

Abstract
Immunotherapy has changed the standard of care for multiple deadly cancers including lung, head and neck, gastric, and some colorectal cancers. However, single agent immunotherapy has had little effect in pancreatic adenocarcinoma (PDAC). Increasing evidence suggests that the PDAC microenvironment is comprised of an intricate network of signals between immune cells, PDAC cells, and stroma, resulting in an immunosuppressive environment resistant to single agent immunotherapies. In this review, we discuss differences between immunotherapy sensitive cancers and PDAC, the complex interactions between PDAC stroma and suppressive tumor infiltrating cells that facilitate PDAC development and progression, the immunologic targets within these complex networks that are drugable, and data supporting combination drug approaches that modulate multiple PDAC signals, which should lead to improved clinical outcomes.
Keywords: pancreatic cancer, immunotherapy, tumor microenvironment
Introduction
Current estimates predict PDAC to overtake breast cancer and become the third most common cause of cancer-related death in the United States (1,2). Only 20–30% of patients with PDAC have resectable disease at diagnosis, and the majority of patients who undergo surgical resection subsequently relapse (3–7). Most patients present with metastatic disease at diagnosis and have only a 2% five-year survival (2). To date, the rate of successful clinical trials in pancreatic cancer remains low (8). Of the many therapies investigated in large clinical trials over the past two decades, only two systemic therapies have demonstrated a statistically significant and clinically meaningful improvement in overall survival (OS) as compared to gemcitabine alone (9,10). As a result, the five year survival rate for PDAC has improved only marginally since the 1970s, from 3% to 7% (2). This highlights the continued need for new and effective therapies in PDAC.
Immune checkpoint immunotherapies have produced unprecedented clinical benefits in a variety of different cancers, including lung cancer, which was previously thought to be non-immune responsive (11). However clinical trials using single agent checkpoint immunotherapy in PDAC have been unsuccessful thus far. This may be explained by increasing evidence which suggests that PDAC creates a potently immunosuppressive microenvironment via activation of multiple regulatory mechanisms (12,13), whereby interactions between the tumor, stroma, and immune cells in the pancreatic tumor microenvironment (TME) result in cancer progression (Figure 1). In this review, we discuss potential approaches to increasing immunogenicity, or immune responsiveness, to PDAC. Specifically, we will (1) examine the challenges in developing successful immunotherapies for PDAC, (2) describe the complex immune components of the TME and discuss how the immune system, pancreatic tumor cells, microbiome, and stromal signals suppress immune-mediated attack, and (3) discuss novel multi-agent therapeutic strategies to target signals within this integrated immunosuppressive network that are under development in clinical trials. Current standard of care therapy and clinical trials in progress are also reviewed by Manji et al. in this CCR Focus issue (14).
Figure 1. Mechanisms within the PDAC TME drives resistance to therapies.
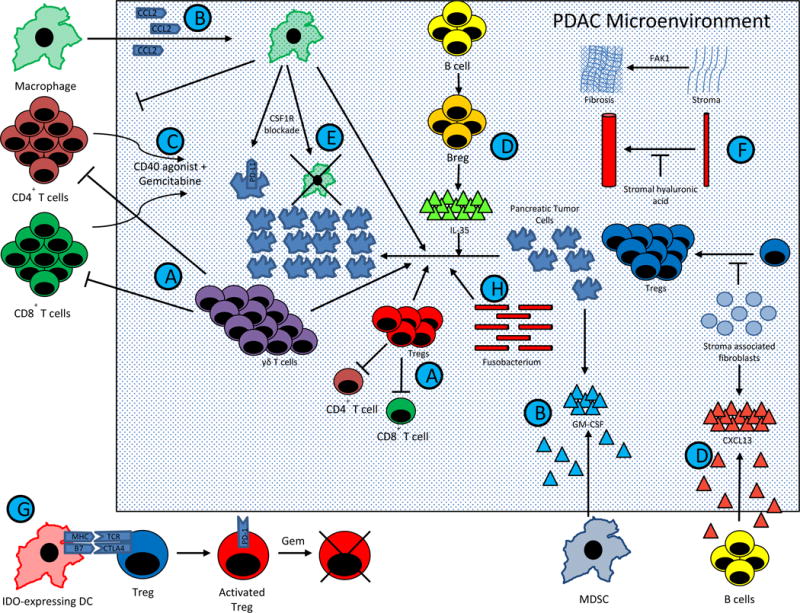
PDAC comprises of complex interactions between T cells, B cells, APCs, pancreatic tumor cells, and stromal elements. These interactions result in a profoundly immunosuppressive tumor microenvironment, and consequently single agent immunotherapy has been largely ineffective. However, emerging preclinical data has suggested that combination therapy may dramatically affect overall survival. Current trial design is being driven largely by this data. The figure summarizes major pathways in PDAC tumorigenesis that are being manipulated in clinical trials for patients with metastatic PDAC. Except for (G.), which represents in part IDO activated Tregs in TDLNs from a melanoma model (40), this figure represents data known exclusively from PDAC models.
(A.) Tregs and γδ T cells block Teff division and drive PDAC growth, while γδ T cells block T cell infiltration (47).
(B.) MDSCs and macrophages are mobilized into the TME by PDAC derived GM-CSF and CCL2, respectively. (145,183,184).
(C.) Macrophages block CD4+ T cell entry into the PDAC microenvironment. CD40 is expressed on these CD4+ T cells, and activation of the CD40 pathway concurrently with gemcitabine can drive T cell infiltration (140).
(D.) Stromal associated fibroblasts produce CXCL13, which recruits regulatory B cells into the TME. These regulatory B cells produce IL-35, which drives PDAC progression (136,185). These Bregs may be inhibited by BTK inhibitors, such as ibrutinib (137).
(E.) Tumor infiltrating macrophages stimulate PDAC progression. Blockade of the CSF1 receptor expressed by macrophages can lead to macrophage depletion, CTLA-4 upregulation on CD8+ T cells, and PD-L1 upregulation on pancreatic tumor cells (146,147).
(F.) Stromal elements create a physical barrier to immune infiltration and therapeutic agents. Stromal fibroblasts block Treg accumulation and PDAC progression (62), but targeting other stromal elements have achieved encouraging results. Stromal hyaluronic acid deposition results in decreased vascular patency (72,73), and FAK1 drives stromal fibrosis (68). Inhibition of either target has led to decreased PDAC progression when combined with chemotherapy in preclinical models.
(G.) IDO induction in DCs by tumors activate Tregs via MHC and CTLA4 pathways (40,131). In phase II studies, gemcitabine based therapy synergizes with IDO inhibition to improve response rates in PDAC (133), possibly via transient depletion of Tregs (39). This provides an immune system reset, allowing for chemotherapy-mediated elimination of previously activated Tregs, followed by indoximod mediated inhibition of subsequent Treg activation.
(H.) Recent evidence suggests the Fusobacterium found within the PDAC microenvironment drives PDAC progression, but the mechanism of this is unknown (91).
Clinical Challenges in Developing Immunotherapies for PDAC
There is mounting evidence that immune mediated inflammation is an integral component of the environment that supports PDAC development and progression (15). Genomic analyses show that PDAC frequently upregulates multiple pathways involved in acquired immune suppression, and upregulation of these pathways is associated with poor survival (16). This may explain why early human clinical studies involving immunotherapy monotherapy in PDAC have been discouraging. While treatment with single agent immune checkpoint inhibitors targeting cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1) cause meaningful objective responses in many tumor types (11,17–20), only 1 of 27 patients with PDAC responded to the CTLA-4 inhibitor ipilimumab (21), and 0 of 14 patients with PDAC had an objective response to anti-PD-L1 therapy (22). Recently completed and planned immunotherapy clinical trials for patients with PDAC have been reviewed in detail elsewhere (23–27). Although single-agent immunotherapies have failed to show benefit in PDAC, increasing data support the testing of combinatorial approaches that target multiple suppressive mechanisms. In addition to examining genetic mutations in PDAC tumor samples, which is reviewed by Dreyer and colleagues in this CCR Focus Issue (28), performing RNA sequencing to determine which immune escape mechanisms are upregulated (e.g. PD-1, IDO) may allow us to further personalize therapy for patients by combining immunotherapy agents with chemotherapy to reset the immune system (29). This may be critical specifically in patients with PDAC as the failure of single agent checkpoint therapy indicates that the PDAC tumor microenvironment is more complicated and suppressive than in other more immunogenic cancers. This would also have the advantage of being able to determine a tumor’s immunogenicity upfront before initiating treatment. As we better understand the role of the multiple immunologic contributors to PDAC growth, it should be possible to design multi-agent immunotherapies that target multiple pathways, leading to increased antitumor immunity.
The multiple immunosuppressive components of the PDAC TME collectively suppress effector T cells (cells that recognize and kill tumor cells), preventing immune mediated destruction (Figure 1). Accumulation of effector CD4+ and CD8+ T cells in human PDAC are associated with improved overall survival (30–32). As pancreatic lesions progress, tumor infiltrating CD8+ effector T cells decrease while suppressive regulatory T cells (Tregs) comprise a higher percentage of the CD4+ T cell compartment (33), leading to a low number of tumor infiltrating effector lymphocytes (TILs) and a high number of immunosuppressive cells (13). Thus PDAC is considered to be a poorly immune responsive cancer. By contrast, highly immune responsive solid tumors are characterized by a high number of TILs at baseline and a high response rate to immune checkpoint inhibitors (34). Although PDAC is poorly immunogenic, that is likely due to having a more complex and suppressive tumor microenvironment, not because the immune system does not recognize the tumor. Discovery of the complex immune pathways involved in PDAC progression and immune escape (summarized in Figure 1) has led to additional novel PDAC immunotherapy targets (Table 1). Increasing data suggest that poorly immune responsive cancers like PDAC require multiagent therapy to elicit an immune response. One multipronged approach involves vaccines, which stimulate accumulation of lymphoid aggregates in PDAC (35) (Figure 2). One likely reason why vaccines have not stimulated effective antitumor responses, despite inducing lymphoid infiltration, is that vaccines also upregulate T cell inhibitory pathways such as the PD-1/PD-L1 pathway (36). Although vaccine therapy has thus far been unsuccessful, we believe that these lymphoid infiltrates represent increased immunogenicity, and speculatively, patients with vaccine-induced infiltration of lymphoid aggregates may benefit from a combination approach involving vaccine plus costimulatory blockade. Also, upregulation of immune checkpoint pathways after vaccine therapy may be a biomarker of increased immunogenicity and suggest these patients may also respond to checkpoint blockade. It is also possible that vaccines upregulate multiple immune escape mechanisms, and elucidation of these would be necessary to ensure vaccine efficacy. As chemotherapy transiently depletes suppressive Tregs in PDAC patients (37–39), chemotherapy should be considered in addition to administration of an immunomodulatory agent to attempt to overcome the potent immunosuppressive TME.
Table 1.
