

Abstract
AIM
To assess the effect of sodium selenite on the severity of dextran sulfate sodium (DSS)-induced colitis in C57BL/6 mice.
METHODS
Mice were randomly divided into four groups (n = 10/group): normal group, selenium (Se) group, chronic colitis group, and Se + chronic colitis group. The mice were sacrificed on day 26. Survival rates, clinical symptoms, colon length, and histological changes were determined. The percentages and absolute numbers of immune system cells in the lamina propria lymphocytes (LPL) of the colon, the expression of mRNA in colon tissue, and the concentrations of Th1, Th17, and Treg cytokines in LPL from the large intestine, were measured.
RESULTS
Se significantly ameliorated the symptoms of colitis and histological injury (P < 0.05 each), increasing the proportions of neutrophils and CD4+ CD25+ T cells (P < 0.05 each) and decreasing the proportions of γδT cells, CD4+, CD4+CD44+, and CD4+ CD69+ T cells in LPL (P < 0.05 each). Moreover, Se reduced the expression of IL-6, IFN-γ, IL-17A, IL-21, T-bet, and RORγt (P < 0.05 each), but enhanced the expression of IL-10 and Foxp3 (P < 0.05 each).
CONCLUSION
These results suggest that Se protects against DSS-induced chronic colitis perhaps by increasing the number of CD4(+)CD25(+) Tregs that suppress the secretion of proinflammatory cytokines and populations of Th1, Th17, and γδT cells.
Keywords: Sodium selenite, Dextran sulfate sodium, Chronic colitis
Core tip: Se significantly ameliorated the symptoms of colitis and histological injury, increasing the proportions of neutrophils and CD4+ CD25+ T cells and decreasing the proportions of γδT cells, CD4+, CD4+CD44+, and CD4+ CD69+ T cells in LPL. Moreover, Se reduced the expression of IL-6, IFN-γ, IL-17A, IL-21, T-bet, and RORγt, but enhanced the expression of IL-10 and Foxp3. The study suggests that Se protects against DSS-induced chronic colitis perhaps by increasing the number of CD4(+)CD25(+) Tregs that suppress the secretion of proinflammatory cytokines and populations of Th1, Th17, and γδT cells.
INTRODUCTION
Inflammatory bowel disease (IBD) is a chronic, remittent-relapsing intestinal inflammatory condition. It consists of two major forms, ulcerative colitis and Crohn’s disease, which are more common in developed countries than in developing countries. The major symptoms of IBD include abdominal pain, diarrhea, weight loss, and intestinal bleeding. Although the pathogenesis of IBD has not been definitively elucidated, factors including genetic mutations, immunological disorders, environmental exposure, oxidative stress, and intestinal flora have been suggested to be involved in this process[1-5]. In addition, there is sufficient evidence linking IBD to the over-response of the mucosal immune system[6]. T cells are important components of the adaptive immune response. The activation and proliferation of colonic lamina propria (LP) T lymphocytes during immune responses are important in maintaining intestinal immune homeostasis. CD4+ T cells are especially important in regulating intestinal inflammation. CD4+ T cells have been divided into subsets, based on the cytokines they produce; these subsets include Th1, Th2, Th17, and Treg cells. Th1 and Th17 cells are involved in the pathogenesis of IBD[7,8], whereas Treg cells ameliorate intestinal inflammation by suppressing Th17 cells[9].
To date, treatment options for IBD have mainly focused on controlling symptoms by suppressing inflammatory responses. Infliximab, a monoclonal antibody against tumor necrosis factor (TNF)-α, has proven effective in IBD by inducing the apoptosis of mucosal T cells[10,11], suggesting that strategies targeting lamina propria lymphocytes (LPL) in the intestinal mucosa may be effective in treating IBD. However, although immunomodulatory therapies are effective in many patients, many become refractory, suggesting the need to develop new agents to treat IBD.
