of specific DNA sequences, we carried out high-throughput SELEX (HT-SELEX) (Zhao et al., 2009) with two randomized sequence libraries, one with a 10 bp and the other with a 20 bp of randomized region. HT-SELEX was performed for two rounds on each of the two libraries. Neither selection enriched for any sequence motif, suggesting that the SMARCAD1 protein used in this study does not recognize specific DNA sequences.
We then asked whether SMARCAD1 recognizes specific histone post-translational modifications. We incubated SMARCAD1 with two MODified™ histone peptide arrays. Each array contained 384 19-mer histone peptides. Each peptide is an N-terminal tail of H2A, H2B, H3, or H4 with a unique combination of post-translational modifications (ActiveMotif, 2014). The two arrays exhibited reproducible binding signals, and they consistently identified the peptide with a single modification (H3R26Cit) as the strongest binding peptide (Figure 2A). We then compared the post-translationally modified peptides to those with the same amino acid sequence without any modification by calculating the ratio of the binding signals between every modified peptide and its unmodified counterpart. H3R26Cit exhibited the largest binding increment (ratio) to the unmodified peptide, followed by H3K27ac, however only the H3R26Cit peptide exhibited 3 fold or larger binding signals than the background peptides in both arrays (Figure 2B, Figure S1). These data prioritize H3R26Cit, followed by H3K27ac, as histone modifications that SMARCAD1 may preferentially associate with in vitro.
Figure 2. SMARCAD1 recognizes H3R26Cit in vitro and co-localizes with H3R26Cit in vivo.

(A) Binding signals of SMARCAD1 on MODified™ histone peptide arrays. Each dot represents the binding intensities to a peptide, that is modified with a unique combination of post-transcriptional modifications, on array 1 (x axis) and array 2 (y axis). (B) Binding signals on post-translationally modified versus unmodified peptide (log ratio, y axis). All the peptides with a single modification and the unmodified peptides are shown (columns). If the raw binding signal to a modified peptide was smaller than the average binding signal to background peptides, the log ratio was assigned to 0 (non-informative, Columns 1 – 50). If the raw binding signal was above background, this binding signal was divided by that of another peptide with identical amino acid sequence without any modification (y axis, in log scale). Error bar: standard deviation of the mean. *: signal > 3-fold background in one array. **: signal > 3-fold background in both arrays. (C) Relative levels (y axis) of co-localization of SMARCAD1 and each epigenetic modification (x axis), using the entire genome (blue bars) or OCT4 (orange bars) as the controls. Odds ratio > 1 or < 1 corresponds to an increased or decreased level of co-localization. (D) Cumulative counts of overlaps (y axis) of SMARCAD1 and H3R26Cit peaks, ordered by the significance (MACS reported p-value) of H3R26Cit peaks (x axis). Red curve shows the overlaps from a permutation analysis where SMARCAD1 peaks were randomly shifted to other genomic locations, while keeping the size of each peak and the locations of the H3R26Cit peaks.
SMARCAD1 co-localizes with H3R26Cit on the genome
The in vitro binding specificity led us to posit that SMARCAD1 binds to chromatin with H3R26Cit or H3K27ac in vivo. To test this, we carried out chromatin immunoprecipitation followed by sequencing (ChIP-seq) on H3R26Cit (Abcam: ab19847, (Zhang et al., 2012)) and SMARCAD1 (Abcam: ab67548) in mouse ES cells. These data were jointly analyzed with our published ChIP-seq datasets for 9 other histone modifications from the same cell line, including H3K27ac, H2A.Z, H3K4me1, H3K4me2, H3K4me3, H3K9me3, H3K27me3, H3K36me3, and 5-mC (MeDIP-seq) (Xiao et al., 2012) (Figure S2A). The extent of H3K27ac-SMARCAD1 co-localization was 4.6 fold greater than expectation (odds ratio = 4.56, blue bar, Figure 2C). Among the 10 histone modifications, H3R26Cit exhibited the strongest co-localization with SMARCAD1 (odds ratio = 9.94, p-value < 10−20, Chi-square test) (blue bars, Figure 2C). These data suggest a genomewide correlation of H3 citrullination and SMARCAD1 binding in ES cells.
We used OCT4 ChIP-seq (Xiao et al., 2012) as another control to further assess the specificity of the genomewide co-localization of SMARCAD1 binding with any the 10 histone modifications. OCT4 and SMARCAD1 exhibited similar degrees of co-localization (odds ratio ≈ 1) with 9 of the 10 histone marks including H3K27ac (orange bars, Figure 2C). In particular, the extent of H3K27ac-SMARCAD1 co-localization does not clearly exceed that of H3K27ac-OCT4 (odds ratio ≈ 1, orange bar in H3K27ac column of Figure 2C), suggesting that H3K27ac may generally enhance protein-chromatin interactions but such an effect is not specific to SMARCAD1. The only exception among the 10 analyzed histone marks was H3R26Cit, which exhibited stronger genomewide co-localization to SMARCAD1 than OCT4 (odds ratio = 4.22, p-value < 10−20, Chi-square test) (orange bar in H3R26Cit column, Figure 2C).
Using MACS (v1.4.0beta) (Zhang et al., 2008) and IgG ChIP-seq as control, we identified a total of 363 H3R26Cit peaks in the genome. Among them, 22% (79 peaks) were bound by SMARCAD1, which correspond to an approximately 300 fold enrichment of overlap than expectation (1,279 SMARCAD1 peaks in the genome, covering ~1 million bases (approximately 1/1800 of the effective genome), odds ratio > 300, p-value < 10−10, Chi-square test, Figure 2D). Moreover, 34%, 42%, and 48% out of the top 150, top 100, and top 50 H3R26Cit peaks overlapped with SMARCAD1 peaks, suggesting that the more significant H3R26Cit peaks were more preferentially bound by SMARCAD1 (Figure 2D).
Phenotypic differences between Smarcad1 KD and naïve ES cells
We asked whether SMARCAD1 expression relates to naïve state pluripotency. To test this, we knocked down Smarcad1 (Smarcad1 KD) in naïve ES cells with two different shRNA constructs. These constructs reduced Smarcad1 mRNA to 42% of the original level on average (Figure 3E), and resulted in decreased protein levels (Figure 3F). We examined cell morphology, alkaline phosphatase (AP) activity, energy metabolism, contribution to ICM in chimeras, switch of culture condition, and the transcriptome of Smarcad1 KD cells.