A list of notable immunotherapies in clinical development for PDAC.
| Therapeutic target and agents under investigation for PDAC | Preclinical rationale | Clinical evidence and ongoing trials |
|---|---|---|
|
PD-1/PD-L1 nivolumab pembrolizumab durvalumab |
PD-1/PD-L1 inhibition has activity in a wide number of tumors. PD-L1 expression is upregulated in a subset of PDAC, and is associated with shortened survival (161,162). | Responses were observed in a subset of patients with MMR-deficient pancreatic cancer (≤ 5% of PDAC) (56), and additional trials in MMR-d disease are ongoing NCT01876511, NCT02465060). None of 14 pancreatic patients responded in a study of single-agent nivolumab (22). Multiple combination immunotherapy trials are ongoing (NCT02558894, NCT02268825, NCT02472977, NCT02243371, NCT02777710). |
|
CTLA-4 ipilimumab tremelimumab |
Anti-CTLA-4 therapy may reduce intratumoral Tregs and shift the threshold needed for T cell activation. A trial of ipilimumab failed to show convincing clinical activity, but a possible delayed response was observed in one patient (21). | Multiple combination trials are ongoing, including combinations with PD-1 inhibition and/or therapeutic vaccines (NCT02558894, NCT01896869). |
|
IDO1 indoximod |
IDO1 mediates tumor immunosuppression in preclinical models (non-PDAC), and PDAC frequently overexpresses IDO as a mechanism of immune escape (132,163,164). | Evidence of clinical activity was observed in combination with chemotherapy (133). A clinical trial is ongoing in combination with gemcitabine-based chemotherapy (NCT02077881). |
|
BTK Ibrutinib |
BTK is involved with B cell receptor signaling and is also expressed by macrophages. In preclinical models ibrutinib synergizes with gemcitabine to increase antitumor immunity (137). | Clinical trials are ongoing in combination with gemcitabine-based chemotherapy in PDAC (NCT02562898, NCT02436668) |
|
CD-40 RO7009789 (CP-870,893) JNJ-64457107 |
CD40 is expressed on B cells, DCs, and other cell types. CD40 agonists inhibit PDAC stroma, increase CCL2 levels and interferon gamma (IFN-g) in the TME, and synergize with chemotherapy (145,165). | Evidence of clinical activity was observed in an early stage clinical trial in PDAC (141). Additional trials of monotherapy or combination with gemcitabine-based chemotherapy are ongoing (NCT02588443, NCT02829099). |
|
CCR2 CCX872 PF-04136309 |
CCR2 recruits suppressive macrophages to the immunosuppressive TME in PDAC, and CCR2 inhibition depletes tumor infiltrating macrophages and improves survival in a preclinical model (145). | CCR2 inhibition has shown safety and possible evidence of clinical activity in combination with chemotherapy. Clinical trials in combination with chemotherapy in PDAC are ongoing (NCT02345408, NCT02732938) |
|
CSF-1R Cabiralizumab (FPA008) Pexidartinib (PLX3397) BLZ945 AMG 820 |
CSF1R inhibition reprograms tumor-associated macrophages and upregulates immune checkpoints. Synergistic activity has been observed with immune checkpoint inhibitors in preclinical models of PDAC (146,147). | Multiple agents are in clinical trials in metastatic PDAC in combination with PD-1 inhibitors (NCT02526017, NCT02777710, NCT02829723, NCT02713529) |
| CXCR4 LY2510924 |
CXCR4 blockade abrogated metastasis in prelclinical models (151), and synergized with PD-L1 therapy to increase antitumor immunity (158) | CXCR4 inhibitor is in clinical trial in combination with PD-L1 blockade to treat advanced solid tumors, including PDAC (NCT27037072). |
Figure 2. Therapeutic vaccine immunotherapy for PDAC requires multiple steps to overcome immunosuppression.
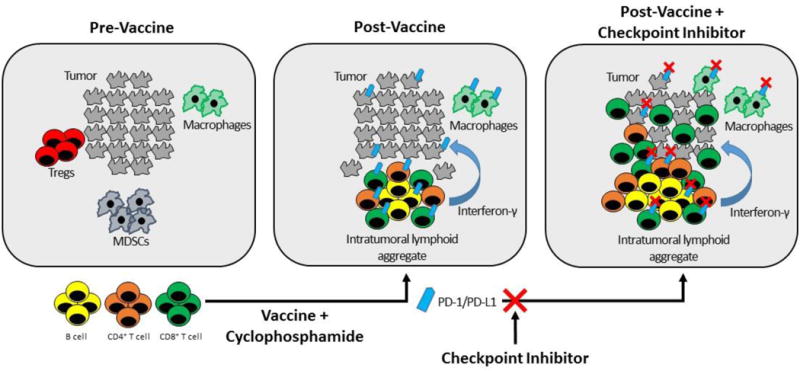
PDAC and other poorly immune responsive cancers are characterized by low numbers of tumor infiltrating lymphocytes (TILs), low levels of PD-L1 expression, and high numbers of immunosuppressive cells such as Tregs and MDSCs at baseline (left panel) (13). Using a vaccine approach will require at least two immunotherapeutics to achieve an immune response. In Step 1 (center panel), a therapeutic vaccine is used to induce accumulation of lymphoid aggregates (35). These lymphocytes secrete interferon gamma and other soluble factors that induce high levels of PD-L1/PD-1 expression on epithelial tumor cells and on immune cells (186). Vaccines can also be combined with other therapies such as cyclophosphamide, to deplete immunosuppressive cells in the TME (29). In Step 2 (right panel), the addition of a PD-pathway inhibitor to a vaccine-primed tumor inhibits PD-L1/PD-1 signaling to increase lymphocyte proliferation and activation and promote tumor eradication (36). The hypothesis that vaccine therapy can synergize with immune checkpoint inhibition is currently under clinical investigation in multiple trials in PDAC.
The TME’s Role in PDAC Development and Progression
Immune Checkpoints andImmune Checkpoint Inhibitors
There are many immune signaling pathways that regulate antitumor immunity, which involve costimulatory and inhibitory receptors (immune checkpoints) on T cells. Most studies of immunomodulatory agents in PDAC have examined the role of the inhibitory costimulatory receptors CTLA-4 and PD-1. Both receptors are critical in activation and suppressive activity of Tregs (40), and exist primarily to prevent autoimmunity and excessive immune responses to infection (41). However, tumors also induce Treg activation and suppression via these pathways, leading to dampened antitumor immune responses (40). This concept is critical, because it suggests that the immune system is not ignorant of PDAC; rather, the immune system detects PDAC, but is instructed by the tumor not to attack it (42). Thus inhibition of immunosuppression, rather than immune activation alone, is critical to achieve durable clinical responses. Consistent with this, CTLA-4 and PD-pathway expression are upregulated in PDAC (43–45), and both are associated with worse survival (44,46). Furthermore, PD-1 is expressed on multiple PDAC infiltrating T cell subsets, including Tregs, and CD4+ and CD8+ effector T cells (37). Additionally, PDAC-infiltrating γδ T cells were recently identified, which represent a subset of suppressive T cells that express PD-L1 and suppress effector T cell activation (47). Collectively these studies indicate that immune checkpoint inhibition may be a target for PDAC related immunotherapy.
A number of principles have emerged that characterize immune checkpoint pathways. First, these pathways develop in response to the genetic changes that occur within developing tumors and are shaped by the evolving inflammatory response to these genetic changes. Second, there are many inhibitory and activating signaling pathways (48,49), but much still needs to be learned about their role in different cancer types. While melanoma, lung carcinoma, and renal cell carcinoma respond to blockade of one checkpoint pathway (i.e. PD-1/PD-L1 or CTLA-4) (11,17–20), most cancers will likely require combination therapy to fully activate T cell responses. Figure 1 depicts a non-exhaustive description ofthe broad range of suppressive mechanisms in PDAC, which account for single agent immunotherapy having limited clinical activity. Increasing preclinical evidence (see below) suggests that combining checkpoint inhibition with other targeted therapy may improve clinical efficacy. Third, additional studies are needed to understand primary (patients who do not respond) and secondary (patients who initially respond but then recur) resistance to these agents.
Although immune checkpoint inhibitors have thus far failed as single agents to demonstrate convincing clinical activity in PDAC, there may be subgroups of PDAC that are more likely to respond to these agents as monotherapy. Predictive biomarkers have now been used in multiple cancer types to identify patients who may be more likely to respond to immune checkpoint inhibitors. For example, expression of PD-L1 is used to identify patients who should receive frontline PD-1 inhibitor immunotherapy instead of chemotherapy in non-small cell lung cancer (NSCLC) (50). In gastrointestinal malignancies including PDAC, one emerging biomarker of response to immune checkpoint inhibitors is mismatch repair deficiency (MMR-d), which results in a failure to repair errors in base pair mismatches in tumor DNA (e.g. C-T instead of C-G), leading to microsatellite instability (MSI) (51). In unselected populations of colorectal cancer, little to no clinical activity was reported in the initial clinical trials of immune checkpoint inhibitors. However, the PD-1 inhibitor pembrolizumab demonstrated significant clinical activity in the small subset of colorectal cancers (≤5% of advanced disease, (52)) with MMR-d (53). This activity is likely due to the high baseline immunogenicity of the MMR-d cancer subtype, as evidenced by the increased lymphoid infiltration in MMR-d colorectal carcinomas at baseline, as well as the high expression of multiple immune checkpoints, including PD-L1 (54,55).
Mismatch repair status is not routinely checked in PDAC, and we are aware of only four reported cases of MMR-d pancreatic cancer treated with a PD-1 inhibitor. Of these four cases, one patient had a partial response to pembrolizumab, and the other three achieved stable disease (56). Additional basket trials of single-agent PD-1 inhibition in MMR-d cancers (including PDAC) are ongoing. Although MMR-d PDAC is a small subset of all PDAC, (13–17.4% in prior studies (57–59)), these preliminary data suggest that single agent immune checkpoint inhibitors may have meaningful clinical activity in such cases. These studies also suggest that it is important to perform genetic sequencing studies on all patient tumors to better define each cancer’s biology and to identify potential therapeutic options that may otherwise be missed.
Stroma
The dense stroma surrounding pancreatic cancers creates a hypovascular environment that can block the penetration of chemotherapeutics and facilitate immune escape. T cells were first demonstrated in the late 1990s to form aggregates in the fibrotic tissue of pancreatic cancer samples (60), leading to the current hypothesis that interactions between stroma, lymphocytes, and antigen presenting cells (APC) create a complex TME that makes overcoming immunosuppression difficult. Initial studies demonstrated that tumor incidence and metastasis increased when an increased proportion of pancreatic stellate cells were co-injected with PDAC cells, identifying the stroma as a potential target for therapeutic intervention (61). However, in preclinical models of PDAC, simple depletion of fibroblasts lead to increased regulatory T cell (Treg) accumulation and decreased survival, suggesting that the relationship between PDAC and stroma may be more complex than previously appreciated (62). This may explain why depletion of fibroblasts via inhibition of Hedgehog signaling, while leading to disease stabilization in some preclinical studies, ultimately failed in other preclinical models and clinical trials (63–65). This conflicting data are described in more detail elsewhere (66), and may reflect heterogeneity between fibroblasts (67) or different systems used.
However, targeting other factors that drive stromal fibrosis have elicited encouraging preclinical data in PDAC that may overcome the limitations of targeting fibroblasts alone and also facilitate effector T cell access and activation the TME. As one example, inhibition of focal adhesion kinase-1 (FAK1), a tyrosine kinase expressed on PDAC cells and stroma that drives stromal fibrosis, with the selective inhibitor VS-4718, can improve responses to chemotherapy and immunotherapy in a preclinical model of PDAC (68). Unfortunately, three previous clinical trials studying single agent FAK inhibition in patients with solid tumors, including PDAC, demonstrated no objective responses (69–71). However, several trials of combination FAK inhibition with gemcitabine and/or PD-1 blockade are now ongoing (NCT02758587, NCT02651727, NCT02546531). Additionally, targeting hyaluronic acid (HA) restored vascular patency in a preclinical model, and improved overall survival in patients with high HA content (72–74). Ongoing trials are examining PEGPH20 plus standard of care chemotherapy for PDAC (NCT02715804, NCT02487277, NCT01959139).
