

Abstract
Keloids are benign fibroproliferative tumors more frequently found among African Americans. Until now, keloid etiopathogenesis is not fully understood. To characterize keloids in African Americans, we performed transcriptional profiling of biopsies from large chronic keloids, adjacent nonlesional (NL) skin (n=3) and a newly formed keloid lesion using Affymetrix HGU133 2.0 plus arrays. Quantitative RT-PCR (qRT-PCR) and immunohistochemistry staining were done to confirm increased expression of relevant genes. We identified 1,202 up-regulated and 961 down-regulated differentially expressed genes (DEGs) between keloid and NL skin; 1,819 up- and 1,867 down-regulated DEGs between newly formed keloid and NL skin; and 492 up- and 775 down-regulated DEGs between chronic and newly formed keloid (Fold change >2, False discovery rate <0.05). Many of the top up-regulated DEGs between chronic keloid and NL skin, and between newly formed keloid and NL skin are involved in bone/cartilage formation including Fibrillin 2(FBN2), Collagen type X alpha1(COL10A1), Asporin(ASPN), Cadherin 11(CDH11), Bone morphogenic protein 1(BMP1), Secreted phosphoprotein 1(SPP1), and Runt-related transcription factor2(RUNX2). qRT-PCR confirmed significant (p<0.05) up-regulation of BMP1, RUNX2, CDH11 and FBN2 in chronic keloid compared to NL skin. Immunohistochemistry staining showed increased protein expression of ASPN, CDH11, BMP1 and RUNX2 on chronic and newly formed keloid compared to NL skin. Our study shows that large keloids in African Americans represent a dysplasia of cutaneous connective tissue towards immature cartilage or bone differentiation. The phenotype is potentially regulated by overexpression of RUNX2. This knowledge may give insights to guide the development of better treatment for the disease in the future.
Keywords: Keloid, cartilage and bone formation, transcriptional profiling, connective tissue differentiation
Introduction
Keloids are benign fibroproliferative dermal tumors that are believed to result from a dysregulated wound-healing process. Lesions can be induced by trauma, surgery, or skin inflammation but can also develop spontaneously. Keloid growth can continue over months to years and may cause considerable pruritus, pain, restriction of mobility, and psychological impairment leading to decreased quality of life1. Keloids can be observed in all races, but are more common in native Africans, Hispanics, and Asians2. In the US, keloid occurs in ∼1/30 of African Americans vs. ∼1/625 of the overall US population3. Although keloid can occur at any age, it is most likely to occur between 10 and 30 years of age4. Keloids range in size from small lesions (<1 cm in diameter) to extensive, sometimes massive, tumors (>25cms in diameter)5.
The etiopathogenesis of keloid is not fully understood. This is in part due to the fact that there are no validated animal models of keloid to study keloid formation or treatment. Many alternative hypotheses exist for keloid formation (all having different therapeutic implications), ranging from dysregulated synthesis of collagen by dermal fibroblasts, to insufficient degradation of deposited matrix, to altered growth factor signaling in disease2. Altered endothelial cells that bind or trap platelets, leading to increased release of PDGF-α or TGFβhas been proposed6. Altered cross-regulation of cell growth between epidermal and dermal compartments has also been suggested, since keratinocytes synthesize and release many growth factors such as PDGF, FGF, and VEGF that regulate underlying connective tissue structure and function7.
Although there have been several gene expression profiling studies on keloid, most were done in cultured fibroblasts8 with only a few in-vivo studies9-11. Interestingly, one study performed in Japanese subjects and with a limited gene array platform found several up-regulated genes associated with chondrogenic and osteogenic connective tissue differentiation10. However, studies done on other populations, with different array series, have failed to detect these alterations11.
To further characterize human keloids, we performed transcriptional profiling of biopsies from large chronic keloid lesions and adjacent non-lesional (NL) skin and a newly formed keloid in African Americans diagnosed with keloids. We studied the differentially expressed genes (DEGs) between chronic keloid and non-lesional skin of the same patient, and compared this to the DEGs of a newly formed keloid. We also performed immunohistochemistry and quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) on relevant up-regulated genes to confirm increased expression on chronic keloid and newly formed keloid compared to NL skin. Our microarray analysis revealed more than 2,000 DEGs and a molecular signal of dysplasia of connective tissue towards immature cartilage and bone differentiation in associated lesions.
Methods
Skin samples
Biopsies from non-lesional, chronic keloid lesion (n=3) and newly formed keloid skin were obtained from African American patients with severe keloids under the Rockefeller University IRB-approved protocols. Informed consent was obtained and the study was performed in adherence with the Principles of the Declaration of Helsinki. Patients who developed a new keloid from the non-lesional biopsy site were treated with intra-lesional steroid injection. The chronic keloid lesions were present on the patients for more than 20 years while the newly formed keloids on the non-lesional biopsy site developed about 6 weeks after the biopsy was done.