Figure 3. Smarcad1 knockdown in mouse ES cells.

Mouse ES cells transfected with a control shRNA (A) and a Smarcad1 targeting shRNA (B). AP staining of control (C) and SMARCAD1 KD cells (D). (E) RT-PCR derived expression fold changes between Smarcad1 KD and control knockdown cells (Control). Error bars were derived from three biological replicates. (F) Western blots. pSuper: empty vector control. Luci: control knockdown. RNAi1, RNAi2: two shRNAs targeting different parts of Smarcad1 mRNA. (G) Xist expression measured by qPCR in control (Luciferase KD) El16.6 (green) and Smarcad1 KD El16.6 ES cells (yellow). (H) Proportions of injected embryos with ICM integration. (I–J) Control (Luciferase KD) (I) and Smarcad1 KD (J) ES cells cultured under EpiSC culture condition. (K) Tissues and cell types identified by histological staining of EBs derived from SMARCAD1 KD E14 ES cells. Scar bar = 100 μm in panels A–D, and I–K.
Within 48 hours of shRNA transfection, mouse ES cells lost their typical morphology of compact, rounded colonies and exhibited a flattened shape (Figure 3A–B). These changes were consistently observed in the two shRNA transfections (Figure S3A–C). We passaged Smarcad1 KD cells for more than 80 passages and could not observe colonies with the appearance of differentiation, but flattened monolayer colonies reminiscent of epiblast stem cells (EpiSCs, primed state) were persistently observed (Figure S3A–C). A total of 27 out of 30 Smarcad1 KD colonies lost AP activity (Figure 3C), a marker of self-renewal which is absent in EpiSCs (Brons et al., 2007; G reber et al., 2010; van Oosten et al., 2012), whereas 9 out 10 control (Luciferase KD) colonies retained AP activity (Figure 3D). Compared to control (Luciferase KD), Smarcad1 KD cells exhibited reduced oxygen consumption rate and increased extracellular acidity rate (Figure S4A–B), suggesting greater dependence of glycolysis for energy metabolism (Zhou et al., 2012). In a female ES cell line (EL16.6, established by Jeannie Lee lab (Zhao et al., 2008)), Smarcad1 KD cells exhibited 2.6 fold increase in Xist expression as compared to Luciferase KD control (Figure 3G).
We compared the frequencies of ICM contribution of chimeric blastocysts using an Oct4-EGFP reporter ES cell line (Ogr1). While 90.0% of the embryos (n=10) injected with Ogr1 exhibited ES cell integration to ICM, 55.8% of Smarcad1 KD Ogr1 injected embryos (n=43) exhibited ICM integration, suggesting a reduction of chimeric formation in Smarcad1 KD cells (P-value = 0.04, Fisher’s exact test) (Figure 3H, Figure S4C–E). In summary, Smarcad1 KD ES cells exhibited phenotypic differences to naïve ES cells in cell morphology, AP activity, energy metabolism, capacity of forming chimeras. Moreover, these phenotypic assays were carried out without transferring the cells to EpiSC culture condition (with FGF2 and ACTIVIN-A, without LIF) (Wu et al., 2015), suggesting the observed phenotypic differences were unlikely due to environmental differences. Therefore, by controlling for the culture condition, Smarcad1 KD cells maintained in ES culture may offer a unique opportunity to dissect the genetic factors responsible for naïve pluripotency.
Finally, we moved cells from ES culture condition (with LIF) into EpiSC culture condition (with FGF2 and ACTIVIN-A, without LIF) (Wu et al., 2015). After changing to the EpiSC culture condition, control (Luciferase KD) E14 ES cells exhibited a differentiated cell morphology at passage 5 (Figure 3I). In comparison, Smarcad1 KD E14 ES cells kept its monolayer, human ES-like morphology at the same passage (Figure 3J). This suggests that Smarcad1 KD and control ES cells respond differently to FGF2/ACTIVIN-A signaling. We note that all other characterizations of the Smarcad1 KD cells in the rest of this manuscript were carried out in the ES culture condition (with LIF) (Li et al., 2011) for the purpose of teasing out the direct effects of Smarcad1 expression change.
Smarcad1 KD ES cells retain pluripotency characteristics
We asked whether Smarcad1 KD ES cells lost pluripotency. Real time quantitative PCR analysis showed no expression differences of pluripotency markers Oct4, Nanog, and Sox2 in Smarcad1 KD cells (passage 50) and in control ES cells (passage 50) (Figure 3E). Subjected to an embryoid body (EB) formation assay, Smarcad1 KD cells (passage 35) formed EBs that were morphologically indistinguishable from those formed by control cells (passage 35) (Figure S5D, E). Marker genes of all three germ layers, including Mtap2, Nestin, Cd31, T, Flk-1, Gata4, Afp, Ihh, and Gata6 were expressed 5 to 1500 times higher in 16-day EBs than in ES cells, but at similar levels in control EBs and in SMARCAD1 knockdown ES cell-derived EBs (Figure S5F). No discernable difference in cell proliferation rates was identified between Smarcad1 KD cells and control (Luciferase KD) cells (Figure S5B–C).
To test teratoma formation, we injected four mice with Smarcad1 KD (passage 55) and control ES cells (passage 55). The total numbers of tumors formed were the same (7) for Smarcad1 KD and control cells (Figure S5G–H, Table S1). In the teratomas originating from Smarcad1 KD cells, we found characteristic cell types and tissues of all 3 germ layers, including neural cells, the neural tube, keratin pearl, cartilage, striated muscle, and ciliated epithelium (Figure 3K). Collectively, the expression of pluripotency markers and the formation of EBs and teratomas suggest that Smarcad1 KD ES cells maintained the capacity for pluripotent lineage specification.