The stroma also produces factors, such as the proinflammatory cytokine IL-6, which are associated with poorer survival when expressed in peripheral blood of patients with PDAC (46,75). Unfortunately no objective responses were noted in any patients who received single agent IL-6 blockade in a phase I trial of patients with solid tumors, including nine patients with PDAC (76). More recently, Lesinski and colleagues demonstrated that blockade of IL-6, which upregulates PD-L1 in viral models (77), synergized with PD-L1 inhibition to increase lymphocyte infiltration and improve CD8+ T cell dependent antitumor immunity (78). This suggests that in addition to the stroma functioning as a physical barrier for immune infiltration, the stroma actively suppresses T cell infiltration via production of soluble factors, and blocking IL-6 may increase PDAC immunogenicity via upregulation of PD-L1. Speculatively, instead of complete stromal depletion, targeting the soluble factors produced may lead to improved outcomes. As IL-6 is known to promote chronic inflammation (79), targeting other mechanisms driving chronic inflammation, such as IL-17, may also be relevant (80,81). Overall the stroma is complex and requires further study to determine which components support and which suppress antitumor immune responses.
The Microbiome
Systemic factors also appear to impact the development and progression of PDAC, and several reviews have examined the relationship between the oral microbiome and PDAC (82,83). Multiple studies have found a possible relationship between tooth loss, self-reported periodontal disease, or clinically documented periodontitis (respectively) and PDAC or PDAC-associated mortality (84–86). This association between periodontitis and PDAC appears to remain even after controlling for multiple risk factors (87,88). Certain bacteria, such as Porphyromonas gingivalis and Actinobacillus actinomycetemcomitans, are frequently linked with the development of periodontal disease (89), and RNA sequencing from pre-diagnostic oral washings have demonstrated that the presence of these two bacteria are also significantly associated with developing PDAC (90). In contrast, oral bacteria of the genus Leptotrichia has been associated with decreased PDAC risk. Notably, P. gingivalis and Leptotrichia levels collected more than 2 years prior to PDAC diagnosis retained their respective positive and negative associations with PDAC, suggesting that the altered oral microbiome may have been present prior to PDAC carcinogenesis (90). Another bacteria, Fusobacterium, was associated with decreased risk of acquiring PDAC when it was found in the oral cavity (90), but was associated with decreased survival when it was found in human PDAC tissues (91). Fusobacterium may therefore have differential effects pre- and post-diagnosis, or its carcinogenic effects may be dependent on its location.
Several explanations have been proposed to explain why certain oral microorganisms correlate with PDAC development. The altered oral microbiota may simply be a consequence of systemic inflammation, as patients with diabetes, a risk factor for PDAC (92), also has a significantly different oral microbiome than normal controls (93). Alternatively, it is biologically plausible that certain microbes may directly facilitate PDAC carcinogenesis. Consistent with this notion, the colonic bacterium Enterotoxigenic Bacteroides fragilis has been implicated in causing colon cancer via IL-17 production (94), and Porphyromonas has been implicated in carcinogenesis of oral squamous cell carcinoma in preclinical models (95). Although IL-17 has been implicated in facilitating PDAC and may emerge as a potential therapeutic target (80,81), further studies are needed to determine whether alterations in the oral microbiome play a role in the development of PDAC, which immune signals are involved,or if these findings are simply correlative. One potential study would be to examine patients with IL-17R overexpressing PDAC, which has been associated with poorer prognosis (80) to see if the microbiome is altered in these patients versus non-IL-17R overexpressing patients, and then colonization of mice predisposed to obtain PDAC with the microbe in question to see if this accelerates PDAC. If the microbiome is conclusively shown to affect PDAC tumorigenesis or progression, prospective clinical studies of novel therapeutic agents that modify the microbiome as a treatment or prevention of PDAC will be warranted. Additionally, understanding the immune mechanisms through which the microbiome affect PDAC development and progression could inform the development of novel immunotherapies.
Vaccine Immunotherapy Strategies for PDAC Treatment
AsPDAC is a a poorly immunogenic cancer for which single agent vaccines have been ineffective, using a vaccine based approach will require at least one additional immunotherapeutic agent to optimally achieve an antitumor immune response (Figure 2). Optimal vaccine design will require knowledge of immune relevant antigens that are recognized by effector T cells that have the potential to be activated, and identification of vaccine approaches that effectively activate them. The second step is determining which immune escape mechanisms (such as checkpoint pathways) are induced by the vaccine itself. Thus, a baseline biopsy before vaccine therapy will not be the best indicator to determine which immune checkpoints require modulation.
Tumor antigens and antigen delivery systems for generating anti-PDAC T cells
A few PDAC tumor antigens capable of inducing an anti-tumor immune response have been identified. An ideal tumor antigen target should be highly expressed in PDAC cells and minimally expressed in normal tissue. Most PDAC antigens fall into one of two categories: 1) tumor-associated antigens (TAAs), which are found mostly on tumor but have limited expression on normal cells, and 2) tumor-specific antigens (TSAs), also called neoantigens, which are expressed exclusively on malignant cells and not expressed on normal cells (96). TAAs have received the most attention as targets for PDAC immunotherapy because of the potential to treat many patients with the same therapy. Epidermal growth factor receptor (HER/EGFR/ERBB) family proteins (97,98), and mesothelin (99–101) are examples of TAAs that are under clinical investigation as therapeutic targets in PDAC. However, because these antigens are also expressed on normal cells, off-target toxicity remains an important clinical concern (102,103). Due to their tumor-specific expression, TSAs are particularly appealing targets for PDAC immunotherapy. However, most TSAs arise from individual tumor mutations and are not shared between most patients. Therefore, while most (if not all) PDACs have TSAs (104), therapies targeting TSAs may need to be personalized.
A notable exception in PDAC is the driver oncogene KRAS, which is mutated at codon 12 in approximately 90% of PDACs and has been explored as a target for immunotherapy (105–108). KRAS is often described to be an ‘undruggable’ protein because despite several decades of intensive efforts, no pharmacologic inhibitors of KRAS have reached the clinic. However, mutated KRAS, like other tumor antigens, is presented on the cell surface of cells and thus is accessible to the immune system. Recently, the Rosenberg group provided proof of principle for KRAS immune targeting by successfully inducing a durable partial response in a patient with KRAS-mutant colorectal cancer by infusing an enriched population of CD8+ T cells that reacted to the specific KRAS mutation expressed by the colorectal cancer (109). Although additional studies are still needed to determine which type of antigen induce the T cells best equipped to eradicate PDAC, increasing data suggests that immune suppressive mechanisms may be more complex and harder to bypass in the case of TAAs and mutated driver gene antigens such as mutated Kras because of the extensive length of time that they are expressed within the TME, which suggest these antigens have undergone immunoediting and subsequent immune escape (96).
Many different platforms are available for inducing TAA and TSA specific T cells, including various vaccine and adoptive T cell strategies. Notable antigen targets in PDAC and the therapies targeting these antigens are reviewed in Table 2. A number of vaccine delivery systems under development include plasmid DNA, polypeptide, and modified viral and bacterial approaches. In addition, new adjuvants under clinical development activate specific innate immune responses, via Toll Like Receptors and STING pathways (110,111). Chimeric antigen receptor (CAR) T cells, which are genetically engineered to express an antigen receptor specific for a malignancy-related target, are a platform for targeted immunotherapy that has shown promise in treating hematologic malignancies (112–114) and is now under clinical investigation in PDAC. Recently CAR T cells have been developed that target MUC1, a cell membrane protein that is overexpressed in PDAC and other cancers (115,116). In preclinical studies, mice harboring pancreatic cancer xenografts had increased OS when they received MUC1-specific CAR T cell therapy (116). CAR T cells targeting MUC1 are currently in clinical trials for solid tumors, including metastatic PDAC (NCT02587689). CAR T cells targeting mesothelin, a glycoprotein overexpressed in PDAC (100), are also being explored in human clinical trials for PDAC (NCT01583686) (117). However, no objective radiographic responses were reported in the initial PDAC clinical trial results for this agent (118). Although additional single-agent studies of these novel targeted immunotherapies are ongoing, it is likely that these targeted approaches will need to be combined with other therapies to overcome the immunosuppressive signals within the TME.
Table 2.
A non-exhaustive list of antigen targets for pancreatic cancer immunotherapies, and notable therapies targeting these antigens.
| Target | Expression | Notable immunotherapies against antigen target |
|---|---|---|
| Mesothelin | Highly overexpressed in virtually all pancreatic cancers and also expressed at lower levels in pleura, peritoneum, and pericardium (100). | CRS-207 (Aduro Biotech); live-attenuated listeria monocytogenes engineered to secrete mesothelin (99,121) Amatuximab (MORAb-009, Morphotek); monoclonal antibody (166) DMOT4039A (Genentech); antibody-drug conjugate (167) Anetumab ravtansine, (BAY 94-9343, Bayer); antibody-drug conjugate (168) Anti-mesothelin CAR-T cells (UPENN, NCI) (118) |
| CEA | Glycosylated homotypic/heterotypic cell surface intracellular adhesion molecule, overexpressed in 56–98% of pancreatic cancers and also expressed on oncofetal tissues (169). | CEA peptide vaccine (CAP1-6D) emulsified in montanide and GM-CSF (170) TRICOM-CEA(6D); poxvirus-based vaccine expressing costimulatory molecules and CEA (171,172) AVX701 (Alphavax); poxvirus-based vaccine expressing costimulatory molecules and CEA (171,173) GI-6207 (GlobeImmune/Celgene); recombinant yeast-CEA vaccine (174) |
| MUC1 | Transmembrane glycoprotein, overexpressed in ~90% of pancreatic tumors. Also low levels of expression on ductal and glandular epithelial cells. However, cancer-associated MUC1 is structurally different from normal MUC1 (hypoglycosylated) and may function as a tumor-specific antigen (175). | MUC1-peptide pulsed dendritic cells (176) Autologous dendritic cell vaccine (177) MUC1-peptide vaccine with SB-AS2 adjuvant (178) Adoptive transfer with MUC1 peptide-pulsed dendritic cells and activated T lymphocytes (179) |
| HER/EGFR/ERBB family proteins (eg HER1, HER2, HER3) | Cell-surface receptors implicated in tumor growth. HER2/neu is overexpressed in approximately 50% and EGFR in approximately 70% of pancreatic cancers and expression correlates with poor survival (97,98). These proteins are also expressed at lower levels in normal tissues. | Cetuximab (Erbitux, Lilly); EGFR antibody previously failed in clinical trials alone or in combination with cytotoxic agents, but may induce innate and adaptive immune responses that could synergize with novel immunotherapies (180,181). MM-141 (Merrimack Pharmaceuticals); bispecific antibody targeting IGF-1R and ErbB3 (HER3) (182) |
| Mutated KRAS | Intracellular GTPase important for cell growth and survival, mutated in up to 90% of pancreatic cancers (16,106). Mutated KRAS is a tumor-specific antigen. | GI-4000 (GlobeImmune); attenuated yeast expressing mutated RAS proteins (107) TG01 (Targovax); mutated RAS peptide vaccine co-administered with GM-CSF as an adjuvant (108) |
While most therapeutic cancer vaccines are categorized by their antigen target, whole cell vaccines deliver many tumor antigens without the need for specific knowledge of the relevant target. Autologous vaccines use the patient’s own tumor as an antigen source, whereas allogeneic vaccines are derived from another patient’s tumor. Allogeneic vaccines are more convenient and pragmatic because a single vaccine can be used to treat many patients, by presenting many relevant PDAC TAAs (119,120), whereas autologous vaccines must be personalized from each patient’s individual tumor. It is usually not feasible to utilize autologous tumor cells due to the lack of adequate tumor specimen.