Gene Array and qRT-PCR
RNA was extracted from non-lesional, chronic keloid lesion and newly formed keloid biopsies using the RNeasy Mini KIT (Qiagen, Valencia, CA). RNA was amplified, labeled, and hybridized to Affymetrix HGU133 2.0 plus arrays. qRT-PCR was performed using EZ PCR core reagents, primers, and probes (Life Technologies, Grand Island, NY, U.S.A.) as previously published12. Sequences of primers and probes used in this study are found in Table S6a. The results were normalized to HARP housekeeping gene and transformed to base 2 logarithmic values before the analysis. The means and the differences between chronic keloid lesional and non-lesional tissue at each biomarker was assessed with a mixed-effect model (Fixed-factor: tissue, random intercept for each patient).
Immunohistochemistry and Immunofluorescence
Standard procedures were used for immunohistochemistry (IHC) and immunofluorescence (IF) as previously described12. For IHC, frozen tissue sections from non-lesional, chronic keloid and newly formed keloid biopsies were stained with Asporin (Abcam, Cambridge, MA), Cadherin 11 (Abcam, Cambridge, MA), Bone morphogenic protein 1 (BMP1) (Novus Biologics, Littleton, CO) and Runt-related transcription factor 2 (RUNX2) (Abcam, Cambridge, MA). Antibody clones and dilutions are shown in Table S6b. Biotin-labeled horse anti-mouse (Vector Laboratories, Burlingame, CA) and Biotin-labeled goat anti-rabbit (Vector Laboratories, Burlingame, CA) was used to detect the mouse monoclonal and rabbit polyclonal antibodies, respectively. The staining signal was amplified with avidin-biotin complex (Vector Laboratories, Burlingame, CA) and developed using chromogen 3-amino-9-ethylcarbazole (Sigma-Aldrich, St. Louis, MO).
For IF, frozen tissue sections from nonlesional skin, chronic keloid and newly formed keloid biopsies were fixed with acetone and blocked in 10% normal chicken serum (Vector Laboratories) for 30 minutes. The tissue sections were incubated with RUNX2 mouse monoclonal antibody (Abcam, Cambridge, MA) and Collagen 10A1 affinity purified goat polyclonal antibody (Santa Cruz, Dallas, TX) overnight at 4°C. The next day, the same sections were amplified with chicken anti-goat IgG Alexa Flour 594 (Invitrogen, Eugene, OR) for 30 minutes and then goat anti-mouse IgG2a Alexa Fluor 488 (Invitrogen, Eugene, OR) for another 30 minutes.
Images were acquired using the appropriate filters of a Zeiss Axioplan 2 widefield fluorescence microscope with a Plan Neofluar 20 x 0.7 numerical aperture lens and a Hamamatsu Orca ERcooled charge-coupled device camera, controlled by METAVUE software (MDS Analytical Technologies, Downington, PA). Alternatively, images were acquired using appropriate filters of an upright confocal microscope Zeiss Axioplan 2 (LSM 510) with C-Apochromat 40x/1.2 W objective using Argon/Krypton laser-488nm and 568nm. Images in each figure are presented both as single color stains (green and red) located above the merged image, so that localization of two markers on similar or different cells can be appreciated. Cells that co-express the two markers in a similar location are yellow in color. A white line denotes the junction between epidermis and the dermis. Some dermal collagen gave green auto-fluorescence, and antibodies conjugated with a fluorochrome often gave background epidermal fluorescence.
Statistical Analysis
Affymetrix (Santa Clara, CA) CEL files were scanned using software packages Harshlight13 and array Quality Metrics from R/Bioconductor (www.bioconductor.org). Expression values (in log2-scale) were obtained using the GCRMA algorithm. Genes with expression higher than three in at least one sample and standard deviation >0.1 were included in the statistical analysis. Gene expression was modeled using mixed effect models with group (LS, NL, newly formed) as a fixed effect and a random intercept for each patient. Resultant p-values from the moderated paired t-test were adjusted for multiple hypotheses using the Benjamini–Hochberg procedure, which controls for the false discovery rate (FDR). The data discussed in this publication have been deposited in the Gene Expression Omnibus of the National Center for Biotechnology Information and are accessible through its series accession number GSEXXXXX.