Selenium (Se) is an important micronutrient for human health and has antioxidative properties. Se helps maintain the catalytic function of selenoproteins that indirectly alleviate oxygen-rich free radicals[4,5]. In addition, Se is necessary for the expression of glutathione peroxidase (GPX)[12] that protects organisms from oxidative damage. Se levels are inversely correlated with cancer risk and IBD[12-14]. Clinical studies have shown that Se[15] and Se protein P[16] contents are significantly lower in patients with IBD than in healthy controls, and that Se supplementation can reduce intestinal symptoms in patients with IBD[17]. Se can also protect against experimental colitis and human ulcerative colitis (UC)[18,19].
Little is known about the mechanism of action of Se in IBD. Activation of NF-κB and AP-1 is important in the pathogenesis of IBD[19,20]. Although Se has been shown to affect NF-κB and AP-1 activation[21-24], the effect of Se on mucosal LPL in the colon remains unclear.
Experimental colitis can be induced in mice by oral administration of dextran sulfate sodium (DSS). The pathogenesis and clinical features of this animal model of IBD resemble those of human UC. This study assessed the effect of sodium selenite on DSS-induced colitis in mice, revealing that sodium selenite pretreatment protected against chronic colitis by reducing the levels of Th1, Th17, and γδT type cytokines and increasing those of Treg cytokines.
MATERIALS AND METHODS
Experimental animals
Eight-week-old male C57BL/6 mice, weighing 20 ± 1 g, were purchased from the Animal Care Facility of China Medical University. The mice were maintained and bred under specific pathogen-free conditions (temperature 24-25 °C, humidity 70%-75%, and a 12-h light/12-h dark lighting regimen). Mice were fed chow, which had a basal selenium content of 0.1 μg/g diet. The study protocol was approved by the Animal Ethics Committee and Animal Care Committee of China Medical University.
Induction of chronic DSS colitis
Mice received oral 1.5% DSS (molecular mass 36-50 kDa; MP Biomedicals, Solon, OH, United States) on days 0-5, 10-15, and 20-25 d and tap water on the other days.
Experimental design
The mice were randomly divided into four groups (n = 10/group): control group, Se group, chronic colitis group, and Se + chronic colitis group. The control group was fed a normal diet (0.1 μg Se/g diet) and tap water + once-daily gavage of 0.2 mL PBS for 25 d. The Se group was fed a normal diet (0.1 μg Se/g diet) and tap water + once-daily gavage of 2 μg Se/g body weight for 25 d. The chronic colitis group was subjected to chronic colitis induction and fed a normal diet (0.1 μg Se/g diet) + once-daily gavage of 0.2 mL PBS for 25 d. The Se + chronic colitis group was subjected to chronic colitis induction and fed a normal diet (0.1 μg Se/g diet) + once-daily gavage of 2 μg Se/g body weight for 25 d. Body weight and disease activity index were observed daily. Each mouse was weighed at the same time daily.
Disease activity index and histopathology
The severity of colitis was assessed using the disease activity index (DAI) based on weight loss, hemoccult or rectal bleeding, and stool consistency; the scores are described in Table 1. After sacrifice, colon tissue was fixed in 4% paraformaldehyde and embedded in paraffin, and sections 4 μm thick were stained with hematoxylin and eosin to evaluate colonic histology, with histological scores determined in a blinded fashion by two independent pathologists (Table 2).
Table 1.
Disease activity index score chart
| Fecal property | Fecal occult blood | Body weight decrease (%) | Integral |
| Normal | Normal | 0 | 0 |
| 1-5 | 1 | ||
| Relaxed | Positive fecal occult blood | > 5-10 | 2 |
| > 10-15 | 3 | ||
| Loose stools | Naked eye fecal occult blood | > 15 | 4 |
Normal stool: shaped stool; Relaxed stool: pasty, unformed stools not attached to the anus; Loose stools: unshaped stools attached to the anus.
Table 2.