The most studied whole cell vaccine platform in human PDAC trials is composed of 2 allogeneic granulocyte macrophage-colony stimulating factor (GM-CSF) secreting pancreatic tumor cell lines (GVAX). The PDAC GVAX has been combined with CRS-207, an attenuated Listeria monocytogenes-based vaccine targeting mesothelin. While the combination of GVAX plus CRS-207 showed encouraging results in early phase II studies (121), unfortunately an interim analysis of Phase 2b data failed to demonstrate improved OS compared with chemotherapy alone. A different whole-cell vaccine, algenpantucel-L, also recently failed to produce a clinical benefit in a recent phase III study, despite promising phase II data (122). These mixed clinical results suggest that although whole cell vaccination monotherapy induces TAA specific T cells, it is likely not enough to overcome the potently immunosuppressive TME of PDAC (35,123–125).
Despite these recent clinical setbacks, whole cell vaccines may be an important component of combination strategies for PDAC immunotherapy. We and others have shown that GVAX and other vaccines may prime the TME for treatment with an immune checkpoint inhibitor by inducing high levels of PD-L1 expression on epithelial tumor cells and intratumoral lymphoid aggregates (35). The upregulation of immunosuppressive regulatory mechanisms by PDAC suggest that whole cell vaccines should be combined with other immune therapies to maximize anti-tumor efficacy. Combination therapy with GVAX and PD-1 blockade improves survival in tumor-bearing mice (36). This hypothesis that whole-cell vaccine therapy can convert an immunosuppressive tumor into a tumor responsive to immune checkpoint blockade is currently being tested with combination PD-1 inhibitor and GVAX in patients with surgically resectable and borderline resectable PDAC (NCT02451982, NCT02648282). Additionally, GVAX and CRS-207 are now in clinical development in combination with the PD-1 inhibitor nivolumab in a phase 2 trial (STELLAR, NCT02243371).
Treating PDAC via combination therapy
Other combination approaches are actively being tested in patients with PDAC. These approaches include combining immunomodulatory agents with each other or with chemotherapy. (Table 1). Gemcitabine-based chemotherapy is often used as the chemotherapy backbone in these combination immunotherapy trials because it has been shown to increase tumor antigen availability, and transiently deplete immunosuppressive Tregs and myeloid derived suppressor cells (MDSC) in the PDAC TME (37–39,126). Lower numbers of intratumoral Tregs are associated with increased disease free survival after pancreatectomy (30), suggesting that Treg accumulation is an important determinant of survival in patients with PDAC. We and others have demonstrated that low dose cyclophosphamide can also deplete Tregs, modulate the TME and maximize clinical responses to immunotherapy (123,127,128). Another approach is combination therapy with epigenetic modulators, as epigenetic therapy appears to be immunomodulatory (129), and epigenetic therapy in PDAC is reviewed by Evan and colleagues in this CCR focus issue (130). Immunotherapies in clinical development for PDAC in combination with standard chemotherapy include the indoleamine 2,3 dioxygenase (IDO) inhibitor indoximod, the bruton tyrosine kinase (BTK) inhibitor ibrutinib, CD-40 agonists, and CCR2 inhibitors (Table 1). IDO is a tryptophan-catabolizing enzyme that, when activated via tumors or another inflammatory stimulus, activates suppressive activity in dendritic cells (DC) and leads to Treg activation (40,131,132). In a phase II study of untreated metastatic PDAC, the combination of indoximod plus gemcitabine/nab-paclitaxel demonstrated a response rate of 45% (133). This appears favorable compared to the 23% historical response rate of patients treated with gemcitabine/nab-paclitaxel alone in phase III studies (10), but must be tempered with phase II data demonstrating a 48% overall response rate with this chemotherapy combination (134). Another suppressive cell involved in Treg generation is the regulatory B cell (Breg), which has been implicated in converting resting CD4+ T cells to Tregs in a breast cancer model (135), and promotes tumorigenesis in PDAC (136). While identifying a specific Breg inhibitor is an area of active study, targeting BTK, which is expressed by tumor infiltrating B cells and myeloid cells, with ibrutinib synergizes with gemcitabine to inhibit murine PDAC growth (137). Ibrutinib is currently in clinical trials in combination with gemcitabine and nab-paclitaxel in the first line setting for metastatic PDAC (NCT02562898, NCT02436668).
CD40 is a TNF receptor superfamily member that is expressed by many cells, including B cells, DCs, monocytes, endothelial cells, and fibroblasts (138). CD40 agonists have been shown to activate APCs and promote tumor regression (139), and synergize with gemcitabine in mice to increase intratumoral effector T cell infiltration and induce T cell dependent PDAC tumor regression (140). CD40 agonists (NCT02588443, NCT02829099) and CCR2 blockade (NCT02732938) are currently being tested in clinical trials in multiple settings (141,142).
The presence of tumor infiltrating macrophages (TIMs) are associated with poorer outcomes in patients with resected PDAC (143,144). CCR2 is a chemokine receptor involved in the recruitment of immunosuppressive macrophages; CCR2 inhibition depletes CCR2 expressing tumor infiltrating macrophages and improves survival in mouse models (145). CCR2 blockade (NCT02732938) is currently being tested in combination with gemcitabine / nab-paclitaxel in a phase Ib/II study (142).
Another receptor whose inhibition facilitates depletion of TIMs in preclinical models is the colony-stimulating factor-1 receptor (CSF1R), which synergized with gemcitabine to increase effector T cell infiltration and slow pancreatic tumor growth (146). CSF1R inhibition also increased expression of checkpoint molecules on PDAC tumor cells and T cells, and when combined with checkpoint blockade and gemcitabine, further slowed murine PDAC growth (147). Multiple human trials are examining whether targeting CSF1R synergizes with PD-pathway blockade in solid tumors, including PDAC (NCT02526017, NCT02777710).
The C-X-C chemokine receptor 4 (CXCR4) is a chemokine receptor whose expression in human pancreatic tissues is associated with a poorer prognosis (148–150). CXCR4 blockade abrogated invasion and metastasis (151–153), and transfection of CXCR4 into pancreatic tumor cells increased their metastatic potential (154). Gemcitabine upregulates CXCR4 expression in human pancreatic cancer cells (155), which may be a mechanism of acquired resistance to gemcitabine (155–157). Inhibiting CXCR4 synergized with anti-PD-L1 blockade to decrease tumor size in a mouse PDAC model (158). Based on this encouraging preclinical data, clinical trials are examining the combination of CXCR4 inhibition with PD-pathway blockade in advanced solid tumors, including PDAC (NCT02737072, NCT02472977, NCT02826486).
Due to the suppressive nature of the PDAC tumor microenvironment, it is likely that multiple suppressive cell types will need to be targeted in order to improve clinical outcomes. The Treg, antigen presenting cell, and speculatively, the Breg are the three cell subtypes that appear to most potently suppress immune responses in PDAC. Chemotherapy should be the backbone of most trials in metastatic PDAC due to its immunomodulatory effect and already proven (although modest) survival benefit. Targeting Treg suppression via the PD-pathway is reasonable if done with chemotherapy (to transiently eliminate already established Tregs to “reset” the immune system) and in combination with at least one other immunomodulatory agent that affects another immune cell type, preferably either suppressive APCs or Bregs. Targeting the IDO pathway is attractive due to its induction of tolerogenic DCs and Treg activation and the encouraging phase II results in PDAC. Synergy with IDO inhibition and PD-pathway inhibitors or chemotherapy in early studies with other tumor types suggests that combination therapy with IDO inhibitor, PD-pathway, and chemotherapy may be efficacious if not overly toxic (159,160). Similarly, promising data in early studies with combination macrophage targeting (via CCR2 inhibition) and FOLFIRINOX in patients with borderline resectable or locally advanced PDAC make CCR2 an appealing target (142).
Future Directions
The failure of single agent immunotherapy in PDAC (21,22) at first glance suggests that immunotherapy may not have a role in future management of PDAC. However, the documented involvement of an integrated suppressive network of immune cells and stroma in PDAC development and progression suggest that a combination approach involving chemotherapy, immunotherapy, targeted therapy against stromal elements, and other modalities will be necessary in order to improve survival. Combination therapy, including strategies to boost adaptive immunity, break systemic tolerance, and increase tumor immunogenicity, has the potential to revolutionize PDAC treatment. Increasing our understanding of the PDAC TME, and how therapies affect the suppressive milieu, will help identify the best potential targets for therapeutic development and testing in clinical trials.
Acknowledgments
This work was supported in part by the Viragh Foundation and the Skip Viragh Pancreatic Cancer Center at Johns Hopkins (D. L. and E.M.J.), the Bloomberg-Kimmel Institute for Cancer Immunotherapy (all authors), an NCI SPORE in Gastrointestinal Cancers P50 CA062924 (E.M.J.), NIH R01 CA184926 (E.M.J.), and NIH T32 CA009071 (B.A.J. and M.Y.). Research supported by a Stand Up To Cancer – Lustgarten Foundation Pancreatic Cancer Convergence Dream Team Translational Research Grant (EMJ; Grant Number: SU2C-AACR-DT14-14). Stand Up To Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research.
Disclosures / Conflict of Interest
Elizabeth M. Jaffee receives research funding from Aduro BioTech and is a consultant with MedImmune. She has the potential to receive royalties from GVAX as a result of a licensing agreement with Aduro BioTech and Johns Hopkins University.