Results
Clinical and histology images
Clinical photos of patients with extensive keloids who enrolled in the study are shown in Figure 1a. Representative histology images of chronic keloid lesion and background non-lesional skin are shown in Figure 1b-d. Non-lesional skin had a normal appearing epidermis and papillary dermis with clearly demarcated larger collagen bundles in the reticular dermis. Some vascular and stromal bundles were present, similar to the appearance of normal skin. The dermis of non-lesional skin extended to a depth of 4-5mm in all cases examined. No overt features of scars were present in non-lesional skin. In contrast, chronic keloid lesions were thicker than normal skin (6-7mm deep) and had a thickened epidermis with deep rete ridges (notable for the absence of dermo-epidermal junction flattening that often appears in scars). The dermis had two distinct zones of pathology. A superficial zone (∼1 mm deep) showed staining of bright pink, homogenized collagen intermixed with stromal cells. Below this zone, a highly cellular infiltrate of spindle-shaped connective tissue cells with appearance of fibroblasts or myofibroblasts organized in bundles or whorls were detected in some regions, while other regions continued markedly thickened, largely acellular collagen bundles. Areas of marked cellularity also contained pink-staining matrix and some smaller collagen bundles.
Figure 1.

Clinical photos of large keloids from patients who enrolled in the study are shown in 1a. The representative histology images of non-lesional and chronic keloid lesion are shown in (1b) lower magnification (4×), (1c) higher magnification (10×) and (1d) deeper dermis. Size bar = 100 um
Microarray analysis showed significant upregulation of genes involved in cartilage and bone development in chronic and newly formed keloids
We observed a wide separation between chronic lesional and non-lesional skin in a PCA plot using the PC-1 axis (Figure S1a). A new keloid that formed at a biopsy site of non-lesional (NL) skin also clustered with the chronic keloid lesions. Using a conservative analysis approach (FCH>2, FDR<0.05) we detected >2000 DEGs between keloid and non-lesional skin; 1202 up-regulated and 961 down-regulated DEGs in chronic keloid tissue and 1819 up- and 1,867 down-regulated DEGs in new keloid tissue compared to NL skin (Figure S1b). The top 100 up-regulated DEGs are characterized by very large expression increases (20-fold to >50-fold increases from non-lesional skin) and also highly significant p-values (p<10-4) after correction for multiplicity (FDR values), as shown in Table 1a/b and fully detailed in Tables S1 and S2. We also identified 1438 up- and 1066 down-regulated DEGs in chronic keloid and 2179 up- and 2360 down-regulated genes in new keloid lesion compared to normal skin; and 492 up- and 775 down-regulated DEGs between chronic keloid and newly formed keloid. Up and down-regulated DEGs between chronic keloid and newly formed keloid vs. normal skin, as well as the chronic vs. newly formed keloid tissue are shown in Tables S3 - S5.
Table Ia. Selected Upregulated DEGs between Chronic keloid Lesion and NL skin.
| GENE | FCH |
|---|---|
| FBN2 – fibrillin 2 | 51.12 |
| COL10A1- collagen, type X, alpha 1 | 48.63 |
| COL12A1 - collagen, type XII, alpha 1 | 47.91 |
| ADAM12 - ADAM metallopeptidase domain 12 | 46.48 |
| IGF2 - insulin-like growth factor 2 (somatomedin A) | INS-IGF2 readthrough | 44.06 |
| ASPN - asporin | 41.39 |
| COL14A1 - collagen, type XIV, alpha 1 | 37.62 |
| CILP2 - cartilage intermediate layer protein 2 | 37.19 |
| LAMP5 - lysosomal-associated membrane protein family, member 5 | 36.22 |
| VCAN - versican | 31.97 |
| COL1A1 - collagen, type I, alpha 1 | 31.42 |
| CDH11 - cadherin 11, type 2, OB-cadherin (osteoblast) | 28.96 |
| ACAN - aggrecan | 28.68 |
| COMP – cartilage oligomeric matrix protein | 27.52 |
| COL6A1 - collagen, type VI, alpha 1 | 26.74 |
| BGN - biglycan | 26.41 |
| BMP1 - bone morphogenetic protein 1 | 25.37 |
| COL11A1 - collagen, type XI, alpha 1 | 25.01 |
| COL3A1 - collagen, type III, alpha 1 | 22.02 |
| PCDH17 – protocadherin 17 | 21.10 |
| RUNX2 - runt-related transcription factor 2 | 21.06 |
| NEFH - neurofilament, heavy polypeptide | 20.37 |
Table Ib. Selected Upregulated DEGs between New keloid lesion and NL skin.