Histology injury score chart
| Grade | 0 | 1 | 2 | 3 | 4 |
| Inflammation | None | Mild | Moderate | Severe | - |
| Mucosal damage | None | Mucous layer | Submucosa | Muscularis and serosa | - |
| Crypt damage | None | 1/3 | 2/3 | 100% | 100% with epithelium loss |
| Pathological change range | None | 0%-25% | 26%-50% | 51%-75% | 76%-100% |
Cell preparation, culture, and stimulation
The large intestine of each mouse was cut into 1-2 mm pieces. The pieces were stirred twice for 15 min each in PBS containing 3 mmol/L EDTA and twice for 20 min each in RPMI1640 (Hyclone), containing 1 mmol/L EGTA, all at 37 °C, to eliminate epithelium. The remaining pieces were stirred for 90 min at 37 °C in RPMI 1640 (Hyclone) containing 20% fetal bovine serum, 100 U/mL collagenase (C2139; Sigma-Aldrich Corp., St. Louis, MO, United States), and 5 U/mL DNase1 (Sigma-Aldrich Corp). The suspensions were centrifuged, and the pellets were washed. LPL were isolated from the lamina propria (LP)-cell preparations by centrifugation through a 45%-66.6% discontinuous Percoll (Solarbio) gradient at 2500 rpm for 20 min.
LPL (1 × 105/well in 0.2 mL RPMI1640 containing 10% fetal bovine serum, 1% penicillin, and 1% streptomycin) were cultured for 48 h in 96-well plates coated with anti-CD3 (10 μg/mL e-Bioscience, San Diego, CA, United States) and soluble anti-CD28 (1 μg/mL, e-Bioscience) mAb at 37 °C in an atmosphere containing 5% CO2[25]. After 48 h, the supernatants were collected and cytokine concentrations assayed by enzyme-linked immunosorbent assay.
Enzyme-linked immunosorbent assay
Supernatants of cell cultures were collected after centrifugation at 1000 rpm for 10 min, and cytokine concentrations were measured using mouse immunoassay kits (R&D Systems Inc., Minneapolis, MN, United States), according to the manufacturer’s protocol.
The levels of IL-6, IL-23, IL-1β, IL-12p70 and TNF-α were measured in supernatants without anti-CD3/anti-CD28 mAbs stimulations. The levels of IFN-γ, IL-17A, IL-21, IL-22 and IL-10 were measured in supernatants with or without anti-CD28/anti-CD3 mAbs stimulations.
RNA extraction and real-time polymerase chain reaction
Total RNA was extracted from colon tissue using Trizol reagent (Takara, Dalian, China), according to the manufacturer’s protocol. RNA was reverse transcribed to cDNA using reverse transcriptase (Takara), followed by PCR assays using primers for β-actin (forward, 5′-TTCCAGCGTTCCTTCTTGGGTAT-3′; reverse, 5′-GTTGGCATAGAGGTGTTTACGG-3′). IL-17F (forward, 5′-TCCCACGTGAATTCCAGAAC-3′; reverse, 5′-ATGGTGCTGTCTTCCTGACC-3′), IL-21 (forward, 5′-TCAGAAGGCCAAACTCAAGC-3′; reverse, 5′-TCACAGGAAGGGCATTTAGC-3′), IL-22 (forward, 5′-GACAGGTTCCAGCCCTACAT-3′; reverse, 5′-CTGGATGTTCTGGTCGTCAC-3′), IL-23 (forward, 5′-TGCCCAGCCTGAGTTCTAGT-3′; reverse, 5′-AGTCAGAGTTGCTGCTCCGT-3′); IL-6 (forward, 5′-GAGGATACCACTCCCAACAGACC-3′; reverse, 5′-AAGTGCATCATCGTTGTTCATACA-3′), IL-10, (forward, 5′-GGTTGCCAAGCCTTATCGGA-3′; reverse, 5′-ACCTGCTCCACTGCCTTGCT-3′), IL-17A (forward, 5′-GCTCCAGAAGGCCCTCAGA-3′; reverse, 5′-AGCTTTCCCTCCGCATTGA-3′), IFN-γ (forward, 5′-AAAGACAATCAGGCCATCAG-3′; reverse, 5′-TGGGTTGTTGACCTCAAACT-3′), T-bet (forward, 5′-CCAGGGAACCGCTTATATGT-3′; reverse, 5′-CTGGGTCACATTGTTGGAAG-3′), Foxp3 (forward, 5′-GGCCCTTCTCCAGGACAGA-3′; reverse, 5′-GCTGATCATGGCTGGGTTGT-3′), and RORγt (forward, 5′-CCACTGCATTCCCAGTTTCT-3′; reverse, 5-CGTAGAAGGTCCTCCAGTCG-3′). The amplification protocol consisted of an initial denaturation at 95 °C for 30 s, followed by 40 cycles of denaturation at 95 °C for 15 s and annealing and extension at 60 °C for 34 s, on an ABI PRISM 7500 Sequence Detection System (Applied Biosystems, Foster City, CA, United States). The level of each gene was normalized relative to that of β-actin mRNA, and relative expression was calculated using the 2-ΔΔCT formula.
Flow cytometry
Briefly, 1 × 106 cells were isolated from the colon of each mouse. For surface antigen detection, the cells were labeled with monoclonal antibodies against Gr-1, F4/80, αβTCR, γδTCR, NK1.1, CD4, CD44, CD25, and CD69 for 30 min at 4 °C. LPL were stimulated with ionomycin (1 mg/mL; Sigma-Aldrich) and PMA (25 ng/mL; Sigma-Aldrich) for 5 h at 37 °C, with brefeldin A (10 mg/mL; Sigma-Aldrich) added after 1 h.
For intracellular staining, cells were fixed and permeabilized with fixation/ permeabilization working solution for 20 min at 4 °C, followed by incubation with monoclonal antibodies against IFN-γ, IL-17A, and IL-10. The cells were analyzed using a Cytofix/Cytoperm Kit Plus (BD Biosciences, San Jose, CA, United States).
Statistical analysis
Data are expressed as mean ± SD and analyzed by one-way ANOVA or t-test. P values < 0.05 were considered statistically significant.
RESULTS
Sodium selenite ameliorates the severity of DSS-induced chronic colitis
The effect of sodium selenite on DSS-induced colitis in mice was assessed by comparing survival rates, clinical symptoms (body weight loss, diarrhea, and rectal bleeding), DAI score, colon length, and colon histology in mice that received and did not receive sodium selenite. Survival rates were similar in the chronic colitis and the Se + chronic colitis group (Figure 1A), and body weight loss, colon length, and macroscopic inflammatory score were similar in the control and Se groups (Figure 1B-G) (P > 0.05 each). Compared with the chronic colitis group, the Se + chronic colitis group showed significant amelioration of body weight loss (Figure 1B) and a significantly lower DAI score (Figure 1C), beginning on day 23 (P < 0.05 each), as well as significantly longer colon (Figure 1D and E) and a significantly lower macroscopic inflammatory score (Figure 1F and G) (P < 0.05 each).
Figure 1.

Sodium selenite ameliorates chronic dextran sulfate sodium-induced colitis in the C57BL/6 mice. A: Survival rate; B: Changes in body weight (%); C: Changes in the disease activity index (DAI); D and E: Colon length; F and G: Colon histopathological injury scores. The data are presented as the mean ± SD (Se + chronic DSS colitis vs chronic DSS colitis, aP < 0.05) (n = 10). DSS: Dextran sulfate sodium.
Cytokine production by LPL
Assessment of cytokine concentrations in the culture supernatants of unstimulated LPL showed that the levels of IL-6, IL-23, IL-1β, TNFα, IFN-γ and IL-17A were significantly lower in the Se + chronic colitis than in the chronic colitis group. There were no significant between-group differences in the levels of IL-12p70, IL-21, IL-22, and IL-10 (Figure 2).