Abbreviations
- PDAC
pancreatic ductal adenocarcinoma
- PanIN
pancreatic intraepithelial neoplasia
- Treg
regulatory T cell
- PD-1
programmed death-1
- PD-L1
programmed death ligand 1
- TIM
tumor infiltrating macrophage
- MDSC
myeloid derived suppressor cell
- BTK
Bruton’s tyrosine kinase
- γδ T cells
gamma delta T cells
- CTLA-4
cytotoxic T lymphocyte associated protein 4
- CSF1R
colony stimulating factor 1 receptor
- GM-CSF
granulocyte macrophage colony stimulating factor
- DC
dendritic cell
- FAK1
focal adhesion kinase 1
- TAA
tumor associated antigen
- TSA
tumor specific antigen
- MMR
mismatch repair
- GVAX
granulocyte macrophage colony stimulating factor secreting pancreatic tumor cell vaccine
- CAR T cells
chimeric antigen receptor T cells
References
- 1.Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. 2016;66:271–89. doi: 10.3322/caac.21349. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 3.Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet. 2004;363:1049–57. doi: 10.1016/S0140-6736(04)15841-8. Available from: [DOI] [PubMed] [Google Scholar]
- 4.Mahipal A, Frakes J, Hoffe S, Kim R. Management of borderline resectable pancreatic cancer. World J Gastrointest Oncol. 2015;7:241–9. doi: 10.4251/wjgo.v7.i10.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Regine WF, Winter KA, Abrams RA, Safran H, Hoffman JP, Konski A, et al. Fluorouracil vs gemcitabine chemotherapy before and after fluorouracil-based chemoradiation following resection of pancreatic adenocarcinoma: a randomized controlled trial. JAMA. 2008;299:1019–26. doi: 10.1001/jama.299.9.1019. [DOI] [PubMed] [Google Scholar]
- 6.Oettle H, Post S, Neuhaus P, Gellert K, Langrehr J, Ridwelski K, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA. 2007;297:267–77. doi: 10.1001/jama.297.3.267. [DOI] [PubMed] [Google Scholar]
- 7.Neoptolemos JP, Stocken DD, Bassi C, Ghaneh P, Cunningham D, Goldstein D, et al. Adjuvant chemotherapy with fluorouracil plus folinic acid vs gemcitabine following pancreatic cancer resection: a randomized controlled trial. JAMA. 2010;304:1073–81. doi: 10.1001/jama.2010.1275. [DOI] [PubMed] [Google Scholar]
- 8.Kundranda M, Kachaamy T. Promising new therapies in advanced pancreatic adenocarcinomas. Future Oncol. 2014;10:2629–41. doi: 10.2217/fon.14.197. [DOI] [PubMed] [Google Scholar]
- 9.Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–25. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 10.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691–703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WEE, Poddubskaya E, et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373:123–35. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laheru D, Jaffee EM. Immunotherapy for pancreatic cancer - science driving clinical progress. Nat Rev Cancer. 2005;5:459–67. doi: 10.1038/nrc1630. [DOI] [PubMed] [Google Scholar]
- 13.Zheng L, Xue J, Jaffee EM, Habtezion A. Role of immune cells and immune-based therapies in pancreatitis and pancreatic ductal adenocarcinoma. Gastroenterology. 2013;144:1230–40. doi: 10.1053/j.gastro.2012.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manji GA, Olive K, Saenger Y, Oberstein P. Current and emerging therapies in metastatic pancreatic cancer. Clin Cancer Res. 2017 doi: 10.1158/1078-0432.CCR-16-2319. [DOI] [PubMed] [Google Scholar]
- 15.Steele CW, Jamieson NB, Evans TRJ, McKay CJ, Sansom OJ, Morton JP, et al. Exploiting inflammation for therapeutic gain in pancreatic cancer. Br J Cancer. 2013;108:997–1003. doi: 10.1038/bjc.2013.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bailey P, Chang DK, Nones K, Johns AL, Patch A-M, Gingras M-C, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47–52. doi: 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- 17.Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375–84. doi: 10.1016/S1470-2045(15)70076-8. [DOI] [PubMed] [Google Scholar]
- 18.Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373:1627–39. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. 2015;373:1803–13. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother. 2010;33:828–33. doi: 10.1097/CJI.0b013e3181eec14c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brahmer JR, Tykodi SS, Chow LQM, Hwu W-J, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–65. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kunk PR, Bauer TW, Slingluff CL, Rahma OE. From bench to bedside a comprehensive review of pancreatic cancer immunotherapy. J Immunother cancer. 2016;4:14. doi: 10.1186/s40425-016-0119-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kotteas E, Saif MW, Syrigos K. Immunotherapy for pancreatic cancer. J Cancer Res Clin Oncol. 2016;142:1795–805. doi: 10.1007/s00432-016-2119-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Foley K, Kim V, Jaffee E, Zheng L. Current progress in immunotherapy for pancreatic cancer. Cancer Lett. 2016;381:244–51. doi: 10.1016/j.canlet.2015.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ko AH. Progress in the treatment of metastatic pancreatic cancer and the search for next opportunities. J Clin Oncol. 2015;33:1779–86. doi: 10.1200/JCO.2014.59.7625. [DOI] [PubMed] [Google Scholar]
- 27.Varghese AM, Lowery MA, Yu KH, O’Reilly EM. Current management and future directions in metastatic pancreatic adenocarcinoma. Cancer. 2016 doi: 10.1002/cncr.30342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dreyer SB, Chang DK, Biankin AV. Pancreatic cancer genomes: implications for clinical management and therapeutic development. Clin Cancer Res. 2017 doi: 10.1158/1078-0432.CCR-16-2411. [DOI] [PubMed] [Google Scholar]
- 29.Ercolini AM, Ladle BH, Manning EA, Pfannenstiel LW, Armstrong TD, Machiels J-PH, et al. Recruitment of latent pools of high-avidity CD8(+) T cells to the antitumor immune response. J Exp Med. 2005;201:1591–602. doi: 10.1084/jem.20042167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu L, Zhao G, Wu W, Rong Y, Jin D, Wang D, et al. Low intratumoral regulatory T cells and high peritumoral CD8(+) T cells relate to long-term survival in patients with pancreatic ductal adenocarcinoma after pancreatectomy. Cancer Immunol Immunother. 2016;65:73–82. doi: 10.1007/s00262-015-1775-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fukunaga A, Miyamoto M, Cho Y, Murakami S, Kawarada Y, Oshikiri T, et al. CD8+ tumor-infiltrating lymphocytes together with CD4+ tumor-infiltrating lymphocytes and dendritic cells improve the prognosis of patients with pancreatic adenocarcinoma. Pancreas. 2004;28:e26–31. doi: 10.1097/00006676-200401000-00023. [DOI] [PubMed] [Google Scholar]
- 32.Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, Kanai Y, et al. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br J Cancer. 2013;108:914–23. doi: 10.1038/bjc.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hiraoka N, Onozato K, Kosuge T, Hirohashi S. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin Cancer Res. 2006;12:5423–34. doi: 10.1158/1078-0432.CCR-06-0369. [DOI] [PubMed] [Google Scholar]
- 34.Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. 2014;20:5064–74. doi: 10.1158/1078-0432.CCR-13-3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lutz ER, Wu AA, Bigelow E, Sharma R, Mo G, Soares K, et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol Res. 2014;2:616–31. doi: 10.1158/2326-6066.CIR-14-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soares KC, Rucki AA, Wu AA, Olino K, Xiao Q, Chai Y, et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J Immunother. 2015;38:1–11. doi: 10.1097/CJI.0000000000000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shibuya KC, Goel VK, Xiong W, Sham JG, Pollack SM, Leahy AM, et al. Pancreatic ductal adenocarcinoma contains an effector and regulatory immune cell infiltrate that is altered by multimodal neoadjuvant treatment. PLoS One. 2014;9:e96565. doi: 10.1371/journal.pone.0096565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Homma Y, Taniguchi K, Nakazawa M, Matsuyama R, Mori R, Takeda K, et al. Changes in the immune cell population and cell proliferation in peripheral blood after gemcitabine-based chemotherapy for pancreatic cancer. Clin Transl Oncol. 2014;16:330–5. doi: 10.1007/s12094-013-1079-0. [DOI] [PubMed] [Google Scholar]
- 39.Eriksson E, Wenthe J, Irenaeus S, Loskog A, Ullenhag G. Gemcitabine reduces MDSCs, tregs and TGFβ-1 while restoring the teff/treg ratio in patients with pancreatic cancer. J Transl Med. 2016;14:282. doi: 10.1186/s12967-016-1037-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sharma MD, Baban B, Chandler P, Hou D-Y, Singh N, Yagita H, et al. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J Clin Invest. 2007;117:2570–82. doi: 10.1172/JCI31911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fife BT, Bluestone JA. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol Rev. 2008;224:166–82. doi: 10.1111/j.1600-065X.2008.00662.x. [DOI] [PubMed] [Google Scholar]
- 42.Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. 2007;117:1147–54. doi: 10.1172/JCI31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Loos M, Giese NA, Kleeff J, Giese T, Gaida MM, Bergmann F, et al. Clinical significance and regulation of the costimulatory molecule B7-H1 in pancreatic cancer. Cancer Lett. 2008;268:98–109. doi: 10.1016/j.canlet.2008.03.056. [DOI] [PubMed] [Google Scholar]
- 44.Nomi T, Sho M, Akahori T, Hamada K, Kubo A, Kanehiro H, et al. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin Cancer Res. 2007;13:2151–7. doi: 10.1158/1078-0432.CCR-06-2746. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Y, Velez-Delgado A, Mathew E, Li D, Mendez FM, Flannagan K, et al. Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut. 2016;66:124–36. doi: 10.1136/gutjnl-2016-312078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Farren MR, Mace TA, Geyer S, Mikhail S, Wu C, Ciombor K, et al. Systemic Immune Activity Predicts Overall Survival in Treatment-Naïve Patients with Metastatic Pancreatic Cancer. Clin Cancer Res. 2016;22:2565–74. doi: 10.1158/1078-0432.CCR-15-1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Daley D, Zambirinis CP, Seifert L, Akkad N, Mohan N, Werba G, et al. γδ T Cells Support Pancreatic Oncogenesis by Restraining αβ T Cell Activation. Cell. 2016;166:1485–1499.e15. doi: 10.1016/j.cell.2016.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 49.Śledzińska A, Menger L, Bergerhoff K, Peggs KS, Quezada SA. Negative immune checkpoints on T lymphocytes and their relevance to cancer immunotherapy. Mol Oncol. 2015;9:1936–65. doi: 10.1016/j.molonc.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N Engl J Med. 2016;375:1823–33. doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 51.Poulogiannis G, Frayling IM, Arends MJ. DNA mismatch repair deficiency in sporadic colorectal cancer and Lynch syndrome. Histopathology. 2010;56:167–79. doi: 10.1111/j.1365-2559.2009.03392.x. [DOI] [PubMed] [Google Scholar]