| GENE | FCH |
|---|---|
| COL12A1 - collagen, type XII, alpha 1 | 77.48 |
| COL10A1 - collagen, type X, alpha 1 | 68.49 |
| ADAM12 - ADAM metallopeptidase domain 12 | 60.18 |
| COL11A1 - collagen, type XI, alpha 1 | 56.3 |
| S100A7A- S100 calcium binding protein A7A | 54.88 |
| SPP1 - secreted phosphoprotein 1 | 54.05 |
| WT1- Wilms tumor 1 | 48.7 |
| FBN2-fibrillin 2 | 48.53 |
| ASPN - asporin | 45.66 |
| MMP16 - matrix metallopeptidase 16 (membrane-inserted) | 42.6 |
| CDH11 - cadherin 11, type 2, OB-cadherin (osteoblast) | 41.82 |
| RUNX2 - runt-related transcription factor 2 | 39.13 |
| COL6A1 - collagen, type VI, alpha 1 | 38.08 |
| VCAN - versican | 38.01 |
| FN1 - fibronectin 1 | 38.07 |
| COL14A1 - collagen, type XIV, alpha 1 | 36.49 |
| SPON1 - spondin 1, extracellular matrix protein | 36.38 |
| NRP2 - neuropilin 2 | 35.19 |
| COL1A1 - collagen, type I, alpha 1 | 33.99 |
| PCDH17 - protocadherin 17 | 31.47 |
| PLOD2 - procollagen-lysine, 2-oxoglutarate 5-dioxygenase 2 | 30.55 |
| EDNRA - endothelin receptor type A | 29.8 |
| NEFH - neurofilament, heavy polypeptide | 29.36 |
| TLL2 - tolloid-like 2 | 29.04 |
| BMP1 - bone morphogenetic protein 1 | 28.01 |
| MMP14 - matrix metallopeptidase 14 (membrane-inserted) | 27.28 |
| TNFRSF9 - tumor necrosis factor receptor superfamily, member 9 | 25.05 |
Many of the upregulated genes that were highly expressed in chronic keloid lesions are involved in cartilage and bone development: Fibrillin2 (FBN2), Collagen type X alpha 1 (COL10A1), Asporin (ASPN), Cadherin11 (CDH11), Secreted phosphoprotein 1 (SPP1), Bone morphogenic protein 1 (BMP1) and Runt-related transcription factor 2 (RUNX2). SPP1, a gene for bone remodeling, and Neuropilin (NRP2), a nerve-associated gene, had increased expressions in new keloid lesion compared to NL and normal skin. Neurofilament, heavy polypeptide (NEFH), another nerve gene, also had an increased expression in chronic and new keloid lesion compared to NL and normal skin. Many unanticipated genes encoding cartilage and bone-related proteins were expressed at levels comparable to or higher than dermal collagen genes, e.g., collagen type I, alpha 1 (COL1A1) was up-regulated 31-fold and collagen type III, alpha 1 (COL3A1) was up-regulated 22-fold, but collagen type X, alpha 1 (COL10A1) was up-regulated by 48-fold (Table 1a).
qRT-PCR analysis confirmed significant upregulation of relevant genes in chronic keloid lesion compared to nonlesional skin
We performed quantitative RT-PCR to confirm upregulation of some DEGs namely BMP1, RUNX2, CDH11, FBN2, ASPN, SPP1, COL10A1, Collagen type X11 alpha 1 (COL12A1), ADAM metallopeptidase domain 12 (ADAM12) and Cartilage Intermediate Layer Protien 2 (CILP2). We found significant increases in BMP1, RUNX2, CDH11 and FBN2 (p<0.05) and COL12A1, ASPN, ADAM12, CILP2 (p<0.1) in chronic lesional keloid compared to NL skin (Figure 2).
Figure 2.
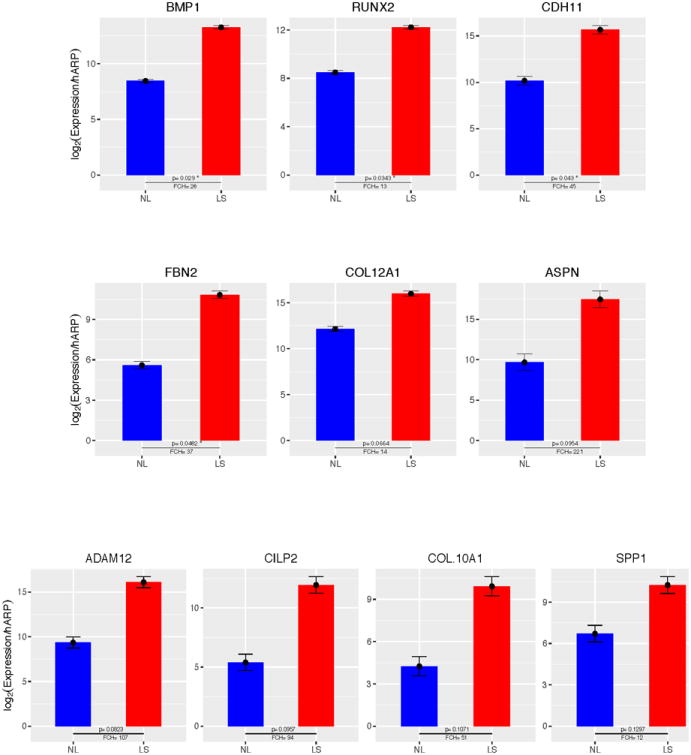
qRT-PCR analysis confirmed significant upregulation of BMP1, RUNX2, CDH11, FBN2 (p<0.05) and COL12A1, ASPN, ADAM12, CILP2 (p<0.1) in chronic keloid lesion compared to non-lesional skin. Shown are average normalized Log expression values for non-lesional(NL) and lesional(LS) (n=3). Error bars represent standard errors of the mean and p values are as indicated.