Figure 2.
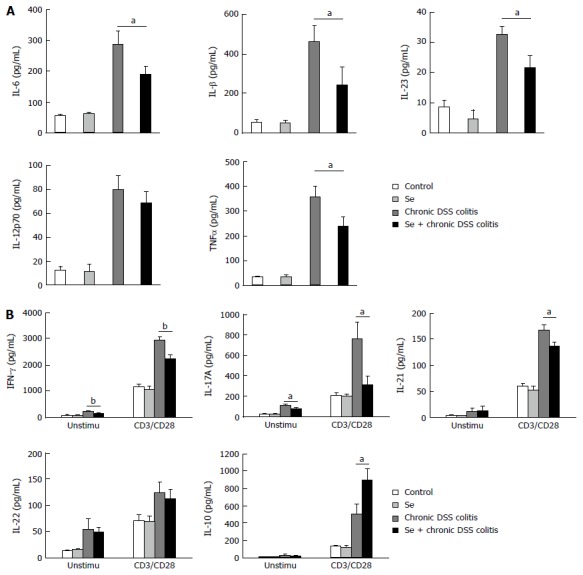
Cytokine production of LPL cells analyzed by ELISA. A: Unstimulated cells; B: LPL cells with or without anti-CD3 and anti-CD28 mAbs (CD3/CD28) stimulations. Each group consisted of three mice. Values represent mean ± SD (aP < 0.05; bP < 0.01) (n = 3).
When cytokine concentrations were assayed in culture supernatants of LPL stimulated with anti-CD3 and anti-CD28 mAbs for 48 h, the concentrations of IFN-γ, IL-17A and IL-21 were found to be significantly lower, while the concentration of IL-10 was significantly higher, in supernatants from the Se + chronic colitis than from the chronic colitis group. The level of IL-22, however, was similar in these two groups (Figure 2).
mRNA expression in colon tissue
RT-PCR assays of mRNA levels in cells showed that the expression levels of IL-6, IFN-γ, IL-17A, IL-21, IL-23, T-bet, and RORγt mRNAs were significantly lower, while the levels of IL-10 and Foxp3 mRNAs were significantly higher, in the Se + chronic colitis group than in the chronic colitis group. There was no significant between-group differences in the expression levels of IL-22 mRNA (Figure 3).
Figure 3.
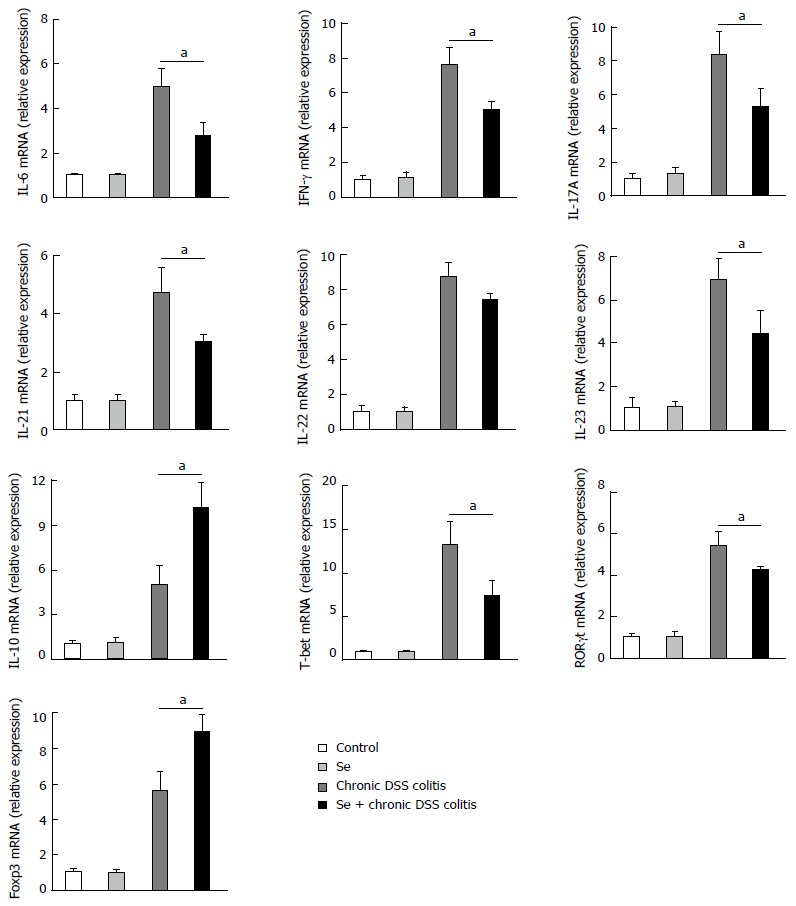
mRNA expression changes in colonic tissue. Values represent mean ± SD (aP < 0.05) (n = 3).