Transcriptome of Smarcad1 KD cells in ES culture
It remains impossible to dissect the genetic regulators of naïve to primed transition from environmental regulators, because in vitro naïve or primed cells have to be maintained in their respective culture condition, and in vivo these cells are exposed to different signals as well (Han et al., 2010). We assayed the transcriptomes of control (naïve) and Smarcad1 KD ES cells cultured in ES medium using microarrays and RNA-seq, and compared with published gene-expression data of EpiSCs cultured in EpiSC medium and wildtype ES cells (naïve) cultured in ES medium (Tesar et al., 2007, Factor et al., 2014, Huang et al., 2014). Because our Smarcad1 KD cells were maintained in ES medium, we did not expect the expression differences between Smarcad1 KD and ES cells to exhibit genomewide correlations to the expression differences between EpiSC (in EpiSC culture) and ES cells (in ES culture), however, the data exhibited a moderate genomewide correlation (Figure S3F–G) (Supplementary Text). In particular, Smarcad1 KD cells in ES culture exhibited reduced expression of naïve pluripotency markers Klb, Tbx3, Bmp4, Tfcp2l1, Tet2, Piwil2, Klf4, Stra8, Fgf4, Wnt6, Esrrb, Pecam1, Zfp42 and increased expression of primed state markers Smad3, Fgf8, Lefty1, Pitx2, Fgf5, Pim2, Fabp7 (Figure S3D). In addition, 9 genes in the ACTIVIN/TGFβ pathway were up-regulated in Smarcad1 KD cells, making the TGFβ pathway the only signaling pathway that was statistically enriched with differentially expressed genes (Fisher’s test p-value < 0.001, Figure S3E). Activation of the ACTIVIN/TGFβ signal is another hallmark of the primed state, but previously reported changes in this pathway were compounded with changes of external signals (Brons et al., 2007, Merrill, 2012). Finally, neither Cdkn1a (a.k.a. P21) nor Cdkn2a (a.k.a. P16) exhibited increased expression which suggested these cells were unlikely to have undergone senescence (Figure S5A).
Histone modification changes induced by Smarcad1 KD
In order to characterize Smarcad1 KD induced histone modification changes, we carried out ChIP-seq on H3K4me3 and H3K27ac in Smarcad1 KD cells (in ES medium), and compared them with published ChIP-seq datasets in EpiSC (in EpiSC medium) and ES cells (in ES medium). Using published H3K4me3 ChIP-seq (GEO: GSM1382218), we identified a total of 27,431 peaks from EpiSC or ES cells, and calculated modification intensity in each peak. We compared the ratio of modification intensities between EpiSC and ES cells (y axis, Figure 4A) to the ratio between Smarcad1 KD and ES cells (x axis, Figure 4A). The ratios between EpiSC and ES are correlated with that between Smarcad1 KD and ES cells in a genomewide manner (Figure 4A, p-value < 10−15). Next, we identified the genomic regions with strong induction of H3K4me3 in Smarcad1 KD (KD induced peaks) and that with strong repression (KD suppressed peaks). H3K4me3 in EpiSC exhibited increased intensities in KD induced peaks (Figure 4B), and reduced intensities in KD suppressed peaks (Figure 4C). Taken together, without changing the culture condition, suppression of Smarcad1 expression in ES cells makes the genomewide distribution of H3K4me3 shift toward that in EpiSC.
Figure 4. Epigenomic difference between Smarcad1 KD and ES cells.

(A) H3K4me3 changes (log ratio) between Smarcad1 KD and control ES cells (x axis), versus H3K4me3 changes (log ratio) between EpiSC and ES cells (y axis). Each dot represents an H3K4me3 peak identified in either EpiSC or ES cells. A total of 27,431 peaks (the union of 16,115 peaks in ES cells and 28,431 peaks in EpiSC) are plotted. (B–C) Average H3K4me3 ChIP-seq intensities (y axis) in Smarcad1 KD cells (red), ES cells (green), and EpiSC cells (yellow), in a total of 565 Smarcad1 KD induced peaks (B), and a total of 496 Smarcad1 KD repressed peaks (C). (D) H3K27ac differences between Smarcad1 KD and ES cells (x axis) versus H3K27ac changes between EpiSC and ES cells (y axis) on the union of H3K27ac peaks (43,797) identified from EpiSC and ES cells. (E) H3K27ac distribution near the Kdm5b locus in ES cells, Smarcad1 KD, and EpiSC, marked with previously (Factor et al.) identified ES-specific peaks (blue) and EpiSC-specific peaks (pink). H3K27ac in Smarcad1 KD exhibited reduced signals in 3 ES-specific peaks and increased signals in the EpiSC-specific peak (marked with arrows). A new H3K27ac peak was identified (yellow), where both EpiSC and Smarcad1 KD exhibited increased signals as compared to ES cells. (F–G) Western blots of H3K9me2 (F) and H3K9me3 (G) in Control (Luciferase KD) and Smarcad1 KD (KD) mouse ES cells. (H) Average H3K9me3 ChIP-Seq signals (read counts per 5×107 reads, color bar) in ES cells (left) and Smarcad1 KD cells (right) are plotted against the distances to peak centers (x axis, peak center = 0) of 9 chromatin binding proteins (rows). A total of 10,000 random genomic locations are also included (last row).
We identified a total of 43,797 H3K27ac peaks in either EpiSC or ES cells (GEO: GSM1382218). The ratio of H3K27ac intensities between EpiSC and ES cells (y axis) is correlated with that between Smarcad1 KD and ES cells in a genomewide manner (x axis, Figure 4D, p-value < 10−15). In particular, we examined the Kdm5b gene locus, which was the main example where difference in H3K27ac was observed between EpiSC and ES cells (see Figure 2 in (Factor et al., 2014)). Factor et al. identified 5 ES-specific and 1 EpiSC-specific H3K27ac peaks. Smarcad1 KD cells exhibited H3K27ac decrease on 3 out of the 5 ES-specific peaks (blue regions, Figure 4E), and H3K27ac increase on the EpiSC-specific peak (pink region, Figure 4E). These data suggest that Smarcad1 KD shared some but not all enhancer switches with EpiSC, as indicated by H3K27ac changes. In addition, we identified a new EpiSC-specific peak at the Kdm5b locus (yellow bar, Figure 4E), which exhibits increased H3K27ac in both EpiSC and Smarcad1 KD cells. Taken together, H3K27ac changes between Smarcad1 KD and ES cells correlate with the changes between EpiSC (in EpiSC medium) and ES cells (in ES medium).