Increased protein expression of chondrogenic genes in chronic and new keloid lesion
To confirm the protein expression of selected up-regulated genes associated with bone and cartilage formation, we performed immunohistochemistry staining of ASPN, CDH11, BMP1 and RUNX2 on non-lesional, chronic, and newly formed keloid tissue. We observed a strong expression of ASPN, CDH11, and BMP1 in the epidermis of non-lesional, chronic and newly formed keloid tissue (Figure 3). There was increased expression of Asporin and Cadherin in some dermal and endothelial cells of chronic and new keloid lesion compared to non-lesional skin (Figure 3a and 3b). BMP1 was also expressed strongly in some dermal cells of lesional skin compared to non-lesional and new keloid lesion (Figure 3c). RUNX2 showed moderate expression in the epidermis of non-lesional, chronic, and newly formed keloid tissue, but had increased expression of distinct dermal cells in chronic keloid and newly formed keloid compared to nonlesional skin (Figure 3d).
Figure 3.

Representative immunohistochemistry images show increased expression of (a) Asporin and (b) Cadherin 11 in dermal cells of chronic and new keloid lesion compared to non-lesional skin. There is also increased expression of (c) BMP1 and (d) RUNX2 in dermal cells of chronic and new keloid compared to non-lesional skin. (e) Representative two-color immunofluorescence images show many COL10A1+ cells contained RUNX2+ cells (yellow) in chronic and new keloid lesion and a few in non-lesional skin. Size bar = 100 um
We also performed two-color immunofluorescence for RUNX2 and COL10A1 to determine co-expression of this transcription factor with the cells producing articular cartilage (Type 10A collagen). Our results showed many COL10A1+ cells contained RUNX2+ nuclei (white arrows) in the dermis of chronic keloid and new keloid lesion compared to non-lesional skin (Figure 3e).
Discussion
Keloids have previously been characterized by an abundance of disorganized type I and III collagen bundles in the extracellular matrix (ECM)6. Through proliferation and production of collagens I/III, dermal fibroblasts have been proposed as the main cellular element implicated in the pathogenesis of keloid14,15. Syed et al observed that keloid fibroblasts from the growing margin of keloid have elevated production of collagen I and III compared with other lesional sites16. Overexpression of TGF-β isoforms and TGF-β receptors has been detected in keloids and keloid-derived fibroblasts, which suggest that excess fibroplasia is regulated by TGF-β cytokines2.
The keloids we profiled in this study were large and hard exophytic growths, corresponding to Stages II-IV in a recent classification scheme5. All lesions had a prominent dermal cellular component of spindle-shaped cells, but also acellular areas with enlarged collagen bundles. A small fraction of the spindle-shaped cells stained for Ki67, indicating ongoing proliferative activation, whereas Ki67+ dermal cells were not common in non-lesional skin biopsies (data not shown). These lesions fit described diagnostic criteria for keloids by histopathology, but they are more cellular than many others described in the literature2. However, cells are not highly proliferative as might be seen in a fibrosarcoma. In molecular terms, our chronic keloids had mean up-regulation of collagen I by 31-fold and collagen III by 22-fold vs. non-lesional controls, along with an increase of TGFβ3 by 9-fold (Supplemental Table S1), fitting with the general concept of excess production of expected dermal collagens. We also detected a marked increase (44-fold) in IGF-2 (somatomedin a) mRNA and a 12-fold increase in IGF-1 (somatomedin c), which are key growth factors for connective tissue and epithelial cells, both acting through the IGF-1 receptor (Table 1a).