- 52.Koopman M, Kortman GAM, Mekenkamp L, Ligtenberg MJL, Hoogerbrugge N, Antonini NF, et al. Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br J Cancer. 2009;100:266–73. doi: 10.1038/sj.bjc.6604867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372:2509–20. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim H, Jen J, Vogelstein B, Hamilton SR. Clinical and pathological characteristics of sporadic colorectal carcinomas with DNA replication errors in microsatellite sequences. Am J Pathol. 1994;145:148–56. [PMC free article] [PubMed] [Google Scholar]
- 55.Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, et al. The Vigorous Immune Microenvironment of Microsatellite Instable Colon Cancer Is Balanced by Multiple Counter-Inhibitory Checkpoints. Cancer Discov. 2015;5:43–51. doi: 10.1158/2159-8290.CD-14-0863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Le D, Uram J, Wang H, Kemberling H, Eyring A, Bartlett B, et al. PD-1 blockade in mismatch repair deficient non-colorectal gastrointestinal cancers. J Clin Oncol. 2016;34:195. [Google Scholar]
- 57.Yamamoto H, Itoh F, Nakamura H, Fukushima H, Sasaki S, Perucho M, et al. Genetic and clinical features of human pancreatic ductal adenocarcinomas with widespread microsatellite instability. Cancer Res. 2001;61:3139–44. [PubMed] [Google Scholar]
- 58.Nakata B, Wang YQ, Yashiro M, Nishioka N, Tanaka H, Ohira M, et al. Prognostic value of microsatellite instability in resectable pancreatic cancer. Clin Cancer Res. 2002;8:2536–40. [PubMed] [Google Scholar]
- 59.Ottenhof NA, Morsink FHM, Ten Kate F, van Noorden CJF, Offerhaus GJA. Multivariate analysis of immunohistochemical evaluation of protein expression in pancreatic ductal adenocarcinoma reveals prognostic significance for persistent Smad4 expression only. Cell Oncol (Dordr) 2012;35:119–26. doi: 10.1007/s13402-012-0072-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ademmer K, Ebert M, Müller-Ostermeyer F, Friess H, Büchler MW, Schubert W, et al. Effector T lymphocyte subsets in human pancreatic cancer: detection of CD8+CD18+ cells and CD8+CD103+ cells by multi-epitope imaging. Clin Exp Immunol. 1998;112:21–6. doi: 10.1046/j.1365-2249.1998.00546.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, et al. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008;68:918–26. doi: 10.1158/0008-5472.CAN-07-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu C-C, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25:719–34. doi: 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–61. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25:735–47. doi: 10.1016/j.ccr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Catenacci DVT, Junttila MR, Karrison T, Bahary N, Horiba MN, Nattam SR, et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J Clin Oncol. 2015;33:4284–92. doi: 10.1200/JCO.2015.62.8719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mei L, Du W, Ma WW. Targeting stromal microenvironment in pancreatic ductal adenocarcinoma: controversies and promises. J Gastrointest Oncol. 2016;7:487–94. doi: 10.21037/jgo.2016.03.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ikenaga N, Ohuchida K, Mizumoto K, Cui L, Kayashima T, Morimatsu K, et al. CD10+ Pancreatic Stellate Cells Enhance the Progression of Pancreatic Cancer. Gastroenterology. 2010;139:1041–1051.e8. doi: 10.1053/j.gastro.2010.05.084. [DOI] [PubMed] [Google Scholar]
- 68.Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. 2016;22:851–60. doi: 10.1038/nm.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Infante JR, Camidge DR, Mileshkin LR, Chen EX, Hicks RJ, Rischin D, et al. Safety, pharmacokinetic, and pharmacodynamic phase I dose-escalation trial of PF-00562271, an inhibitor of focal adhesion kinase, in advanced solid tumors. J Clin Oncol. 2012;30:1527–33. doi: 10.1200/JCO.2011.38.9346. [DOI] [PubMed] [Google Scholar]
- 70.Jones SF, Siu LL, Bendell JC, Cleary JM, Razak ARA, Infante JR, et al. A phase I study of VS-6063, a second-generation focal adhesion kinase inhibitor, in patients with advanced solid tumors. Invest New Drugs. 2015;33:1100–7. doi: 10.1007/s10637-015-0282-y. [DOI] [PubMed] [Google Scholar]
- 71.Soria JC, Gan HK, Blagden SP, Plummer R, Arkenau HT, Ranson M, et al. A phase I, pharmacokinetic and pharmacodynamic study of GSK2256098, a focal adhesion kinase inhibitor, in patients with advanced solid tumors. Ann Oncol. 2016;27:2268–2274. doi: 10.1093/annonc/mdw427. [DOI] [PubMed] [Google Scholar]
- 72.Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418–29. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2013;62:112–20. doi: 10.1136/gutjnl-2012-302529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hingorani SR, Harris WP, Beck JT, Berdov BA, Wagner SA, Pshevlotsky EM, et al. Phase Ib Study of PEGylated Recombinant Human Hyaluronidase and Gemcitabine in Patients with Advanced Pancreatic Cancer. Clin Cancer Res. 2016;22:2848–54. doi: 10.1158/1078-0432.CCR-15-2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, Young GS, et al. Pancreatic Cancer-Associated Stellate Cells Promote Differentiation of Myeloid-Derived Suppressor Cells in a STAT3-Dependent Manner. Cancer Res. 2013;73:3007–18. doi: 10.1158/0008-5472.CAN-12-4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Angevin E, Tabernero J, Elez E, Cohen SJ, Bahleda R, van Laethem J-L, et al. A Phase I/II, Multiple-Dose, Dose-Escalation Study of Siltuximab, an Anti-Interleukin-6 Monoclonal Antibody, in Patients with Advanced Solid Tumors. Clin Cancer Res. 2014;20:2192–204. doi: 10.1158/1078-0432.CCR-13-2200. [DOI] [PubMed] [Google Scholar]
- 77.Jin Y-H, Hou W, Kang HS, Koh C-S, Kim BS. The Role of Interleukin-6 in the Expression of PD-1 and PDL-1 on Central Nervous System Cells following Infection with Theiler’s Murine Encephalomyelitis Virus. J Virol. 2013;87:11538–51. doi: 10.1128/JVI.01967-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mace TA, Shakya R, Pitarresi JR, Swanson B, McQuinn CW, Loftus S, et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut. 2016 doi: 10.1136/gutjnl-2016-311585. gutjnl-2016-311585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Scheller J, Garbers C, Rose-John S. Interleukin-6: From basic biology to selective blockade of pro-inflammatory activities. Semin Immunol. 2014;26:2–12. doi: 10.1016/j.smim.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 80.Wu H-H, Hwang-Verslues WW, Lee W-H, Huang C-K, Wei P-C, Chen C-L, et al. Targeting IL-17B–IL-17RB signaling with an anti–IL-17RB antibody blocks pancreatic cancer metastasis by silencing multiple chemokines. J Exp Med. 2015;212:333–49. doi: 10.1084/jem.20141702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McAllister F, Bailey JM, Alsina J, Nirschl CJ, Sharma R, Fan H, et al. Oncogenic Kras activates a hematopoietic-to-epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia. Cancer Cell. 2014;25:621–37. doi: 10.1016/j.ccr.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Michaud DS, Izard J. Microbiota, oral microbiome, and pancreatic cancer. Cancer J. 20:203–6. doi: 10.1097/PPO.0000000000000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zambirinis CP, Pushalkar S, Saxena D, Miller G. Pancreatic cancer, inflammation, and microbiome. Cancer J. 20:195–202. doi: 10.1097/PPO.0000000000000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stolzenberg-Solomon RZ, Dodd KW, Blaser MJ, Virtamo J, Taylor PR, Albanes D. Tooth loss, pancreatic cancer, and Helicobacter pylori. Am J Clin Nutr. 2003;78:176–81. doi: 10.1093/ajcn/78.1.176. [DOI] [PubMed] [Google Scholar]
- 85.Hujoel PP, Drangsholt M, Spiekerman C, Weiss NS. An exploration of the periodontitis-cancer association. Ann Epidemiol. 2003;13:312–6. doi: 10.1016/s1047-2797(02)00425-8. [DOI] [PubMed] [Google Scholar]
- 86.Ahn J, Segers S, Hayes RB. Periodontal disease, Porphyromonas gingivalis serum antibody levels and orodigestive cancer mortality. Carcinogenesis. 2012;33:1055–8. doi: 10.1093/carcin/bgs112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Michaud DS, Joshipura K, Giovannucci E, Fuchs CS. A prospective study of periodontal disease and pancreatic cancer in US male health professionals. J Natl Cancer Inst. 2007;99:171–5. doi: 10.1093/jnci/djk021. [DOI] [PubMed] [Google Scholar]
- 88.Michaud DS, Liu Y, Meyer M, Giovannucci E, Joshipura K. Periodontal disease, tooth loss, and cancer risk in male health professionals: a prospective cohort study. Lancet Oncol. 2008;9:550–8. doi: 10.1016/S1470-2045(08)70106-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pihlstrom BL, Michalowicz BS, Johnson NW. Periodontal diseases. Lancet (London, England) 2005;366:1809–20. doi: 10.1016/S0140-6736(05)67728-8. [DOI] [PubMed] [Google Scholar]
- 90.Fan X, Alekseyenko AV, Wu J, Peters BA, Jacobs EJ, Gapstur SM, et al. Human oral microbiome and prospective risk for pancreatic cancer: a population-based nested case-control study. Gut. 2016 doi: 10.1136/gutjnl-2016-312580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mitsuhashi K, Nosho K, Sukawa Y, Matsunaga Y, Ito M, Kurihara H, et al. Association of Fusobacterium species in pancreatic cancer tissues with molecular features and prognosis. Oncotarget. 2015;6:7209–20. doi: 10.18632/oncotarget.3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Maisonneuve P, Lowenfels AB. Risk factors for pancreatic cancer: a summary review of meta-analytical studies. Int J Epidemiol. 2015;44:186–98. doi: 10.1093/ije/dyu240. [DOI] [PubMed] [Google Scholar]
- 93.Wang C, Li J. Pathogenic Microorganisms and Pancreatic Cancer. Gastrointest tumors [Internet] 2015;2:41–7. doi: 10.1159/000380896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wu S, Rhee K-J, Albesiano E, Rabizadeh S, Wu X, Yen H-R, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med. 2009;15:1016–22. doi: 10.1038/nm.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Perera M, Al-Hebshi NN, Speicher DJ, Perera I, Johnson NW. Emerging role of bacteria in oral carcinogenesis: a review with special reference to perio-pathogenic bacteria. J Oral Microbiol. 2016;8:32762. doi: 10.3402/jom.v8.32762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ward JP, Gubin MM, Schreiber RD. The Role of Neoantigens in Naturally Occurring and Therapeutically Induced Immune Responses to Cancer. Adv Immunol. 2016;130:25–74. doi: 10.1016/bs.ai.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yamanaka Y, Friess H, Kobrin MS, Büchler M, Kunz J, Beger HG, et al. Overexpression of HER2/neu oncogene in human pancreatic carcinoma. Hum Pathol. 1993;24:1127–34. doi: 10.1016/0046-8177(93)90194-l. [DOI] [PubMed] [Google Scholar]
- 98.Lei S, Appert HE, Nakata B, Domenico DR, Kim K, Howard JM. Overexpression of HER2/neu oncogene in pancreatic cancer correlates with shortened survival. Int J Pancreatol. 1995;17:15–21. doi: 10.1007/BF02788354. [DOI] [PubMed] [Google Scholar]
