

Abstract
The sweetpotato whitefly Bemisia tabaci is a highly destructive agricultural and ornamental crop pest. It damages host plants through both phloem feeding and vectoring plant pathogens. Introductions of B. tabaci are difficult to quarantine and eradicate because of its high reproductive rates, broad host plant range, and insecticide resistance. A total of 791 Gb of raw DNA sequence from whole genome shotgun sequencing, and 13 BAC pooling libraries were generated by Illumina sequencing using different combinations of mate-pair and pair-end libraries. Assembly gave a final genome with a scaffold N50 of 437 kb, and a total length of 658 Mb. Annotation of repetitive elements and coding regions resulted in 265.0 Mb TEs (40.3%) and 20 786 protein-coding genes with putative gene family expansions, respectively. Phylogenetic analysis based on orthologs across 14 arthropod taxa suggested that MED/Q is clustered into a hemipteran clade containing A. pisum and is a sister lineage to a clade containing both R. prolixus and N. lugens. Genome completeness, as estimated using the CEGMA and Benchmarking Universal Single-Copy Orthologs pipelines, reached 96% and 79%. These MED/Q genomic resources lay a foundation for future ‘pan-genomic’ comparisons of invasive vs. noninvasive, invasive vs. invasive, and native vs. exotic Bemisia, which, in return, will open up new avenues of investigation into whitefly biology, evolution, and management.
Keywords: Whitefly Bemisia tabaci, Genomics, Assembly, Annotation
Introduction
Samples and libraries construction
As a globally invasive species, the phloem-feeding whitefly Bemisia tabaci (Genn.; hereafter ‘Bemisia’) has been found on all continents except Antarctica [1,2]. Taxonomically, B. tabaci is considered a species complex that contains several morphologically indistinguishable but genetically distinct ‘cryptic species’ [2–7]. The Bemisia Middle East-Asia Minor 1 (MEAM1, or ‘B’) cryptic species is highly invasive and has emerged as a major pest in the United States, Caribbean Basin, Latin America, Middle East [1], and East Asia [8]. Similarly, the invasive Bemisia Mediterranean (MED, or ‘Q’) cryptic species has been introduced into several geographic locations and has become established throughout China [9,10]. Despite substantial research and the recently published whitefly B. tabaci MEAM1/B genome [11], however, the genetic or genomic basis of MED/Q remains obscure.
The MED/Q B. tabaci adult whitefly females (2n) and males (1n) were initially collected from infested field-grown cucumber plants in Beijing, China during 2011 and used to establish a laboratory colony (MED/Q) at the Institute of Vegetable and Flowers, Chinese Academy of Agriculture Science by transferring adult males and females to caged pepper plants (10–12 leaf stage). Results of mtCOI gene PCR-RFLP assays [12] and direct DNA sequencing followed by phylogenetic evaluation against reference sequences [13] both confirmed that the Bemisia in the MED/Q colony belonged to the Q1 haplotype group, or western Mediterranean region clade (data not shown).
The MED/Q whitefly colony was used as the source initial short shotgun Illumina sequencing. Adult whiteflies fed using Parafilm membrane sachets containing a 25% sucrose solution for 48 hours prior to collection of ∼5000 male and female adults (∼50:50). Samples were immediately frozen in liquid nitrogen for 3 hours prior to transfer to a −80°C freezer. This genomic DNA was used to construct Illumina TruSeq paired end (PE) sequencing libraries (170-, 250-, 300-, 500-, and 800-bp insert sizes) and mate pair (MP) libraries (2, 5, 10, 20, and 40 kb in size) according to the manufacturer's instructions. Additionally, two Illumina PE sequencing libraries (∼500-bp and 800-bp inserts) were constructed from whole genome amplification (WGA) reactions carried out on genomic DNA isolated from two adult male whiteflies. We also constructed 13 BAC libraries with pooling of clones and Illumina library construction according to the manufacturer's instructions.