Flow cytometry analysis of LPL populations in mouse colon
There were no significant differences in the percentages and absolute numbers of neutrophils (CD11b+Gr1+F4/80-), macrophages (CD11b+Gr1- F4/80+), γδT cells, NK cells, NKT cells, CD4+, CD4+CD44+, CD4+CD25+, and CD4+CD69+ T cells in the colons of control and Se group mice. The percentages and absolute numbers of neutrophils and CD4+CD25+ T cells in LPL were significantly higher, while the percentages and absolute numbers of γδT and CD4+CD44+ and CD4+CD69+ T cells in LPL were significantly lower, in the Se+ chronic colitis group than in the chronic colitis group. These two groups did not differ in the percentages and absolute numbers of macrophages (CD11b+Gr1-F4/80+), NK cells, and NKT cells (Figure 4).
Figure 4.
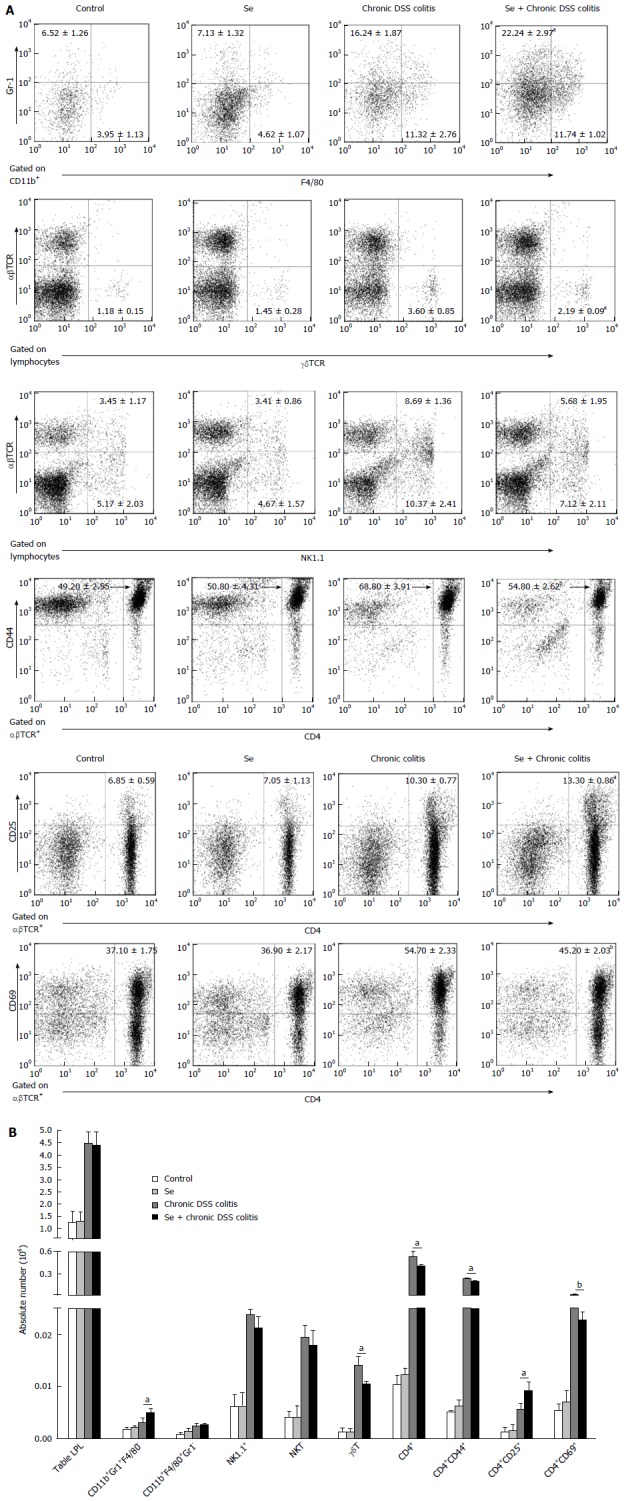
Flow cytometry of the populations of lamina propria lymphocytes in the colon in each group. A: The frequencies of neutrophils (CD11b+Gr1+F4/80-), macrophages (CD11b+Gr1+F4/80+), γδT cells (γδTCR), NK cells (NK1.1+), NKT cells (NK1.1+αβTCR+), CD4+, CD4+CD44+ (effector T cells), CD4+CD25+ (regulatory T cells), CD4+CD69+ (activated T cells) T cells in LPL of the colon in