SMARCAD1 binding modulates H3K9me3 at a subset of SMARCAD1-binding sites
We explored the downstream components of the proposed SMARCAD1-chromatin interaction. ChIP-seq data suggested moderate co-localization of SMARCAD1 and H3K9me3 (H3K9me3 columns, Figure 2C). To assess the biological significance, we used SETDB1-H3K9me3 co-localization as a positive control because H3K9 methyltransferase SETDB1 was thought to co-localize with H3K9me3 in mouse ES cells (Schultz et al., 2002, Karimi et al.). Compared to SETDB1, SMARCAD1 ChIP-seq peaks exhibited larger overlaps with H3K9me3 peaks (Figure S2C–D), suggesting the extent of SMARCAD1-H3K9me3 co-localization was not trivial. Considering that SMARCAD1 does not recognize H3 tail with H3K9me3 in vitro (Column 17 in Figure 2B), we posited that H3K9me3 is downstream to SMARCAD1 binding to chromatin.
To test this, we started by comparing the total amount of H3K9me2 and H3K9me3 in wildtype and Smarcad1 KD ES cells, which revealed no discernable difference (Figure 4F–G, Figure S2G). Next, we carried out H3K9me3 ChIP-seq in Smarcad1 KD cells and compared to H3K9me3 ChIP-seq data in ES cells. There was no discernable difference of H3K9me3 modification levels in 10,000 randomly selected genomic regions (Figure 4H), which is consistent with the western blot data (Figure 4F–G). In addition, there was no discernable difference in the ChIP-seq peaks of OCT4, NANOG, and CTCF (Figure 4H). However, in the 1,279 SMARCAD1 peaks (called with ChIP-seq in ES cells using MACS v1.4.0beta (Zhang et al., 2008)), H3K9me3 modification levels were substantially increased in Smarcad1 KD cells compared to ES cells (p-value < 10−15, two-tailed T test) (Figure 4H). Furthermore, the largest changes of H3K9me3 precisely appeared at the centers of the Smarcad1 peaks (Figure 4H, x = 0). These data suggest that although there are no global changes of H3K9me3 in Smarcad1 KD cells, there is increase of H3K9me3 in SMARCAD1 binding regions. These data are incompatible with an alternative model, where Smarcad1 KD caused differentiation, which in turn resulted in global chromatin condensation and global H3K9me3 increases.
To further assess whether the H3K9me3 changes in SMARCAD1 peaks were due to reduced SMARCAD1 binding in the Smarcad1 KD condition, we did SMARCAD1 ChIP-seq in Smarcad1 KD E14 ES cells. Out of the 1,279 SMACARD1 peaks detected in ES cells, 1207 (94.4%) exhibited decrease of ChIP-seq signal in Smarcad1 KD cells, and 897 (70%) exhibited strong decrease (KD-sensitive). Among these 897 KD-sensitive SMARCAD1 peaks, 166 (18.5%) exhibited increase of H3K9me3 levels in Smarcad1 KD, 3 (0.33%) exhibited decrease, and the rest 728 did not exhibit significant change of H3K9me3 levels (Figure S6A). Therefore, although the total amount of H3K9me3 did not appear to change with Smarcad1 KD, Smarcad1 KD is associated with increased H3K9me3 in a subset of SMARCAD1 binding regions.
H3Cit is negatively associated with H3K9me3 in SMARCAD1 binding regions
The data led us to speculate a model that SMARCAD1 binding prevents the formation of extremely dense H3K9me3 regions. Considering the in vitro binding (Figure 2A–B) and in vivo (Figure 2C–D) correlation of SMARCAD1 and H3Cit, the above model is consistent with the idea that H3Cit interferes with H3K9me3 mediated heterochromatin formation (Sharma et al., 2012), and the latter is a characteristic of ES cell differentiation. However, the data do not exclude H3Cit independent mechanisms of SMARCAD1 recruitment to chromatin.
The above model would predict a negative correlation of H3Cit and H3K9me3 in SMARCAD1 binding regions. Among the 79 H3R26Cit peaks bound by SMARCAD1 in the entire genome, 73 (92.4%) exhibited at least 1.5 fold increase of H3K9me3 in Smarcad1 KD cells (Figure 5E). In particular, the genomic loci of naïve state markers Klf4 and Wnt6 exhibited 2 fold of more increases of H3K9me3 in Smarcad1 KD cells (Figure S6B–C).
Figure 5. SMARCAD1 and H3K9me3 changes in Cl-Amidine treatment.

(A) SMARCAD1 ChIP-seq intensities in Cl-Amidine treated (Cl+) and untreated ES cells. (B–F) H3K9me3 ChIP-seq intensities in Cl+ and ES cells in 10,000 random genomic regions (B), OCT4, NANOG, CTCF, H3K4me3, H3K4me3 ChIP-seq peaks (C), H3R26Cit peaks (D), SMARCAD1 peaks (E), and the overlaps of H3R26Cit and SMARCAD1 peaks (F). All peaks were defined by ChIP-seq in ES cells. (G) A genome browser view of H3R26Cit, SMARCAD1, H3K9me3 ChIP-seq in ES cells, H3K9me3 in Smarcad1 KD cells, H3K9me3 in Cl+ ES cells, and IgG ChIP-seq and MNase-seq in ES cells.
To further test the speculated model, we inhibited H3Cit by Cl-Amidine treatment (Cl+) (Christophorou et al., 2014), and carried out SMARCAD1 ChIP-seq and H3K9me3 ChIP-seq in Cl+ cells. Compared to untreated ES cells, SMARCAD1 ChIP-seq signals exhibited mild decreases near H3R26Cit peaks (H3R26Cit peaks were defined by ChIP-seq in untreated ES cells) (Figure 5A). Next, we compared H3K9me3 ChIP-seq signals between Cl+ and untreated ES cells. No discernable H3K9me3 changes were identified in 10,000 randomly selected regions (Figure 5B). In addition, H3K9me3 ChIP-seq barely exhibited any changes in OCT4, NANOG, CTCF, H3K4me1, H3K4me3 peaks (defined in untreated ES cells) either (Figure 5C). However, H3K9me3 exhibited strong increases in H3R26Cit peaks (defined in untreated ES cells) and in SMARCAD1 peaks (defined in untreated ES cells), lending additional support to the hypothesized roles of H3Cit and SMARCAD1 (Figure 5D–E). Furthermore, in the overlapped peaks of H3R26Cit and SMARCAD1 (defined in untreated ES cells), H3K9me3 exhibited even greater increases than that in H3R26Cit peaks or in SMARCAD1 peaks (Figure 5F). The specific H3K9me3 changes in SMARCAD1 peaks are also incompatible with the alternative model mentioned above.