Genome sequencing and assembly
All libraries were sequenced on an Illumina Hiseq 2000 using 100-bp reads from both fragment ends, and raw data processed and assembled as shown (Supplemental Table S1; Supplemental Fig. S1). Briefly, a series of filtering steps was performed on the raw reads to filter out the following: (1) reads with >10% Ns, >40% low-quality bases, >10 bp overlapping with adapter sequences, allowing no more than 3-bp mismatches; (2) paired-end reads that overlapped >10 bp between two ends, with insert size >200-bp libraries; and (3) duplicated reads generated by PCR amplification during the construction of the large-insert library. Filtered reads were used for K-mer determination within subsequent assembly steps. The frequency of each K-mer was calculated from the genome-sequence reads. K-mer frequencies along the sequence depth gradient follow a Poisson distribution in a given data set except for a high proportion at low frequency due to sequencing errors, as K-mers that contain such sequencing errors may be orphans among all splitting K-mers. The genome size, G, was estimated as G = K_num/K_depth, where K_num is the total number of K-mers and K_depth is the maximal frequency. Initial contigs were assembled from filtered 500- and 800-bp insert-size WGA PE libraries using SOAPdenovo. The sequencing reads obtained for 2-k to 40-kb MP libraries were used to connect the contigs and to generate the scaffolds as described by Li et al. (2010) [14] with a K-mer size of 65.
Individual BAC pools were assembled independently using SOAPdenovo and the whole genome shotgun reads from PE and MP libraries were used to fill gaps in the BAC scaffolds. After sequencing, the raw reads were filtered as described above. In addition, reads representing contamination by Escherichia coli or the plasmid vector were filtered. The pooled reads were separated according to the BAC-reads index, and each BAC was assembled using a combination of “hierarchical assembly” and “de Bruijn graph assembly.” First, the reads linked to each BAC were assembled using SOAPdenovo [14], with various combinations of parameters with a K-mer range from 27 to 63 and a step size of 6. The assembly with the longest scaffold N50 was defined as the “best” for each BAC. The resulting BACs were mapped with the large shotgun MP read data to optimize the assembly for each BAC.
The final draft assembly was produced by integrating sequences that overlapped among the scaffolds independently assembled from genome shotgun and BAC reads, and in doing so eliminated the redundant scaffolds using the following steps. To integrate the two assemblies, the software Rabbit [15] was applied to identify any relationship between scaffolds, to connect the overlapping regions that shared at least 90% similarity, and to remove redundancy based on a 17-mer frequency. Finally, SSPACE [16] was used to construct super-scaffolds containing 800-bp to 40-kb whole genome sequence (WGS) reads, and the 170- to 800-bp genome shotgun read data were used to fill the gaps using GapCloser [14]. Postassembly processing included removal of contaminating bacterial and viral DNA sequences by aligning all assembled sequences to the genome sequences of viruses and bacteria, obtained from previous local BLASTn alignments and by NCBI upload filter. Aligned sequences that shared >90% identity and were >200 bp in size were filtered from the final assembly. The assembled sequences that were covered by at least one expressed sequence tag (EST) sequence were retained. Process read data were mapped to the draft MED/Q genome using SOAPaligner software and read counts were made from .bam files and the average depth was computed from all bases in the window. The relation graph of base pair percentages, and each given sequencing depth along the genome, was obtained.
Using genomic DNA from the MED/Q colony, a total of 20 WGS shotgun sequencing libraries was generated (18 pooled male and female PE and MP libraries, and two haploid male-derived WGA PE libraries), from which sequences were generated on an Illumina Hiseq2500 platform. Library sequencing produced a total of 428.2 Gb or an approximate 594.7-fold genome coverage assuming a 0.72-Gbp genome size (based on 17-mer analysis). For the 10 short-insert PE libraries, there were a total of 229.4 Gb (100-bp or 150-bp read length, approximately 318.6-fold genome coverage). Sequencing the eight large-insert (>1 kb) MP libraries produced 80.3 Gb of reads (49 bp read length, 111.5-fold coverage) for use in scaffold construction (Supplemental Table S1). The two male WGA libraries produced a total of 118.5 Gb of data (Supplemental Table S1) or approximately 164.6-fold genome coverage. Sequencing of 13 BAC pools generated 362.6 Gbp of raw data (288.4 Gbp processed data; results not shown). The subsequent assembly of this sequence data using our pipeline (Supplemental Fig. S1) generated a 658-Mbp draft genome assembly for MED/Q consistent with recent flow cytometry estimates [17]. The mean read depth across 10-kb windows indicated that all genome regions were highly represented within the read data, with <1.5% having a depth of <10× (remaining data not shown).