While these are impressive changes, the scope of gene alterations and the magnitude of change were even greater for connective tissue molecules not normally associated with dermal wound healing. These include: (1) Collagen 10A1, a short-chain minor collagen of cartilage formation17 that is associated with events in the later stages of endochondral bone formation; (2) Asporin, also known as periodontal ligament-associated protein 1 (PLAP1) belongs to a family of leucine-rich repeat (LRR) proteins associated with the cartilage matrix18; (3) Cadherin 11, a cell surface glycoprotein that mediate Ca+2 dependent cell-cell adhesion. Expression of this gene in the osteoblastic cell line was up-regulated during differentiation, suggesting a specific function in bone cell differentiation and bone formation19; (4) Bone morphogenic protein 1 (BMP1), this locus encodes a protein that is capable of inducing formation of cartilage in vivo20; (5) Secreted phosphoprotein 1 (SPP1), which belongs to the small integrin-binding ligand N-linked glycoprotein (SIBLING) family of secreted phosphoproteins, are involved in the anchoring of osteoclasts to the mineral of bone matrix21, (6) Runt-related transcription factor 2 (RUNX2), which is expressed in adult bone marrow, thymus, and peripheral lymphoid organs22, has a primary role in the differentiation of osteoblasts23 and hypertrophy of cartilage at the growth plate, cell migration, and vascular invasion of bone. In chondrogenic and osteoblastic cell lines, RUNX2 has been demonstrated to transactivate Type 1 Collagen and Type X Collagen (Col10A1) 24. Upstream activation of chondrogenic differentiation has been found to be mediated by RUNX225,26.
Thus the molecular fingerprint of cellular activation in keloids is more related to differentiation of cartilage and bone connective tissues than dermal connective tissue and may be mediated by RUNX2 overexpression. The association with IGF-1 and IGF-2 over-expression is intriguing, since the IGF-1 receptor mediates skeletal growth and development as a primary action (IGF-1 and IGF-2 mutations are responsible for growth restriction27). Previous work has identified over-expression of IGF-1 receptors on connective tissue cells in keloid lesions28.
The gene expression changes we observe lead to excess production of bone and cartilage proteins in the extracellular matrix of keloids, as we show by IHC. However, no cartilage or bone differentiation is evident at the level of tissue histopathology. Hence, we consider that keloid lesions represent aberrant differentiation of cutaneous connective tissue towards bone and cartilage cellular lineages, but with incomplete differentiation of activated cells, i.e., dysplasia of immature connective tissue. A prior study of keloids in a Japanese population found over-expression of periostin, Cadherin 11 and COL10A1, among 32 upregulated DEGs10, also pointing to aberrant chondrogenic differentiation. The high expression of RUNX2 is a likely key inducer of this aberrant differentiation program24 and it might be a targetable axis for future therapy.
Supplementary Material
Figure S1. PCA plot and Table of Up-regulated and Down-regulated DEGS for Comparisons between Chronic Lesional, nonlesional, New Keloid and normal skin
Table S1. Table for Comparison between Chronic Keloid lesional and nonlesional skin
Table S2. Table for Comparison between New Keloid lesional and nonlesional skin
Table S3. Table for Comparison between Chronic Keloid lesion and normal skin
Table S4. Table for Comparison between New Keloid lesion and normal skin
Table S5. Table for Comparison between Chronic Keloid and New Keloid lesional skin
Table S6. Table for qRT-PCR Primers and IHC antibodies
Acknowledgments
This research was supported by National Institutes of Health (NIH) grant UL1 RR024143 from the National Center for Research Resources (NCRR) and in part grant # UL1 TR000043 and grant #UL1 TR001866 from the National Center for Advancing Translational Sciences (NCATS), National Institutes of Health (NIH) Clinical and Translational Science Award (CTSA) program. The content of this study is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations Used
- FBN2
Fibrillin 2
- COL10A1
Collagen type X alpha 1, ASPN, Asporin
- CDH11
Cadherin 11
- BMP1
Bone morphogenic protein 1
- SPP1
Secreted phosphoprotein 1
- RUNX2
Runt-related transcription factor 2
Footnotes
Conflict of Interest: The authors state no conflict of interest.
Author Contributions: JGK designed the research study. JFD, KB, MT, JGK wrote the paper. JFD, KB, NK, TC, CQFW, HX, PG, MSW performed the research. MSF, SG analysed the data.
Supporting Information: Additional supporting data may be found in the supplementary information of this article.