Reproducibility of genome-wide SMARCAD1 binding in male and female ES cells
To assess the variation of ChIP-seq data in different ES lines, we generated SMARCAD1 ChIP-seq from a female ES cell line derived by Wu et al. (Wu1) from C57BL mice (Wu et al., 2015), and compared to E14 (derived from a male 129×1 mouse) data. In order to make an unbiased comparison, we fragmented the mouse genome (mm9) into 500 nt bins. After removing the bins with low mappability, we retained 4,854,116 bins. We calculated the ratio between SMARCAD1 and IgG ChIP-seq reads (SMARCAD1/IgG) in every bin and compared these ratios between the two ChIP-seq datasets. SMARCAD1 ChIP-seq in E14 and Wu1 cells exhibited a comparable degree of correlation (Pearson correlation = 0.378) to that of two H3K27me3 ChIP-experiments done in the same cell line (Pearson correlation = 0.359) (Kaneko et al., 2013, Xiao et al., 2012) and that of two ES cell ESET (H3K9 methyltransferase) ChIP-seq experiments (Pearson correlation = 0.344) (Yuan et al., 2009, Bilodeau et al., 2009) (Figure 6). A caveat of this analysis is that Wu1 ES cells were cultured in 2i condition (Wu et al., 2015). We transferred Wu1 cells from 2i medium into LIF-based culture condition (with LIF, without 2i, as described in (Li et al., 2011)) and cultured for 48 hours, and then performed SMARCAD1 ChIP-seq.
Figure 6. Variation of SMARCAD1 ChIP-seq in male (E14) and female (Wu1) ES cells.

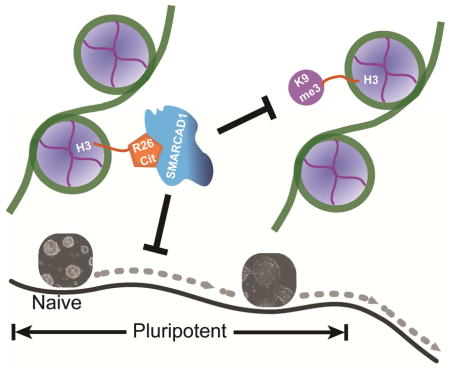
The mappable portion of the genome (mm9) was separated into a total of 4,854,116 non-overlapping 500-bp bins. A subset of 5,000 bins were drawn at random for plotting (A–C). (A) Scatter plot of the ratio of SMARCAD1 and IgG ChIP-seq reads in 500-bp bins in E14 (x axis) and in Wu1 (y axis). (B) Scatter plot between two biological replicates (GSM1199184 on y axis, GSM2111307 on x axis). Each dot is a ratio of H3K27me3 and IgG ChIP-seq reads in a 500 bp bin. (C) Scatter plot of ratio between ESET ChIP-seq and IgG ChIP-seq reads in E14 (GSM440256) (x axis) and V6.5 ES cells (GSM459273) (y axis). (D) Genome browser view of SMARCAD1 ChIP-seq in E14 and Wu1 cells, and H3K9me3 in E14 and in Smarcad1 KD cells (E14-KD). MNase-seq: sequencing of input DNA fragmented by MNase. (E) A speculated model for the role of SMARCAD1 in regulating the naïve pluripotent state. Naïve pluripotent, primed pluripotent, and differentiated cells are situated on an epigenetic landscape (dark curve). In this model, SMARCAD1 contributes to keep cells at the highest position on the epigenetic landscape, by translating H3Cit into an inhibitory signal of H3K9me3.
DISCUSSION
A candidate system for dissecting genetic and environmental regulators of naïve to primed transition
It has been 10 years since the initial derivation and characterization of cultured primed state pluripotent stem cells (Brons et al., 2007, Tesar et al., 2007). It remains extremely difficult to analyze the earliest genetic factors that underscore the naïve to primed transition, because naïve and primed cells are exposed to different signals either in vivo or in vitro (Han et al., 2010, Weinberger et al., 2016).
H3Cit was reported to correlate with the naïve pluripotent state (Christophorou et al., 2014), but inhibition of H3Cit may not be an ideal system for studying naïve to primed transition, because it resulted in cell differentiation as seen in global chromatin compaction, induction of differentiation markers, and reduction of Nanog mRNA expression to 1/3 of the control ES cells (Christophorou et al., 2014). Compared to H3Cit inhibition, Smarcad1 KD phenotypes were more delicate. On one hand, Smarcad1 KD cells remain capable of pluripotent lineage specification, on the other hand Smarcad1 KD ES cells lost representative cellular features of the naïve state even when they remain in ES culture medium. This unique property makes Smarcad1 KD ES cells a candidate for the desired in vitro system.
H3Cit was thought to prevent heterochromatin formation by weakening H3K9me3 (Sharma et al., 2012), however the mechanism was unknown. The histone modification changes in Smarcad1 repression and H3Cit inhibition conditions, especially the stepwise increases of H3K9me3 from other genomic regions to H3R26Cit peaks and SMARCAD1 peaks and then to H3R26Cit/SMARCAD1 overlapping peaks, suggest SMARCAD1 could be a missing link of H3Cit mediated suppression of H3K9me3 (Figure 6E).
Other possible mechanisms for SMARCAD1 recruitment to chromatin
The data above do not rule out H3Cit independent mechanisms for SMARCAD1 recruitment to chromatin. The lack of DNA binding specificity in vitro does not rule out the scenario where SMARCAD1 is tethered to chromatin by co-factors. LTR retrotransposons and satellite repeats appear more frequently in SMARCAD1 peaks than in the entire genome (see Supplementary Text). In addition, SMARCAD1 may interact with histone deacetylase HDAC1 (Rowbotham et al., 2011), which may explain why SMARCAD1 binds to H3K27ac in vitro but SMARCAD1 binding only moderately correlates with H3K27ac in vivo. Furthermore, the peptide array data suggest that H3 S28 phosphorylation (H3S28P) inhibits SMARCAD1 binding to H3 in vitro (Figure S1), which suggests that combinatorial histone modifications could affect SMARCAD1 interaction with chromatin. Finally, we did not find a correlation between nucleosome positions (Chen et al., 2013) and SMARCAD1 binding. Nucleosome phase lengths were slightly reduced in SMARCAD1 peaks but they did not exhibit clear differences between SMARCAD1 associated H3R26Cit peaks and the entire genome (Figure S2E).