Through statistical comparison of genome assembly and annotation between MED/Q and MEAM1/B (Table 1), we found the draft genome of MED/Q consisted of a genome size of 658 Mb with contig N50 size 44 kb, while MEAM1/B assembly was 615 Mb with contig N50 of 30 kb. They have similar G+C content of about 39%, while higher TEs existed in MEAM1/B (44%) than MED/Q (40%). After combining several annotation methods, 20 748 genes were predicted in MED/Q, whereas 15 664 genes in MEAM1/B, and about 80% of both two gene sets were supported by several public functional databases.
Table 1:
Statistics comparison of genome assembly and annotation between MED/Q and MEAM1/B
| MED/Qa | MEAM1/Bb | |||
|---|---|---|---|---|
| Sequencing summary | Scaffoldc | Contigc | Scaffoldc | Contigc |
| Total number | 4954 | 29 618 | 19 761 | 52 036 |
| Total length of (bp) | 658 272 463 | 638 061 971 | 615 029 878 | 599 923 598 |
| Gap number (bp) | 19 828 575 | 0 | 14 380 491 | 0 |
| Average length (bp) | 132 877 | 21 543 | 31 123 | 11 529 |
| N50 length (bp) | 436 791 | 44 366 | 3 232 964 | 29 918 |
| N90 length (bp) | 111 835 | 11 504 | 381 346 | 6117 |
| Maximum length (bp) | 2 857 362 | 362 835 | 11 178 615 | 269 706 |
| Minimum length (bp) | 501 | 500 | 500 | 500 |
| GC content (%) | 39.46 | 39.46 | 39.64 | 39.64 |
| TEs proportion (%) | 265 Mb (0.40) | 269 Mb (0.44) | ||
| CEGMA evaluation (%) | 96 | 100 | ||
| BUSCO evaluation | 78 | 96.8 | ||
| Gene number | 20 786 | 15 664 | ||
| Average gene length (bp) | 10 065 | 22 762 | ||
| Average CDS length (bp) | 1952 | 1470 | ||
| Average exon per gene | 6 | 6 | ||
| Average exon length (bp) | 351 | 234 | ||
| Average intron length (bp) | 1776 | 3125 | ||
| Annotation gene (%) | 79.97 | 81 | ||
| Assemble software | SOAPdenovo | Platanus | ||
From this study.
From the published MEAM1/B genome [11].
Only contigs and scaffolds ≧500 bp were included in the genome assembly.
Annotation of repetitive elements
Repetitive elements were searched for and identified using Repbase [18] implemented in TRF software [19], and a de novo approach implemented in Piler [20]. For the Repbase-based method, two software programs named RepeatMasker [21] and RepeatProteinMask were used to identify repetitive sequences. In the de novo approach, Piler-DF-1.0 [20], RepeatScout-1.0.5 [22], and LTR-FINDER-1.0.5 [23] were used to build de novo repeat libraries from the genome sequences. Finally, the repeated sequences were searched for and classified using the RepeatMasker software. Homology-based annotation of MED/Q repetitive elements was queried against Repbase v.20.05 [18] with RepeatMasker [21]. We found a total of 265.0 Mb TEs, or 40.3% of the MED/Q genome size. This was about 10% higher than the repeat contents of Acyrthosiphon pisum and Rhodnius prolixus, but similar to that of Nilaparvata lugens (39.8%) (Supplemental Table S2). This suggests that long terminal repeat (LTR) (18.5%) are more abundant and contain more nucleotides than all other TE classes. This proliferation of LTR retrotransposons has been found in only one other Hemipteran genome, that of N. lugens (12.29%). The MED/Q genome also contains the high proportion of the DNA-transposon TEs (12.92%) found in other fully described Hemipteran genomes. As with both N. lugens (0.5%) and R. prolixus (0.01%), the MED/Q genome also appears devoid of short interspersed nuclear elements (0.96 %). These other Hemipteran genomes also contain a small amount of long interspersed nuclear elements (A. pisum: 2.6%; MED/Q: 3.18%; R. prolixus: 3.2%), but N. lugens (12.84%). This suggests that MED/Q-specific TEs, especially the LTRs, have evolved relatively recently and contribute to the large number of gene sets.