References
- 1.Bock O, Schmid-Ott G, Malewski P, Mrowietz U. Quality of life of patients with keloid and hypertrophic scarring. Arch Dermatol Res. 2006 Apr;297(10):433–8. doi: 10.1007/s00403-006-0651-7. [DOI] [PubMed] [Google Scholar]
- 2.Tuan TL, Nichter LS. The molecular basis of keloid and hypertrophic scar formation. Mol Med Today. 1998 Jan;4(1):19–24. doi: 10.1016/S1357-4310(97)80541-2. [DOI] [PubMed] [Google Scholar]
- 3.Smith JC, Boone BE, Opalenik SR, Williams SM, Russell SB. Gene profiling of keloid fibroblasts shows altered expression in multiple fibrosis-associated pathways. J Invest Dermatol. 2008 May;128(5):1298–310. doi: 10.1038/sj.jid.5701149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu WS, Zheng XD, Yao XH, Zhang LF. Clinical and epidemiological analysis of keloids in Chinese patients. Arch Dermatol Res. 2015 Mar;307(2):109–14. doi: 10.1007/s00403-014-1507-1. [DOI] [PubMed] [Google Scholar]
- 5.Tirgan MH. Neck keloids: evaluation of risk factors and recommendations for keloid staging system [version 1; referees: 1 approved with reservations] F1000Research. 2016 Jun;5:1528. doi: 10.12688/f1000research.9086.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gauglitz GG, Korting HC, Pavicic T, Ruzicka T, Jeschke MG. Hypertrophic scarring and keloids: pathomechanisms and current and emerging treatment strategies. Mol Med. 2011 Jan-Feb;17(1-2):113–25. doi: 10.2119/molmed.2009.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Funayama E, Chodon T, Oyama A, Sugihara T. Keratinocytes promote proliferation and inhibit apoptosis of the underlying fibroblasts: an important role in the pathogenesis of keloid. J Invest Dermatol. 2003 Dec;121(6):1326–31. doi: 10.1111/j.1523-1747.2003.12572.x. [DOI] [PubMed] [Google Scholar]
- 8.Shih B, McGrouther DA, Bayat A. Identification of novel keloid biomarkers through profiling of tissue biopsies versus cell cultures in keloid margin specimens compared to adjacent normal skin. Eplasty. 2010 Apr;10:e24. [PMC free article] [PubMed] [Google Scholar]
- 9.Nassiri M, Woolery-Lloyd H, Ramos S, Jacob SE, Gugic D, Viciana A, Romanelli P, Elgart G, Berman B, Vincek V. Gene expression profiling reveals alteration of caspase 6 and 14 transcripts in normal skin of keloid-prone patients. Arch Dermatol Res. 2009 Feb;301(2):183–8. doi: 10.1007/s00403-008-0880-z. [DOI] [PubMed] [Google Scholar]
- 10.Naitoh M, Kubota H, Ikeda M, Tanaka T, Shirane H, Suzuki S, Nagata K. Gene expression in human keloids is altered from dermal to chondrocytic and osteogenic lineage. Genes Cells. 2005 Nov;10(11):1081–91. doi: 10.1111/j.1365-2443.2005.00902.x. [DOI] [PubMed] [Google Scholar]
- 11.Na GY, Seo SK, Lee SJ, Kim DW, Kim MK, Kim JC. Upregulation of the NNP-1 (novel nuclear protein-1, D21S2056E) gene in keloid tissue determined by cDNA microarray and in situ hybridization. Br J Dermatol. 2004 Dec;151(6):1143–9. doi: 10.1111/j.1365-2133.2004.06284.x. [DOI] [PubMed] [Google Scholar]
- 12.Fuentes-Duculan J, Suárez-Fariñas M, Zaba LC, Nograles KE, Pierson KC, Mitsui H, Pensabene CA, Kzhyshkowska J, Krueger JG, Lowes MA. A subpopulation of CD13-positive macrophages is classically activated in psoriasis. J Invest Dermatol. 2010 Oct;130(10):2412–22. doi: 10.1038/jid.2010.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suárez-Fariñas M, Pellegrino M, Wittkowski KM, Magnasco MO. Harhslight: a “corrective make-up” program for microarray chips. BMC Bioinformatics. 2005 Dec;6:294. doi: 10.1186/1471-2105-6-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ashcroft KJ, Syed F, Bayat A. Site-Specific Keloid Fibroblasts Alter the Behaviour of Normal Skin and Normal Scar Fibroblasts through Paracrine Signalling. PLoS One. 2013 Dec;8(12):e75600. doi: 10.1371/journal.pone.0075600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Uitto J, Perejda AJ, Abergel RP, Chu ML, Ramirez F. Altered steady-state ratio of type I/III procollagen mRNAs correlates with selectively increased type I procollagen biosynthesis in cultured keloid fibroblasts. Proc Natl Acad Sci U S A. 1985 Sep;82(17):5935–9. doi: 10.1073/pnas.82.17.5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Syed F, Ahmadi E, Iqbal SA, Singh S, McGrouther DA, Bayat A. Fibroblasts from the growing margin of keloid scars produce higher levels of collagen I and III compared with intralesional and extralesional sites: clinical implications for lesional site-directed therapy. Br J Dermatol. 2011 Jan;164(1):83–96. doi: 10.1111/j.1365-2133.2010.10048.x. [DOI] [PubMed] [Google Scholar]