Open chromatin versus closed chromatin hypotheses
H3Cit was thought to interfere with H3K9me3 mediated heterochromatin formation (open chromatin hypothesis) (Sharma et al., 2012), whereas SMARCAD1 was thought to associate with and promote H3K9me3, a mark of closed chromatin (closed chromatin hypothesis) (Rowbotham et al., 2011). At a first glance, the opposite effects do not seem to allow a simple model that directly puts H3K9me3 downstream to the proposed H3R26Cit/SMARCAD1 pathway. The increase of H3K9me3 in Smarcad1 KD cells on H3R26Cit peaks is better aligned with the “open chromatin” hypothesis. In addition, the “closed chromatin” hypothesis primarily relied on the observation of strong decreases of the total amounts of H3K9me2 and H3K9me3 in Smarcad1 KD condition (Rowbotham et al., 2011). We carried out the same test in ES cells. The total amounts of H3K9me2 and H3K9me3 did not decrease in Smarcad1 KD ES cells as compared to control ES cells in multiple independent experiments (Figure 4F–G). These different results are potentially attributable to cell type differences. In summary, at least in naïve ES cells and on H3R26Cit peaks, SMARCAD1 binding may function as a checkpoint, which prevents the formation of overly dense H3K9me3 regions.
H3K9me3 changes in the genomic loci of naïve pluripotency markers may not be the only plausible explanation of Smarcad1 KD induced change of pluripotent state. An non-exclusive alternative hypothesis is that SMARCAD1 binding mediated suppression of H3K9me3 keeps certain retrotransposons transcriptionally active, which is a feature of increased developmental potency and is correlated to an earlier developmental stage (Macfarlan et al., 2012). Consistent to this hypothesis, LTR retrotransposons were enriched in SMARCAD1 binding regions, and also enriched in Smarcad1 KD induced H3K9me3 peaks (Supplementary Text). Interestingly, deletion of an H3K9 methyltransferase upregulated a set of LTR retrotransposons and their nearby genes (Karimi et al., 2011).
EXPERIMENTAL PROCEDURES
Institutional permission and oversight
The analysis of mouse embryos was approved by UC San Diego Institutional Animal Care and Use Committee (IACUC) and University of Illinois Urbana-Champaign IACUC. All analyzed embryos were at blastocyst stage. The genders were not identified. Additional mouse gene expression data were retrieved from public records (GEO: GSE20187 (Tang et al., 2010) and GEO: GSE18290 (Xie et al., 2010)). All human data were retrieved from public records (GEO: GSE36552, (Yan et al., 2013)). The ES cell analyses were approved by UC San Diego Human Research Protection Program.
Change of culture condition
Under ES culture condition with LIF (Li et al., 2011), we carried out shRNA mediated Smarcad1 KD and Luciferase KD (control). After 6 days of puromycin selection in ES culture, the cells were transferred onto matrigel coated plates with EpiSC culture medium as previously described (Wu et al., 2015). Briefly, the EpiSC medium is composed of N2B27 basal medium and 20% KSR, 12ng/ml FGF2 and 2ng/ml ACTIVIN-A.
Quantifying genomewide co-localization of two histone modifications
Odds ratio was used to measure the extent of co-localization between two histone modifications, as previously described (Xiao et al., 2012). Briefly, the genome was split into 200nt non-overlapping bins. Each histone modification was judged as either present or absent on each bin based on ChIP-seq data. A contingency table was built for the distribution of genomic bins, reflecting the presence and absence of each histone modification. Odds ratio was calculated based on this contingency table. The odds ratio larger or smaller than one reflects more or less overlaps of the two histone modifications than random expectation. The odds ratio between a chromatin binding protein and a histone modification was calculated by the same approach, where one ChIP-seq dataset of histone modification was replaced by that of the chromatin binding protein.
CI-Amidine treatment
Cl-Amidine treatment to ES cells were carried out as previously described (Christophorou et al., 2014). Briefly, CI-Amidine (200uM) were added to ES culture medium and E14 ES cells were maintained in this treatment condition for 48 hours. Approximately 10 million treated cells were crosslinked for each ChIP-seq experiment. Approximately 3 million treated cells were obtained at the same time for western blot experiments.
Additional materials and methods are available in Supplementary Text.
Supplementary Material
HIGHLIGHTS.
SMARCAD1 preferentially binds histone peptides carrying the H3R26Cit modification.
Smarcad1 KD embryonic stem cells lose naïve state features but remain pluripotent.
Suppression of Smarcad1 induces H3K9me3 at SMARCAD1 binding regions.
Inhibition of H3Cit induces H3K9me3 at SMARCAD1 binding regions.
Acknowledgments
We thank Dr. Azim Surani for providing EpiSC cells, Drs. Jun Wu and Juan Carlos Izpisua Belmonte for providing Wu1 ES cells. We thank Drs. Sua Myong, Taekjip Ha, Wei Wang, Fernando H. Biase, David Granas, Ankur Jain, Xue Zou, Fabian Hertel, Jin Zhang, Lucie Hebert and Norman Huang for carrying out related work and comments to the manuscript. This work is supported by NIH grants DP1HD087990 and DP2OD007417 to S.Z., R01CA217642 and R01EY025090 to K.Z., and R01HG008135 to S.Z. and K.Z.
Footnotes
AUTHOR CONTRIBUTIONS
S.X., B.S. and S.Z. designed the experiments. S.X., B.S., D.M, L.S., Y.W., B.P.T., J.Z., T.S.T., and G.S. performed the experiments. All authors analyzed the data and wrote the manuscript.
DATA ACCESS
All sequencing data are accessible on GEO (accession number GSE45338).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- ACTIVEMOTIF. MODified(TM) Histone peptide Array Manual. 2014 Available: http://www.activemotif.com/documents/1691.pdf.