Annotation of coding regions
Initial evaluation of the gene coverage rate in the draft MED/Q genome assembly was assessed by comparing against 248 core eukaryotic genes obtained using CEGMA 2.4 [24] and Benchmarking Universal Single-Copy Orthologs (BUSCO) [25]. Additionally, 105 067 B. tabaci transcript sequences, ESTs, of >200 bp were used as BLASTn queries against the assembled genome to estimate the representation (cutoff E-value ≥ 10−40). Protein-coding gene de novo predictions using GENEWISE [26] and ab initio gene predictions using GENSCAN [27] and AUGUSTUS [28] were made in combination with 13.7 Gbp of transcriptome (RNA-Seq) data including published MED/Q B. tabaci body, guts, and salivary glands [29–31] and additional, previously unpublished data from females and males [32], to obtain consensus gene sets using GLEAN [33].
For homolog-based prediction, protein sequences from nine species (A. pisum, A. mellifera, D. melanogaster, R. prolixus, Z. nevadensis, A. gambiae, B. mori, P. humanus, and T. castaneum) were aligned with the MED/Q genome scaffolds using TblastN (E-value <1e-5). Target sequences were used to search for accurate gene structures implementing the GeneWise software [26]. For the RNA-Seq datasets, the transcriptome reads were first aligned against the genome using TopHat [33] to identify candidate exon regions. Then, the Cufflinks software [34] was used to assemble the aligned reads into transcripts, and the open reading frames were predicted to obtain reliable transcripts using a Hidden Markov Model-based training parameter. Finally, GLEAN [33] was used to integrate the predicted genes with the de novo, homologous, and RNAseq data to produce the final gene set. The functional annotation of genes was performed using BLASTP alignment to KEGG [35], SwissProt, and TrEMBL [36] databases. Motifs and domains were determined by InterProScan [37] and protein database searches against ProDom, PRINTS, Pfam, SMART, PANTHER, and PROSITE.
Preliminary evaluation of transcribed regions within the draft MED/Q genome assembly coverage found that ∼95.2% of B. tabaci ESTs > 200 bp were present, with 90 652 ESTs showing ≥90% length coverage on one scaffold (Supplemental Table S7). This alignment encompassed 92.9% of nucleotides within the EST dataset. Analogously, 229 (96%) of the 248 sequences in the CEGMA gene set and 79% complete and fragmented BUSCOs were present in the MED/Q genome assembly (remaining data not shown). The final GLEAN gene models predicted a reference gene set of 20 786 protein-coding genes, a consensus result derived from de novo, orthology, and evidence (RNA-seq)-based prediction methods (Supplemental Table S3) and integrated into GLEAN gene models (Supplemental Table S4). Among the GLEAN gene models, 16 622 (79.97%) received functional gene annotations using the various databases queried in our analysis pipeline (Supplemental Table S5).