- 99.Le DT, Brockstedt DG, Nir-Paz R, Hampl J, Mathur S, Nemunaitis J, et al. A live-attenuated Listeria vaccine (ANZ-100) and a live-attenuated Listeria vaccine expressing mesothelin (CRS-207) for advanced cancers: phase I studies of safety and immune induction. Clin Cancer Res. 2012;18:858–68. doi: 10.1158/1078-0432.CCR-11-2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Argani P, Iacobuzio-Donahue C, Ryu B, Rosty C, Goggins M, Wilentz RE, et al. Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas: identification of a new pancreatic cancer marker by serial analysis of gene expression (SAGE) Clin Cancer Res. 2001;7:3862–8. [PubMed] [Google Scholar]
- 101.Thomas AM, Santarsiero LM, Lutz ER, Armstrong TD, Chen Y-C, Huang L-Q, et al. Mesothelin-specific CD8(+) T cell responses provide evidence of in vivo cross-priming by antigen-presenting cells in vaccinated pancreatic cancer patients. J Exp Med. 2004;200:297–306. doi: 10.1084/jem.20031435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther. 2013;21:904–12. doi: 10.1038/mt.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–51. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tran E, Ahmadzadeh M, Lu Y-C, Gros A, Turcotte S, Robbins PF, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. 2015;350:1387–90. doi: 10.1126/science.aad1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Smit VT, Boot AJ, Smits AM, Fleuren GJ, Cornelisse CJ, Bos JL. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res. 1988;16:7773–82. doi: 10.1093/nar/16.16.7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wedén S, Klemp M, Gladhaug IP, Møller M, Eriksen JA, Gaudernack G, et al. Long-term follow-up of patients with resected pancreatic cancer following vaccination against mutant K-ras. Int J cancer. 2011;128:1120–8. doi: 10.1002/ijc.25449. [DOI] [PubMed] [Google Scholar]
- 107.Muscarella P, Wilfong L, Ross S, Richards D, Raynov J, Fisher W, et al. A randomized, placebo-controlled, double blind, multicenter phase II adjuvant trial of the efficacy, immunogenicity, and safety of GI-4000 plus gem versus gem alone in patients with resected pancreas cancer with activating RAS mutations/survival and immun. J Clin Oncol. 2012;30 Abstract e14501. [Google Scholar]
- 108.Palmer D, Dueland S, Valle J, Otterhaug T, Eriksen J, Muller H, et al. A prospective, single-arm, phase I/II trial of RAS peptide vaccine TG01/GM-CSF and gemcitabine as adjuvant therapy for patients with resected pancreatic adenocarcinoma. J Clin Oncol. 2015;33 Abstract 4121. [Google Scholar]
- 109.Tran E, Robbins PF, Lu Y-C, Prickett TD, Gartner JJ, Jia L, et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med. 2016;375:2255–62. doi: 10.1056/NEJMoa1609279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Baird JR, Friedman D, Cottam B, Dubensky TW, Kanne DB, Bambina S, et al. Radiotherapy Combined with Novel STING-Targeting Oligonucleotides Results in Regression of Established Tumors. Cancer Res. 2016;76:50–61. doi: 10.1158/0008-5472.CAN-14-3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Vaz J, Andersson R. Intervention on toll-like receptors in pancreatic cancer. World J Gastroenterol. 2014;20:5808–17. doi: 10.3748/wjg.v20.i19.5808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Brentjens RJ, Rivière I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–28. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–33. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Vlad AM, Kettel JC, Alajez NM, Carlos CA, Finn OJ. MUC1 immunobiology: from discovery to clinical applications. Adv Immunol. 2004;82:249–93. doi: 10.1016/S0065-2776(04)82006-6. [DOI] [PubMed] [Google Scholar]
- 116.Posey AD, Schwab RD, Boesteanu AC, Steentoft C, Mandel U, Engels B, et al. Engineered CAR T Cells Targeting the Cancer-Associated Tn-Glycoform of the Membrane Mucin MUC1 Control Adenocarcinoma. Immunity. 2016;44:1444–54. doi: 10.1016/j.immuni.2016.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Morello A, Sadelain M, Adusumilli PS. Mesothelin-Targeted CARs: Driving T Cells to Solid Tumors. Cancer Discov. 2016;6:133–46. doi: 10.1158/2159-8290.CD-15-0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Beatty G, O’Hara M, Nelson A, McGarvey M, Torigian D, Lacey S, et al. Safety and antitumor activity of chimeric antigen receptor modified T cells in patients with chemotherapy refractory metastatic pancreatic cancer. J Clin Oncol. 2015;33 Abstract 3007. [Google Scholar]
- 119.Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Rivoltini L, Topalian SL, et al. Cloning of the gene coding for a shared human melanoma antigen recognized by autologous T cells infiltrating into tumor. Proc Natl Acad Sci U S A. 1994;91:3515–9. doi: 10.1073/pnas.91.9.3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Huang AY, Golumbek P, Ahmadzadeh M, Jaffee E, Pardoll D, Levitsky H. Role of bone marrow-derived cells in presenting MHC class I-restricted tumor antigens. Science. 1994;264:961–5. doi: 10.1126/science.7513904. [DOI] [PubMed] [Google Scholar]
- 121.Le DT, Wang-Gillam A, Picozzi V, Greten TF, Crocenzi T, Springett G, et al. Safety and survival with GVAX pancreas prime and Listeria Monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J Clin Oncol. 2015;33:1325–33. doi: 10.1200/JCO.2014.57.4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hardacre JM, Mulcahy M, Small W, Talamonti M, Obel J, Krishnamurthi S, et al. Addition of algenpantucel-L immunotherapy to standard adjuvant therapy for pancreatic cancer: a phase 2 study. J Gastrointest Surg. 2013;17:94–100. 100–1. doi: 10.1007/s11605-012-2064-6. [DOI] [PubMed] [Google Scholar]
- 123.Laheru D, Lutz E, Burke J, Biedrzycki B, Solt S, Onners B, et al. Allogeneic granulocyte macrophage colony-stimulating factor-secreting tumor immunotherapy alone or in sequence with cyclophosphamide for metastatic pancreatic cancer: a pilot study of safety, feasibility, and immune activation. Clin Cancer Res. 2008;14:1455–63. doi: 10.1158/1078-0432.CCR-07-0371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Murata S, Ladle BH, Kim PS, Lutz ER, Wolpoe ME, Ivie SE, et al. OX40 costimulation synergizes with GM-CSF whole-cell vaccination to overcome established CD8+ T cell tolerance to an endogenous tumor antigen. J Immunol. 2006;176:974–83. doi: 10.4049/jimmunol.176.2.974. [DOI] [PubMed] [Google Scholar]
- 125.Keenan BP, Saenger Y, Kafrouni MI, Leubner A, Lauer P, Maitra A, et al. A Listeria vaccine and depletion of T-regulatory cells activate immunity against early stage pancreatic intraepithelial neoplasms and prolong survival of mice. Gastroenterology. 2014;146:1784–94.e6. doi: 10.1053/j.gastro.2014.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Shevchenko I, Karakhanova S, Soltek S, Link J, Bayry J, Werner J, et al. Low-dose gemcitabine depletes regulatory T cells and improves survival in the orthotopic Panc02 model of pancreatic cancer. Int J cancer. 2013;133:98–107. doi: 10.1002/ijc.27990. [DOI] [PubMed] [Google Scholar]
- 127.Leao IC, Ganesan P, Armstrong TD, Jaffee EM. Effective depletion of regulatory T cells allows the recruitment of mesothelin-specific CD8 T cells to the antitumor immune response against a mesothelin-expressing mouse pancreatic adenocarcinoma. Clin Transl Sci. 2008;1:228–39. doi: 10.1111/j.1752-8062.2008.00070.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hermans IF, Chong TW, Palmowski MJ, Harris AL, Cerundolo V. Synergistic effect of metronomic dosing of cyclophosphamide combined with specific antitumor immunotherapy in a murine melanoma model. Cancer Res. 2003;63:8408–13. [PubMed] [Google Scholar]
- 129.Chiappinelli KB, Zahnow CA, Ahuja N, Baylin SB. Combining Epigenetic and Immunotherapy to Combat Cancer. Cancer Res. 2016;76:1683–9. doi: 10.1158/0008-5472.CAN-15-2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Evan GI, Hah N, Littlewood TD, Sodir N, Vidal TC, Downes M, et al. Re-engineering the pancreas tumor microenvironment: A “regenerative program” hacked. Clin Cancer Res. 2017 doi: 10.1158/1078-0432.CCR-16-3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Munn DH, Sharma MD, Hou D, Baban B, Lee JR, Antonia SJ, et al. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J Clin Invest. 2004;114:280–90. doi: 10.1172/JCI21583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Johnson BA, Baban B, Mellor AL. Targeting the immunoregulatory indoleamine 2,3 dioxygenase pathway in immunotherapy. Immunotherapy. 2009;1:645–61. doi: 10.2217/IMT.09.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bahary N, Garrido-Laguna I, Cinar P, O’Rourke MA, Somer BG, Nayak-Kapoor A, et al. Phase 2 trial of the indoleamine 2,3-dioxygenase pathway (IDO) inhibitor indoximod plus gemcitabine/nab-paclitaxel for the treatment of metastatic pancreas cancer: Interim analysis. J Clin Oncol. 2016;34 Abstract 3020. [Google Scholar]
- 134.Von Hoff DD, Ramanathan RK, Borad MJ, Laheru DA, Smith LS, Wood TE, et al. Gemcitabine Plus nab-Paclitaxel Is an Active Regimen in Patients With Advanced Pancreatic Cancer: A Phase I/II Trial. J Clin Oncol. 2011;29:4548–54. doi: 10.1200/JCO.2011.36.5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Olkhanud PB, Damdinsuren B, Bodogai M, Gress RE, Sen R, Wejksza K, et al. Tumor-Evoked Regulatory B Cells Promote Breast Cancer Metastasis by Converting Resting CD4+ T Cells to T-Regulatory Cells. Cancer Res. 2011;71:3505–15. doi: 10.1158/0008-5472.CAN-10-4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Pylayeva-Gupta Y, Das S, Handler JS, Hajdu CH, Coffre M, Koralov SB, et al. IL35-Producing B Cells Promote the Development of Pancreatic Neoplasia. Cancer Discov. 2016;6:247–55. doi: 10.1158/2159-8290.CD-15-0843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, Ruffell B, et al. Bruton Tyrosine Kinase-Dependent Immune Cell Cross-talk Drives Pancreas Cancer. Cancer Discov. 2016;6:270–85. doi: 10.1158/2159-8290.CD-15-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Vonderheide RH. Prospect of targeting the CD40 pathway for cancer therapy. Clin Cancer Res. 2007;13:1083–8. doi: 10.1158/1078-0432.CCR-06-1893. [DOI] [PubMed] [Google Scholar]
- 139.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–6. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Beatty GL, Winograd R, Evans RA, Long KB, Luque SL, Lee JW, et al. Exclusion of T Cells From Pancreatic Carcinomas in Mice Is Regulated by Ly6C(low) F4/80(+) Extratumoral Macrophages. Gastroenterology. 2015;149:201–10. doi: 10.1053/j.gastro.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Beatty GL, Torigian DA, Chiorean EG, Saboury B, Brothers A, Alavi A, et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res. 2013;19:6286–95. doi: 10.1158/1078-0432.CCR-13-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016;17:651–62. doi: 10.1016/S1470-2045(16)00078-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kurahara H, Shinchi H, Mataki Y, Maemura K, Noma H, Kubo F, et al. Significance of M2-polarized tumor-associated macrophage in pancreatic cancer. J Surg Res. 2011;167:e211–9. doi: 10.1016/j.jss.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 144.Kurahara H, Takao S, Maemura K, Mataki Y, Kuwahata T, Maeda K, et al. M2-polarized tumor-associated macrophage infiltration of regional lymph nodes is associated with nodal lymphangiogenesis and occult nodal involvement in pN0 pancreatic cancer. Pancreas. 2013;42:155–9. doi: 10.1097/MPA.0b013e318254f2d1. [DOI] [PubMed] [Google Scholar]