- ADRA CN, DONATO JL, BADOVINAC R, SYED F, KHERAJ R, CAI H, MORAN C, KOLKER MT, TURNER H, WEREMOWICZ S, SHIRAKAWA T, MORTON CC, SCHNIPPER LE, DREWS R. SMARCAD1, a novel human helicase family-defining member associated with genetic instability: cloning, expression, and mapping to 4q22-q23, a band rich in breakpoints and deletion mutants involved in several human diseases. Genomics. 2000;69:162–73. doi: 10.1006/geno.2000.6281. [DOI] [PubMed] [Google Scholar]
- BILODEAU S, KAGEY MH, FRAMPTON GM, RAHL PB, YOUNG RA. SetDB1 contributes to repression of genes encoding developmental regulators and maintenance of ES cell state. Genes Dev. 2009;23:2484–9. doi: 10.1101/gad.1837309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRONS IG, SMITHERS LE, TROTTER MW, RUGG-GUNN P, SUN B, CHUVA DE SOUSA LOPES SM, HOWLETT SK, CLARKSON A, AHRLUND-RICHTER L, PEDERSEN RA, VALLIER L. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature. 2007;448:191–5. doi: 10.1038/nature05950. [DOI] [PubMed] [Google Scholar]
- CHEN K, XI Y, PAN X, LI Z, KAESTNER K, TYLER J, DENT S, HE X, LI W. DANPOS: dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res. 2013;23:341–51. doi: 10.1101/gr.142067.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHRISTOPHOROU MA, CASTELO-BRANCO G, HALLEY-STOTT RP, OLIVEIRA CS, LOOS R, RADZISHEUSKAYA A, MOWEN KA, BERTONE P, SILVA JC, ZERNICKA-GOETZ M, NIELSEN ML, GURDON JB, KOUZARIDES T. Citrullination regulates pluripotency and histone H1 binding to chromatin. Nature. 2014;507:104–8. doi: 10.1038/nature12942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COCKBURN K, ROSSANT J. Making the blastocyst: lessons from the mouse. J Clin Invest. 2010;120:995–1003. doi: 10.1172/JCI41229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CUTHBERT GL, DAUJAT S, SNOWDEN AW, ERDJUMENT-BROMAGE H, HAGIWARA T, YAMADA M, SCHNEIDER R, GREGORY PD, TEMPST P, BANNISTER AJ, KOUZARIDES T. Histone deimination antagonizes arginine methylation. Cell. 2004;118:545–53. doi: 10.1016/j.cell.2004.08.020. [DOI] [PubMed] [Google Scholar]
- EZASHI T, TELUGU BP, ALEXENKO AP, SACHDEV S, SINHA S, ROBERTS RM. Derivation of induced pluripotent stem cells from pig somatic cells. Proc Natl Acad Sci U S A. 2009;106:10993–8. doi: 10.1073/pnas.0905284106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FACTOR DC, CORRADIN O, ZENTNER GE, SAIAKHOVA A, SONG L, CHENOWETH JG, MCKAY RD, CRAWFORD GE, SCACHERI PC, TESAR PJ. Epigenomic comparison reveals activation of “seed” enhancers during transition from naive to primed pluripotency. Cell Stem Cell. 2014;14:854–63. doi: 10.1016/j.stem.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAN DW, TAPIA N, JOO JY, GREBER B, ARAUZO-BRAVO MJ, BERNEMANN C, KO K, WU G, STEHLING M, DO JT, SCHOLER HR. Epiblast stem cell subpopulations represent mouse embryos of distinct pregastrulation stages. Cell. 2010;143:617–27. doi: 10.1016/j.cell.2010.10.015. [DOI] [PubMed] [Google Scholar]
- HUANG K, MARUYAMA T, FAN G. The naive state of human pluripotent stem cells: a synthesis of stem cell and preimplantation embryo transcriptome analyses. Cell Stem Cell. 2014;15:410–5. doi: 10.1016/j.stem.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KANEKO S, SON J, SHEN SS, REINBERG D, BONASIO R. PRC2 binds active promoters and contacts nascent RNAs in embryonic stem cells. Nat Struct Mol Biol. 2013;20:1258–64. doi: 10.1038/nsmb.2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KARIMI MM, GOYAL P, MAKSAKOVA IA, BILENKY M, LEUNG D, TANG JX, SHINKAI Y, MAGER DL, JONES S, HIRST M, LORINCZ MC. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell. 8:676–87. doi: 10.1016/j.stem.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KARIMI MM, GOYAL P, MAKSAKOVA IA, BILENKY M, LEUNG D, TANG JX, SHINKAI Y, MAGER DL, JONES S, HIRST M, LORINCZ MC. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell. 2011;8:676–87. doi: 10.1016/j.stem.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIDDER BL, PALMER S, KNOTT JG. SWI/SNF-Brg1 regulates self-renewal and occupies core pluripotency-related genes in embryonic stem cells. Stem Cells. 2009;27:317–28. doi: 10.1634/stemcells.2008-0710. [DOI] [PubMed] [Google Scholar]
- KLOSE RJ, ZHANG Y. Regulation of histone methylation by demethylimination and demethylation. Nat Rev Mol Cell Biol. 2007;8:307–18. doi: 10.1038/nrm2143. [DOI] [PubMed] [Google Scholar]
- LI Y, YOKOHAMA-TAMAKI T, TANAKA TS. Short-term serum-free culture reveals that inhibition of Gsk3beta induces the tumor-like growth of mouse embryonic stem cells. PLoS One. 2011;6:e21355. doi: 10.1371/journal.pone.0021355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MACFARLAN TS, GIFFORD WD, DRISCOLL S, LETTIERI K, ROWE HM, BONANOMI D, FIRTH A, SINGER O, TRONO D, PFAFF SL. Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature. 2012;487:57–63. doi: 10.1038/nature11244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MERRILL BJ. Wnt pathway regulation of embryonic stem cell self-renewal. Cold Spring Harb Perspect Biol. 2012;4:a007971. doi: 10.1101/cshperspect.a007971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NEELI I, KHAN SN, RADIC M. Histone deimination as a response to inflammatory stimuli in neutrophils. J Immunol. 2008;180:1895–902. doi: 10.4049/jimmunol.180.3.1895. [DOI] [PubMed] [Google Scholar]
- OKAZAKI N, IKEDA S, OHARA R, SHIMADA K, YANAGAWA T, NAGASE T, OHARA O, KOGA H. The novel protein complex with SMARCAD1/KIAA1122 binds to the vicinity of TSS. J Mol Biol. 2008;382:257–65. doi: 10.1016/j.jmb.2008.07.031. [DOI] [PubMed] [Google Scholar]