Prediction of gene orthology
Twelve insect species including B. tabaci (Genn.) (Gennadius, 1889) (Hemiptera: Aleyrodidae), Acyrthosiphon pisum (Harris, 1776) (Hemiptera: Aphididae), Rhodnius prolixus (Stal, 1859) (Hemiptera: Triatominae), Nilaparvata lugens (Stål, 1854) (Hemiptera: Delphacidae), Pediculus humanus (Linnaeus, 1758) (Phthiraptera: Pediculidae), Apis mellifera (Linnaeus, 1758) (Hymenoptera, Apidae), Nasonia vitripennis (Ashmead, 1904) (Hymenoptera, Pteromalidae), Tribolium castaneum (Herbst, 1797) (Coleoptera, Tenebrionidae), Anopheles gambiae (Giles, 1902) (Diptera, Culicidae), Drosophila melanogaster (Meigen, 1830) (Diptera, Drosophilidae), Bombyx mori (Linnaeus, 1758) (Lepidoptera, Bombycidae) and Danaus plexippus (Kluk, 1802) (Lepidoptera, Nymphalidae), and two divergent arthropods, Daphnia pulex (Müller, 1785) (O. Cladocera, Daphniidae) and Tetranychus urticae (C. L. Koch, 1836) (O. Arachnida, Tetranychidae), were used to predict orthologs and to reconstruct the phylogenetic tree. Gene families were identified using TreeFam [38,39], and single-copy gene families were assembled to reconstruct phylogenetic relationships. Coding sequences of each single-copy family were concatenated to form one super gene group for each species. All of the nucleotides at codon position 2 of these concatenated genes were extracted to construct the phylogenetic tree by PhyML [40], with a gamma distribution across sites and an HKY85 substitution model. The same set of sequences at codon position 2 was used to estimate divergence times among lineages. The fossil calibrations were set with two previous node data [41,42]. The PAML mcmctree program (v.4.5) [43,44] was used to compute split times using the approximate likelihood calculation algorithm. The software Tracer (v.1.5.0) was utilized to examine the extent of convergence for two independent runs.
Phylogenetic analysis based on orthologs across 14 arthropod taxa (Supplemental Table S6) suggested that MED/Q is clustered into a hemipteran clade containing A. pisum and is a sister lineage to a clade containing both R. prolixus and N. lugens (Fig. 1A). The range of species-specific genes within the four hemipteran genomes ranged from 38% to 60%, with higher values for the three phloem-feeding specialists. This led us to investigate interspecific changes in the number and diversity of gene family members (orthologs and paralogs) within this group of Hemiptera (Fig. 1C; Supplemental Fig. S2).
Figure 1:

Phylogenetic relationships and genomic comparisons between Bemisia tabaci and other insect species (A) Phylogenetic relationships of B. tabaci (BEMTA) to insects and other arthropods based on single-copy orthologous genes present in their complete genomes. The following 12 insect species were used for this analysis: Acyrthosiphon pisum (ACYPI), Anopheles gambiae (ANOGA), Apis mellifera (APIME), BEMTA, Bombyx mori (BOMMO), Danaus plexippus (DANPL), Drosophila melanogaster (DROME), Nasonia vitripennis (NASVI), Nilaparvata lugens (NILLU), Pediculus humanus (PEDHU), Rhodnius prolixus (RHOPR), and Tribolium castaneum (TRICA). The two arthropods Daphnia pulex (DAPPU) and Tetranychus urticae (TETUR) were used as outgroup taxa. Branch lengths represent divergence times estimated for the second codon position of 308 single-copy genes, using PhyML with a gamma distribution across sites and a HKY85 substitution model. The branch supports were inferred based on the approximate likelihood ratio test (aLRT). Gene orthology was determined by comparing the genomes of these 14 arthropod species. The use of 1:1:1 refers to single-copy gene orthologs found across all 14 lineages. The use of N:N:N refers to multi-copy gene paralogs found across the 14 lineages. Diptera, Hemiptera, Hymenoptera, Lepidoptera, and Insecta refer to taxon-specific genes present only in the particular lineage. SD indicates species-specific duplicated genes, and ND indicates species-specific unclustered genes. (B) Image of adult MED/Q. (C) A Venn diagram showing the orthologous groups shared among the hemipteran genomes of A. pisum, B. tabaci, N. lugens, and R. prolixus. Our analysis found 3341 gene families common to all four hemipteran genomes, and 2921 common to the genomes of the six vascular (blood and phloem) feeders.
In summary, we report the first genome sequencing, assembly, and annotation of the MEQ/Q B. tabaci. This genome assembly will provide a valuable resource for studying climatic and host plant adaptations, invasive-invasive and native-exotic interactions, insecticide resistance, vector competence, and its relationships with bacterial endosymbionts.
Availability of supporting data
This whole genome shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession LIED00000000. The version described in this paper is version LIED01000000 accessible at NCBI. Further data, including annotation files and assembled transcripts, are available in the GigaScience GigaDB repository [32].