- 17.Schmid TM, Linsenmayer TF. Immunohistochemical localization of short chain cartilage collagen (type X) in avian tissues. J Cell Biol. 1985 Feb;100(2):598–605. doi: 10.1083/jcb.100.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lorenzo P, Aspberg A, Onnerfjord P, Bayliss MT, Neame PJ, Heinegard D. Identification and characterization of asporin. a novel member of the leucine-rich repeat protein family closely related to decorin and biglycan. J Biol Chem. 2001 Apr;276(15):12201–11. doi: 10.1074/jbc.M010932200. [DOI] [PubMed] [Google Scholar]
- 19.Okazaki M, Takeshita S, Kawai S, Kikuno R, Tsujimura A, Kudo A, Amann E. Molecular cloning and characterization of OB-cadherin, a new member of cadherin family expressed in osteoblasts. J Biol Chem. 1994 Apr;269(16):12092–8. [PubMed] [Google Scholar]
- 20.Wozney JM, Rosen V, Celeste AJ, Mitsock LM, Whitters MJ, Kriz RW, Hewick RM, Wang EA. Novel regulators of bone formation: molecular clones and activities. Science. 1988 Dec;242(4885):1528–34. doi: 10.1126/science.3201241. [DOI] [PubMed] [Google Scholar]
- 21.Reinholt FP, Hultenby K, Oldberg A, Heinegård D. Osteopontin – a possible anchor of osteoclasts to bone. Proc Natl Acad Sci USA. 1990 Jun;87(12):4473–5. doi: 10.1073/pnas.87.12.4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cohen MM., Jr Perspectives on RUNX genes: an update. Am J Med Genet A. 2009 Dec;149A(12):2629–46. doi: 10.1002/ajmg.a.33021. [DOI] [PubMed] [Google Scholar]
- 23.Komori T. Regulation of osteoblast differentiation by RUNX2. Adv Exp Med Biol. 2010;658:43–9. doi: 10.1007/978-1-4419-1050-9_5. [DOI] [PubMed] [Google Scholar]
- 24.Zheng Q, Zhou G, Morello R, Chen Y, Garcia-Rojas X, Lee B. Type X collagen gene regulation by Runx2 contributes directly to its hypertrophic chondrocyte-specific expression in vivo. J Cell Biol. 2003 Sep;162(5):833–42. doi: 10.1083/jcb.200211089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dalagiorgou G, Piperi C, Adamopoulos C, Georgopoulou U, Gargalionis AN, Spyropoulou A, Zoi I, Nokhbehsaim M, Damanaki A, Deschner J, Basdra EK, Papavassiliou AG. Mechanosensor polycystin-1 potentiates differentiation of human osteoblastic cells by upregulating Runx2 expression via induction of JAK2/STAT3 signaling axis. Cell Mol Life Sci. 2016 Oct 3; doi: 10.1007/s00018-016-2394-8. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsumoto Y, La Rose J, Kent OA, Wagner MJ, Narimatsu M, Levy AD, Omar MH, Tong J, Krieger JR, Riggs E, Storozhuk Y, Pasquale J, Ventura M, Yeganeh B, Post M, Moran MF, Grynpas MD, Wrana JL, Superti-Furga G, Koleske AJ, Pendergast AM, Rottapel R. Reciprocal stabilization of ABL and TAZ regulates osteoblastogenesis through transcription factor RUNX2. J Clin Invest. 2016 Oct 31; doi: 10.1172/JCI87802. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Begemann M, Zirn B, Santen G, Wirthgen E, Soellner L, Büttel HM, Schweizer R, van Workum W, Binder G, Eggerman T. Paternally inherited IGF2 mutation and growth restriction. N Engl J Med. 2015 Jul;373(4):349–56. doi: 10.1056/NEJMoa1415227. [DOI] [PubMed] [Google Scholar]
- 28.Hu ZC, Tang B, Guo D, Zhang J, Liang YY, Ma D, Zhu JY. Expression of insulin-like growth factor-1 receptor in keloid and hypertrophic scar. Clin Exp Dermatol. 2014 Oct;39(7):822–8. doi: 10.1111/ced.12407. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. PCA plot and Table of Up-regulated and Down-regulated DEGS for Comparisons between Chronic Lesional, nonlesional, New Keloid and normal skin
Table S1. Table for Comparison between Chronic Keloid lesional and nonlesional skin
Table S2. Table for Comparison between New Keloid lesional and nonlesional skin
Table S3. Table for Comparison between Chronic Keloid lesion and normal skin
Table S4. Table for Comparison between New Keloid lesion and normal skin
Table S5. Table for Comparison between Chronic Keloid and New Keloid lesional skin
Table S6. Table for qRT-PCR Primers and IHC antibodies