- 145.Sanford DE, Belt BA, Panni RZ, Mayer A, Deshpande AD, Carpenter D, et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clin Cancer Res. 2013;19:3404–15. doi: 10.1158/1078-0432.CCR-13-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013;73:1128–41. doi: 10.1158/0008-5472.CAN-12-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74:5057–69. doi: 10.1158/0008-5472.CAN-13-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Heinrich EL, Lee W, Lu J, Lowy AM, Kim J. Chemokine CXCL12 activates dual CXCR4 and CXCR7-mediated signaling pathways in pancreatic cancer cells. J Transl Med. 2012;10:68. doi: 10.1186/1479-5876-10-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wang Z, Ma Q, Liu Q, Yu H, Zhao L, Shen S, et al. Blockade of SDF-1/CXCR4 signalling inhibits pancreatic cancer progression in vitro via inactivation of canonical Wnt pathway. Br J Cancer. 2008;99:1695–703. doi: 10.1038/sj.bjc.6604745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Krieg A, Riemer JC, Telan LA, Gabbert HE, Knoefel WT. CXCR4–A Prognostic and Clinicopathological Biomarker for Pancreatic Ductal Adenocarcinoma: A Meta-Analysis. Batra SK, editor. PLoS One. 2015;10:e0130192. doi: 10.1371/journal.pone.0130192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313–23. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 152.Xu Q, Wang Z, Chen X, Duan W, Lei J, Zong L, et al. Stromal-derived factor-1α/CXCL12-CXCR4 chemotactic pathway promotes perineural invasion in pancreatic cancer. Oncotarget. 2015;6:4717–32. doi: 10.18632/oncotarget.3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Mori T, Doi R, Koizumi M, Toyoda E, Ito D, Kami K, et al. CXCR4 antagonist inhibits stromal cell-derived factor 1-induced migration and invasion of human pancreatic cancer. Mol Cancer Ther. 2004;3:29–37. [PubMed] [Google Scholar]
- 154.Saur D, Seidler B, Schneider G, Algül H, Beck R, Senekowitsch-Schmidtke R, et al. CXCR4 expression increases liver and lung metastasis in a mouse model of pancreatic cancer. Gastroenterology. 2005;129:1237–50. doi: 10.1053/j.gastro.2005.06.056. [DOI] [PubMed] [Google Scholar]
- 155.Arora S, Bhardwaj A, Singh S, Srivastava SK, McClellan S, Nirodi CS, et al. An undesired effect of chemotherapy: gemcitabine promotes pancreatic cancer cell invasiveness through reactive oxygen species-dependent, nuclear factor κB- and hypoxia-inducible factor 1α-mediated up-regulation of CXCR4. J Biol Chem. 2013;288:21197–207. doi: 10.1074/jbc.M113.484576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Balic A, Sørensen MD, Trabulo SM, Sainz B, Cioffi M, Vieira CR, et al. Chloroquine targets pancreatic cancer stem cells via inhibition of CXCR4 and hedgehog signaling. Mol Cancer Ther. 2014;13:1758–71. doi: 10.1158/1535-7163.MCT-13-0948. [DOI] [PubMed] [Google Scholar]
- 157.Singh S, Srivastava SK, Bhardwaj A, Owen LB, Singh AP. CXCL12-CXCR4 signalling axis confers gemcitabine resistance to pancreatic cancer cells: a novel target for therapy. Br J Cancer. 2010;103:1671–9. doi: 10.1038/sj.bjc.6605968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Feig C, Jones JO, Kraman M, Wells RJB, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110:20212–7. doi: 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zakharia Y, Drabick JJ, Khleif S, McWilliams RR, Munn D, Link CJ, et al. Updates on phase 1b/2 trial of the indoleamine 2,3-dioxygenase pathway (IDO) inhibitor indoximod plus checkpoint inhibitors for the treatment of unresectable stage 3 or 4 melanoma. J Clin Oncol. 2016;34 Abstract 3075. [Google Scholar]
- 160.Colman H, Mott F, Spira AI, Johnson TS, Zakharia Y, Vahanian NN, et al. A phase Ib/2 study of the combination of the IDO pathway inhibitor indoximod and temozolomide for adult patients with temozolomide-refractory primary malignant brain tumors: Safety analysis and preliminary efficacy of the phase Ib component. J Clin Oncol. 2015;33 Abstract 2070. [Google Scholar]
- 161.Loos M, Giese NA, Kleeff J, Giese T, Gaida MM, Bergmann F, et al. Clinical significance and regulation of the costimulatory molecule B7-H1 in pancreatic cancer. Cancer Lett. 2008;268:98–109. doi: 10.1016/j.canlet.2008.03.056. [DOI] [PubMed] [Google Scholar]
- 162.Birnbaum DJ, Finetti P, Lopresti A, Gilabert M, Poizat F, Turrini O, et al. Prognostic value of PDL1 expression in pancreatic cancer. Oncotarget. 2016;7:71198–71210. doi: 10.18632/oncotarget.11685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Munn DH, Mellor AL. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016;37:193–207. doi: 10.1016/j.it.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Witkiewicz A, Williams TK, Cozzitorto J, Durkan B, Showalter SL, Yeo CJ, et al. Expression of Indoleamine 2,3-Dioxygenase in Metastatic Pancreatic Ductal Adenocarcinoma Recruits Regulatory T Cells to Avoid Immune Detection. J Am Coll Surg. 2008;206:849–54. doi: 10.1016/j.jamcollsurg.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 165.Long KB, Gladney WL, Tooker GM, Graham K, Fraietta JA, Beatty GL. IFNγ and CCL2 Cooperate to Redirect Tumor-Infiltrating Monocytes to Degrade Fibrosis and Enhance Chemotherapy Efficacy in Pancreatic Carcinoma. Cancer Discov. 2016;6:400–13. doi: 10.1158/2159-8290.CD-15-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Fujisaka Y, Kurata T, Tanaka K, Kudo T, Okamoto K, Tsurutani J, et al. Phase I study of amatuximab, a novel monoclonal antibody to mesothelin, in Japanese patients with advanced solid tumors. Invest New Drugs. 2015;33:380–8. doi: 10.1007/s10637-014-0196-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Weekes CD, Lamberts LE, Borad MJ, Voortman J, McWilliams RR, Diamond JR, et al. Phase I Study of DMOT4039A, an Antibody-Drug Conjugate Targeting Mesothelin, in Patients with Unresectable Pancreatic or Platinum-Resistant Ovarian Cancer. Mol Cancer Ther. 2016;15:439–47. doi: 10.1158/1535-7163.MCT-15-0693. [DOI] [PubMed] [Google Scholar]
- 168.Golfier S, Kopitz C, Kahnert A, Heisler I, Schatz CA, Stelte-Ludwig B, et al. Anetumab ravtansine: a novel mesothelin-targeting antibody-drug conjugate cures tumors with heterogeneous target expression favored by bystander effect. Mol Cancer Ther. 2014;13:1537–48. doi: 10.1158/1535-7163.MCT-13-0926. [DOI] [PubMed] [Google Scholar]
- 169.Yamaguchi K, Enjoji M, Tsuneyoshi M. Pancreatoduodenal carcinoma: a clinicopathologic study of 304 patients and immunohistochemical observation for CEA and CA19-9. J Surg Oncol. 1991;47:148–54. doi: 10.1002/jso.2930470303. [DOI] [PubMed] [Google Scholar]
- 170.Geynisman DM, Zha Y, Kunnavakkam R, Aklilu M, Catenacci DV, Polite BN, et al. A randomized pilot phase I study of modified carcinoembryonic antigen (CEA) peptide (CAP1-6D)/montanide/GM-CSF-vaccine in patients with pancreatic adenocarcinoma. J Immunother cancer. 2013;1:8. doi: 10.1186/2051-1426-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Huang EH, Kaufman HL. CEA-based vaccines. Expert Rev Vaccines. 2002;1:49–63. doi: 10.1586/14760584.1.1.49. [DOI] [PubMed] [Google Scholar]
- 172.Marshall JL, Gulley JL, Arlen PM, Beetham PK, Tsang K-Y, Slack R, et al. Phase I study of sequential vaccinations with fowlpox-CEA(6D)-TRICOM alone and sequentially with vaccinia-CEA(6D)-TRICOM, with and without granulocyte-macrophage colony-stimulating factor, in patients with carcinoembryonic antigen-expressing carcinomas. J Clin Oncol. 2005;23:720–31. doi: 10.1200/JCO.2005.10.206. [DOI] [PubMed] [Google Scholar]
- 173.Morse M, Hobeika A, Gwin W, Osada T, Gelles J, Rushing C, et al. Phase I study of alphaviral vector (AVX701) in colorectal cancer patients: comparison of immune responses in stage III and stage IV patients. J Immunother cancer. 2015;3:444. [Google Scholar]
- 174.Bilusic M, Heery CR, Arlen PM, Rauckhorst M, Apelian D, Tsang KY, et al. Phase I trial of a recombinant yeast-CEA vaccine (GI-6207) in adults with metastatic CEA-expressing carcinoma. Cancer Immunol Immunother. 2014;63:225–34. doi: 10.1007/s00262-013-1505-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Qu CF, Li Y, Song YJ, Rizvi SMA, Raja C, Zhang D, et al. MUC1 expression in primary and metastatic pancreatic cancer cells for in vitro treatment by (213)Bi-C595 radioimmunoconjugate. Br J Cancer. 2004;91:2086–93. doi: 10.1038/sj.bjc.6602232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Rong Y, Qin X, Jin D, Lou W, Wu L, Wang D, et al. A phase I pilot trial of MUC1-peptide-pulsed dendritic cells in the treatment of advanced pancreatic cancer. Clin Exp Med. 2012;12:173–80. doi: 10.1007/s10238-011-0159-0. [DOI] [PubMed] [Google Scholar]
- 177.Kimura Y, Tsukada J, Tomoda T, Takahashi H, Imai K, Shimamura K, et al. Clinical and immunologic evaluation of dendritic cell-based immunotherapy in combination with gemcitabine and/or S-1 in patients with advanced pancreatic carcinoma. Pancreas. 2012;41:195–205. doi: 10.1097/MPA.0b013e31822398c6. [DOI] [PubMed] [Google Scholar]
- 178.Ramanathan RK, Lee KM, McKolanis J, Hitbold E, Schraut W, Moser AJ, et al. Phase I study of a MUC1 vaccine composed of different doses of MUC1 peptide with SB-AS2 adjuvant in resected and locally advanced pancreatic cancer. Cancer Immunol Immunother. 2005;54:254–64. doi: 10.1007/s00262-004-0581-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kondo H, Hazama S, Kawaoka T, Yoshino S, Yoshida S, Tokuno K, et al. Adoptive immunotherapy for pancreatic cancer using MUC1 peptide-pulsed dendritic cells and activated T lymphocytes. Anticancer Res. 2008;28:379–87. [PubMed] [Google Scholar]
- 180.Bauman JE, Grandis JR. Targeting secondary immune responses to cetuximab: CD137 and the outside story. J Clin Invest. 2014;124:2371–5. doi: 10.1172/JCI76264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Yang X, Zhang X, Mortenson ED, Radkevich-Brown O, Wang Y, Fu Y-X. Cetuximab-mediated tumor regression depends on innate and adaptive immune responses. Mol Ther. 2013;21:91–100. doi: 10.1038/mt.2012.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Fitzgerald JB, Johnson BW, Baum J, Adams S, Iadevaia S, Tang J, et al. MM-141, an IGF-IR- and ErbB3-directed bispecific antibody, overcomes network adaptations that limit activity of IGF-IR inhibitors. Mol Cancer Ther. 2014;13:410–25. doi: 10.1158/1535-7163.MCT-13-0255. [DOI] [PubMed] [Google Scholar]
- 183.Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21:822–35. doi: 10.1016/j.ccr.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 2012;21:836–47. doi: 10.1016/j.ccr.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Nicholl MB, Ledgewood CL, Chen X, Bai Q, Qin C, Cook KM, et al. IL-35 promotes pancreas cancer growth through enhancement of proliferation and inhibition of apoptosis: evidence for a role as an autocrine growth factor. Cytokine. 2014;70:126–33. doi: 10.1016/j.cyto.2014.06.020. [DOI] [PubMed] [Google Scholar]
- 186.Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol. 2012;24:207–12. doi: 10.1016/j.coi.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]