Additional files
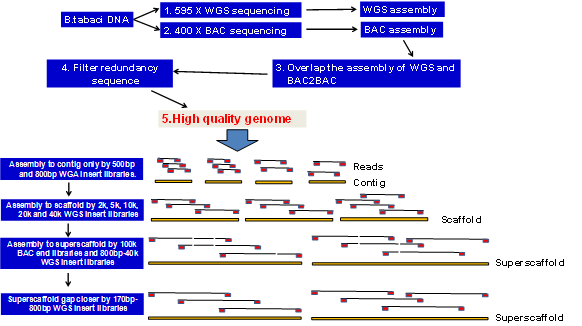
Figure S1. Schematic illustration of the assembly pipeline for MED/Q genome based on the combined assemblies from WGS and BACs.
Table S1. Statistics of the whole genome sequencing data.
Table S2. Repeat Masker analysis in four hemiptera species.
Table S3. Evidenced use within GLEAN MED/Q protein-coding genes.
Table S4. Summary of GLEAN gene models.
Table S5. Functional annotation of the MED/Q genome.
Table S6. Orthologous gene comparison among genomes of 14 arthropod species.
Table S7. Quality control of assembled genome.
Abbreviations
BAC: Bacterial artificial chromosome; BUSCO: Benchmarking Universal Single-Copy Orthologs; CEGMA: Core Eukaryotic Genes Mapping Approach; EST: Express sequence tag; HMW: high molecular weight; MED/Q: Mediterranean Bemisia tabaci Q; mtCOI: mitochondria cytochrome oxidase I; TEs: transposable elements; WGA: whole-genome amplified; WGS: whole genome shotgun.
Author contributions
YJZ is the leader of the project and the first corresponding author. WX, YJZ, XGZ, YY, JKB, and YL were involved in the project design. XGZ, BYX, JYZ, QG, XCL, XQT, MG, HPP, SXR, and BLQ coordinated the related research works of the MED/Q genome project. DW performed genome assembly. DW performed protein-coding gene annotation. MC and CHC performed gene orthology and phylogenomics. XY performed insecticide targets annotation. YTL performed putative sex determination genes annotation. WX performed putative phloem specialization genes identification. LTG, LXT, YNW, YZ, QJW, SLW, and HYC performed metabolic detoxification systems annotation. ZZY performed immune signaling pathway components annotation. ZZY, JQX, and JQH performed nutrient partitioning between invasive MED/Q and its primary endosymbiont. LTG performed PCR validation. WX, XGZ, DC, JKB, HD, MNM, FG, XPZ, XWW, FHW, YZD, CL, FMY, ELP, and XGJ were involved in writing and editing. All authors read and approved the final manuscript.
Competing interests
The authors declare no competing interests defined by Giga Science.
Supplementary Material
Schematic illustration of the assembly pipeline for MED/Q genome based on the combined assemblies from WGS and BACs.
{kind=link}
Statistics of the whole genome sequencing data.
Repeat Masker analysis in four hemiptera species.
Evidenced use within GLEAN MED/Q protein-coding genes.
Summary of GLEAN gene models.
Functional annotation of the MED/Q genome.
Orthologous gene comparison among genomes of 14 arthropod species.
Quality control of assembled genome.