- ROWBOTHAM SP, BARKI L, NEVES-COSTA A, SANTOS F, DEAN W, HAWKES N, CHOUDHARY P, WILL WR, WEBSTER J, OXLEY D, GREEN CM, VARGA-WEISZ P, MERMOUD JE. Maintenance of silent chromatin through replication requires SWI/SNF-like chromatin remodeler SMARCAD1. Mol Cell. 2011;42:285–96. doi: 10.1016/j.molcel.2011.02.036. [DOI] [PubMed] [Google Scholar]
- SCHOOR M, SCHUSTER-GOSSLER K, GOSSLER A. The Etl-1 gene encodes a nuclear protein differentially expressed during early mouse development. Dev Dyn. 1993;197:227–37. doi: 10.1002/aja.1001970307. [DOI] [PubMed] [Google Scholar]
- SCHULTZ DC, AYYANATHAN K, NEGOREV D, MAUL GG, RAUSCHER FJ., 3RD SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002;16:919–32. doi: 10.1101/gad.973302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHARMA P, AZEBI S, ENGLAND P, CHRISTENSEN T, MOLLER-LARSEN A, PETERSEN T, BATSCHE E, MUCHARDT C. Citrullination of histone H3 interferes with HP1-mediated transcriptional repression. PLoS Genet. 2012;8:e1002934. doi: 10.1371/journal.pgen.1002934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOININEN R, SCHOOR M, HENSELING U, TEPE C, KISTERS-WOIKE B, ROSSANT J, GOSSLER A. The mouse Enhancer trap locus 1 (Etl-1): a novel mammalian gene related to Drosophila and yeast transcriptional regulator genes. Mech Dev. 1992;39:111–23. doi: 10.1016/0925-4773(92)90030-n. [DOI] [PubMed] [Google Scholar]
- TAKASHIMA Y, GUO G, LOOS R, NICHOLS J, FICZ G, KRUEGER F, OXLEY D, SANTOS F, CLARKE J, MANSFIELD W, REIK W, BERTONE P, SMITH A. Resetting transcription factor control circuitry toward ground-state pluripotency in human. Cell. 2014;158:1254–69. doi: 10.1016/j.cell.2014.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TANG F, BARBACIORU C, BAO S, LEE C, NORDMAN E, WANG X, LAO K, SURANI MA. Tracing the derivation of embryonic stem cells from the inner cell mass by single-cell RNA-Seq analysis. Cell Stem Cell. 2010;6:468–78. doi: 10.1016/j.stem.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TELUGU BP, EZASHI T, SINHA S, ALEXENKO AP, SPATE L, PRATHER RS, ROBERTS RM. Leukemia inhibitory factor (LIF)-dependent, pluripotent stem cells established from inner cell mass of porcine embryos. J Biol Chem. 2011;286:28948–53. doi: 10.1074/jbc.M111.229468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TESAR PJ, CHENOWETH JG, BROOK FA, DAVIES TJ, EVANS EP, MACK DL, GARDNER RL, MCKAY RD. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature. 2007;448:196–9. doi: 10.1038/nature05972. [DOI] [PubMed] [Google Scholar]
- WEINBERGER L, AYYASH M, NOVERSHTERN N, HANNA JH. Dynamic stem cell states: naive to primed pluripotency in rodents and humans. Nat Rev Mol Cell Biol. 2016;17:155–69. doi: 10.1038/nrm.2015.28. [DOI] [PubMed] [Google Scholar]
- WU J, OKAMURA D, LI M, SUZUKI K, LUO C, MA L, HE Y, LI Z, BENNER C, TAMURA I, KRAUSE MN, NERY JR, DU T, ZHANG Z, HISHIDA T, TAKAHASHI Y, AIZAWA E, KIM NY, LAJARA J, GUILLEN P, CAMPISTOL JM, ESTEBAN CR, ROSS PJ, SAGHATELIAN A, REN B, ECKER JR, BELMONTE JCI. An alternative pluripotent state confers interspecies chimaeric competency. Nature. 2015;521:316–321. doi: 10.1038/nature14413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XIAO S, XIE D, CAO X, YU P, XING X, CHEN CC, MUSSELMAN M, XIE M, WEST FD, LEWIN HA, WANG T, ZHONG S. Comparative epigenomic annotation of regulatory DNA. Cell. 2012;149:1381–92. doi: 10.1016/j.cell.2012.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XIE D, CHEN CC, PTASZEK LM, XIAO S, CAO X, FANG F, NG HH, LEWIN HA, COWAN C, ZHONG S. Rewirable gene regulatory networks in the preimplantation embryonic development of three mammalian species. Genome Res. 2010;20:804–15. doi: 10.1101/gr.100594.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAN L, YANG M, GUO H, YANG L, WU J, LI R, LIU P, LIAN Y, ZHENG X, YAN J, HUANG J, LI M, WU X, WEN L, LAO K, LI R, QIAO J, TANG F. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat Struct Mol Biol. 2013;20:1131–9. doi: 10.1038/nsmb.2660. [DOI] [PubMed] [Google Scholar]
- YUAN P, HAN J, GUO G, ORLOV YL, HUSS M, LOH YH, YAW LP, ROBSON P, LIM B, NG HH. Eset partners with Oct4 to restrict extraembryonic trophoblast lineage potential in embryonic stem cells. Genes Dev. 2009;23:2507–20. doi: 10.1101/gad.1831909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG X, BOLT M, GUERTIN MJ, CHEN W, ZHANG S, CHERRINGTON BD, SLADE DJ, DREYTON CJ, SUBRAMANIAN V, BICKER KL, THOMPSON PR, MANCINI MA, LIS JT, COONROD SA. Peptidylarginine deiminase 2-catalyzed histone H3 arginine 26 citrullination facilitates estrogen receptor alpha target gene activation. Proc Natl Acad Sci U S A. 2012;109:13331–6. doi: 10.1073/pnas.1203280109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG Y, LIU T, MEYER CA, EECKHOUTE J, JOHNSON DS, BERNSTEIN BE, NUSBAUM C, MYERS RM, BROWN M, LI W, LIU XS. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHAO J, SUN BK, ERWIN JA, SONG JJ, LEE JT. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science. 2008;322:750–6. doi: 10.1126/science.1163045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHAO Y, GRANAS D, STORMO GD. Inferring binding energies from selected binding sites. PLoS Comput Biol. 2009;5:e1000590. doi: 10.1371/journal.pcbi.1000590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHOU W, CHOI M, MARGINEANTU D, MARGARETHA L, HESSON J, CAVANAUGH C, BLAU CA, HORWITZ MS, HOCKENBERY D, WARE C, RUOHOLA-BAKER H. HIF1alpha induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/hESC transition. EMBO J. 2012;31:2103–16. doi: 10.1038/emboj.2012.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.