each group; B: The absolute cell numbers of all kinds of cells in each group. Data indicate mean ± SD of six mice of obtained from a representative of three independent experiments (aP < 0.05, bP < 0.01).
Cytokine production by T-LPL cells in mice
There were no significant differences in the percentages and absolute numbers of CD4+IL-17A+, CD4+IFN-γ+, and CD4+IL-10+ cells in LPL of the control and Se groups. The percentages and absolute numbers of CD4+IL-17A+ and CD4+IFN-γ+ cells in LPL were significantly lower, while the percentages and absolute numbers of CD4+IL-10+ cells in LPL were significantly higher, in the Se + chronic colitis group than in the chronic colitis group (Figure 5).
Figure 5.
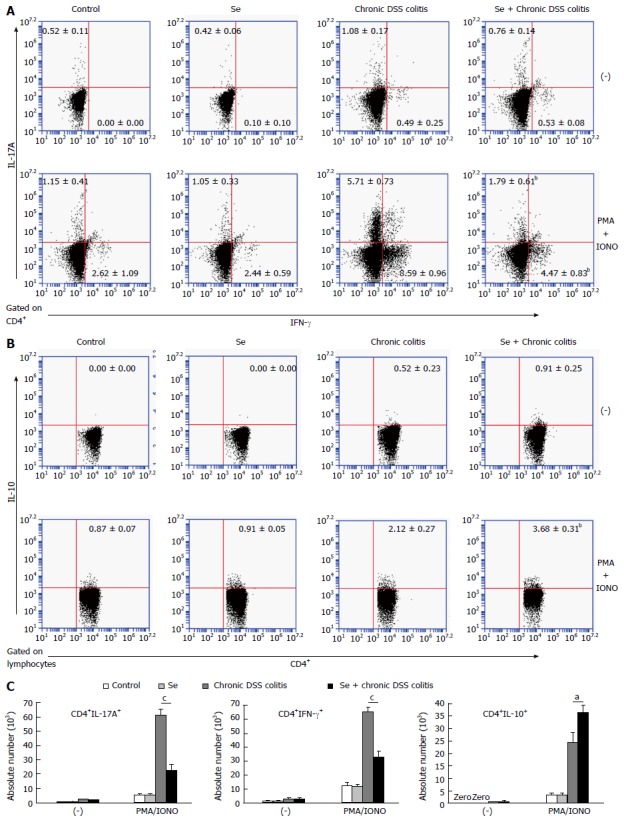
Cytokine-producing T cells in the lamina propria lymphocytes of the colon in each group. A: The frequencies of CD4+IL-17A+ and CD4+IFN-γ+ T LPL. B: The frequency of CD4+IL-10+T LPL. C: The absolute numbers of cytokine producing CD4+IL-17A+, CD4+IFN-γ+ T LPL and CD4+IL-10+T LPL with and without stimulation. Data indicate mean ± SD of six mice of obtained from a representative of three independent experiments (aP < 0.05, bP < 0.01, cP < 0.001).