Acknowledgements
The authors would like to thank Dr. Paul De Barro for his comments on an earlier draft. This research was supported by the National Natural Science Foundation of China (31420103919 and 31672032), the Science and Technology Innovation Program of the Chinese Academy of Agricultural Sciences (CAAS-ASTIP-IVFCAAS) the China Agriculture Research System (CARS-26-10), Beijing Nova Program (Z171100001117039), Beijing Training Project for the Leading Talents in S & T (LJRC201412) and the Beijing Key Laboratory for Pest Control and Sustainable Cultivation of Vegetables. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Brown JK, Frohlich DR, Rosell RC. The sweetpotato or silverleaf whiteflies: biotypes of Bemisia tabaci or a species complex? Ann Rev Entomol 1995;40:511–34, doi: 10.1146/annurev.en.40.010195.002455. [Google Scholar]
- 2. De Barro PJ, Liu SS, Boykin LM, et al. . Bemisia tabaci: astatement of species status. Ann Rev Entomol 2011;56:1–19, doi: 10.1146/annurev-ento-112408-085504. [DOI] [PubMed] [Google Scholar]
- 3. Liu SS, Colvin J, De Barro P. Species concepts as applied to the whitefly Bemisia tabaci systematics: how many species are there? J Inter Agric 2012;11:176–86, doi: 10.1016/S2095-3119(12)60002-1. [Google Scholar]
- 4. Wang HL, Yang J, Boykin LM et al. . Developing conversed microsatellite markers and their implications in evolutionary analysis of the Bemisia tabaci complex. Sci Rep 2014;4:6351, doi: 10.1038/srep06351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tay WT, Evans GA, Boykin LM, et al. . Will the real Bemisia tabaci please stand up? PLoS One 2012;7:e50550, doi: 10.1371/journal.pone.0050550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boykin LM, Armstrong KF, Kubatko L, et al. . Species delimitation and global biosecurity. Evol Bioinform Online 2012;8:1–37, doi: 10.4137/EBO.S8532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Boykin LM. Bemisia tabaci nomenclature: lessons learned. Pest Manag Sci 2014;70:1454–9, doi: 10.1002/ps.3709. [DOI] [PubMed] [Google Scholar]
- 8. Zhang LP, Zhang YJ, Zhang WJ, et al. . Analysis of genetic diversity among different geographical populations and determination of biotypes of Bemisia tabaci in China. J Appl Entomol 2005;129:121–8, doi: 10.1111/j.1439-0418.2005.00950.x. [Google Scholar]
- 9. Pan HP, Preisser EL, Chu D, et al. . Insecticides promote viral outbreaks by altering herbivore competition. Ecol Appl 2015;25:1585–95, doi: 10.1890/14-0752.1. [DOI] [PubMed] [Google Scholar]
- 10. Liu BM, Yan FM, Chu D, et al. . Multiple forms of vector manipulation by a plant-infecting virus: Bemisia tabaci and tomato yellow leaf curl virus. J Virol 2013;87:4929–37, doi: 10.1128/JVI.03571-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen W, Hasegawa DK, Kaur N, et al. . The draft genome of whitefly Bemisia tabaci MEAM1, a global crop pest, provides novel insights into virus transmission, host adaptation, and insecticide resistance. BMC Biol 2016;14:110, doi 10.1186/s12915-016-0321-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chu D, Hu X, Gao C, et al. . Use of mitochondrial cytochrome oxidase I polymerase chain reaction-restriction fragment length polymorphism for identifying subclades of Bemisia tabaci Mediterranean group. J Econ Entomol 2012;105:242–51, doi: http://dx.doi.org/10.1603/EC11039. [DOI] [PubMed] [Google Scholar]
- 13. Frohlich DR, Torres-Jerez II, Bedford ID, et al. . A phylogeographical analysis of the Bemisia tabaci species complex based on mitochondrial DNA markers. Mol Ecol 1999;8:1683–91, doi: 10.1046/j.1365-294x.1999.00754.x. [DOI] [PubMed] [Google Scholar]
- 14. Li R, Fan W, Tian G, et al. . The sequence and de novo assembly of the giant panda genome. Nature 2010;463:311–7, doi: 10.1038/nature08696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. You M, Yue Z, He W, et al. . A heterozygous moth genome provides insights into herbivory and detoxification. Nat Genet 2013;45:220–25, doi: 10.1038/ng.2524. [DOI] [PubMed] [Google Scholar]
- 16. Boetzer M, Henkel CV, Jansen HJ, et al. . Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011;27:578–9, doi: 10.1093/bioinformatics/btq683. [DOI] [PubMed] [Google Scholar]
- 17. Guo LT, Wang SL, Wu QJ, et al. . Flow cytometry and K-mer analysis estimates of the genome sizes of Bemisia tabaci B and Q (Hemiptera: Aleyrodidae). Front Physiol 2015;6:144, doi: 10.3389/fphys.2015.00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jurka J, Kapitonov VV, Pavlicek A, et al. . Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 2005;110:462–7, doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 19. Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 1999;27:573–80, doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Edgar RC, Myers EW. PILER: identification and classification of genomic repeats. Bioinformatics 2005;21:152–8, doi: 10.1093/bioinformatics/bti1003. [DOI] [PubMed] [Google Scholar]
- 21. Smit AFA, Hubley R, Green P. RepeatMasker. 1999;http://www.repeatmasker.org.
- 22. Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes. Bioinformatics 2005;21:351–8, doi: 10.1093/bioinformatics/bti1018. [DOI] [PubMed] [Google Scholar]
- 23. Xu Z, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res 2007;35:265–8, doi: 10.1093/nar/gkm286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Parra G, Bradnam K, Ning Z, et al. . Assessing the gene space in draft genomes. Nucleic Acids Res 2009;37:289–97, doi: 10.1093/nar/gkn916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Simão FA, Waterhouse RM, Ioannidis P, et al. . BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015;btv351 doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 26. Birney E, Clamp M, Durbin R. GeneWise and Genomewise. Genome Res 2004;14:988–95, doi: 10.1101/gr.1865504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol 1997;268:78–94, doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- 28. Stanke M, Keller O, Gunduz I, et al. . AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 2006;34: W435–9, doi: https://doi.org/10.1093/nar/gkl200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang XW, Luan JB, Li JM, et al. . De novo characterization of a whitefly transcriptome and analysis of its gene expression during development. BMC Genom 2010;11:400, doi: 10.1186/1471-2164-11-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ye XD, Su YL, Zhao QY, et al. . Transcriptomic analyses reveal the adaptive features and biological differences of guts from two invasive whitefly species. BMC Genom 2014;15:370, doi: 10.1186/1471-2164-15-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Su YL, Li JM, Li M, et al. . Transcriptomic analysis of the salivary glands of an invasive whitefly. PLoS One 2012;7:e39303, doi: 10.1371/journal.pone.0039303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xie W, Chen C, Yang Z, et al. . Supporting data for “Genome sequencing of the sweetpotato whitefly Bemisia tabaci MED/Q”. GigaScience Database 2017;http://dx.doi.org/10.5524/100286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Elsik CG, Mackey AJ, Reese JT, et al. . Creating a honeybee consensus gene set. Genome Biol 2007;8:R13, doi: 10.1186/gb-2007-8-1-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Trapnell C, Williams BA, Pertea G, et al. . Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010;28:511–5, doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000;28:27–30, doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bairoch A, Apweiler R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res 2000;28:45–8, doi: 10.1093/nar/28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zdobnov EM, Apweiler R. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 2001;17:847–8, doi:10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
- 38. Li H, Coghlan A, Ruan J, et al. . TreeFam: a curated database of phylogenetic trees of animal gene families. Nucleic Acids Res 2006;34:572–80, doi: 10.1093/nar/gkj118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ruan J, Li H, Chen Z, et al. . TreeFam: 2008 update. Nucleic Acids Res 2008;36:735–40, doi: 10.1093/nar/gkm1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Guindon S, Dufayard JF, Lefort V, et al. . New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 2010;59:307–21, doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 41. Benton MJ, Donoghue PC. Paleontological evidence to date the tree of life. Mol Biol Evol 2007;24:26–53, doi: 10.1093/molbev/msl150. [DOI] [PubMed] [Google Scholar]
- 42. Donoghue PCJ, Benton MJ. Rocks and clocks: calibrating the Tree of Life using fossils and molecules. Trends Ecol Evol 2007;22:424–31, doi: 10.1016/j.tree.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 43. Yang Z. PAML: a program package for phylogenetic analyses by maximum likelihood. Comp Appl BioSci 1997;13:555–6, doi: 10.1099/0022-1317-79-8-1951. [DOI] [PubMed] [Google Scholar]
- 44. Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 2007;24:1586–91, doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Schematic illustration of the assembly pipeline for MED/Q genome based on the combined assemblies from WGS and BACs.
Statistics of the whole genome sequencing data.
Repeat Masker analysis in four hemiptera species.
Evidenced use within GLEAN MED/Q protein-coding genes.
Summary of GLEAN gene models.
Functional annotation of the MED/Q genome.
Orthologous gene comparison among genomes of 14 arthropod species.
Quality control of assembled genome.