DISCUSSION
Many diseases have been associated with lack of the essential trace element Se in the body, including asthma, rheumatoid arthritis, and cancer[26-28]. In these diseases, Se deficiency is abnormal, making supplementation with exogenous Se a reasonable method of treatment. Se may be useful for the prevention and/or amelioration of several autoimmune diseases, including IBD[29]. In support of this concept, our results demonstrate that Se sufficiency protects against DSS-induced chronic colitis by attenuating the symptoms of colitis, as shown by measurements of DAI, body weight, and colon length and by histological assessment of mucosal injury. Se may alleviate chronic colitis by enhancing the activity of Tregs, which suppress the secretion of proinflammatory cytokines and populations of Th1, Th17, and γδT cells.
Although neutrophils have been reported to have a proinflammatory effect[30], these cells maybe play a protective role in intestinal colitis[31,32]. Depletion of neutrophils has been shown to exacerbate colonic inflammation, suggesting that neutrophils be involved in mucosal repair processes[33]. Neutrophils produce IL-22 in response to coordinated signaling by IL-23 and TNF-α[34]. IL-22 is up-regulated in chronic colitis and considered beneficial for intestinal epithelial barrier function[35-37]. Neutrophil recruitment was shown to ameliorate experimental colitis by genetic ablation of IL-21[38]. Our results showed that Se reduced IL-21 and IL-23 expression, but had no effect on the IL-22 level, suggesting that Se may enhance neutrophil populations, leading to reductions in IL-21 and IL-23 expression.
LP macrophages are activated in many animal models of experimental colitis[39]. These macrophages produced TNF-α in DSS-induced colitis. IL-10 suppresses macrophage-derived proinflammatory cytokines and downregulates NO and ROS production[40]. Elimination of local macrophages in the intestine was shown to prevent chronic colitis in IL-10-deficient mice[41]. Macrophages may also ameliorate colitis by secreting immunosuppressive factors[42], suggesting that macrophages may play a dual role in different stages of IBD. Our study found that Se did not alter the number of macrophages, but alterations in function were not determined. Selenoproteins in macrophages protect mice from DSS-colitis by enhancing 15-hydroxy-prostaglandin dehydrogenase (15-PGDH)-dependent oxidation of prostaglandin E2 (PGE2) to alleviate inflammation[43]. The study showed that Se through its incorporation into selenoproteins suppressed inflammation and decreased the production of TNF-α and PGE2[44]. We speculate that macrophages in selenoproteins may affect the differentiation of T cells. Therefore, further research is needed.
Mouse intestinal LP T lymphocytes include αβ and γδT cells. γδT cells are a minor T-cell subset present in the LP, but are active in inflammatory processes[45], for example, in patients with IBD[46,47]. Changes in the repertoire and function of human mucosal γδT cells have been associated with the disease process in IBD[46]. γδT cells play a protective role in acute DSS colitis[45], but are involved in the exacerbation of chronic colitis[48], suggesting that γδT cells may represent a promising target in the treatment of human IBD. Our study showed that the absolute numbers and percentages of γδT cells in T-LPL were significantly lower in the Se + chronic colitis group than in the chronic colitis group. Se has been found to upregulate CD4(+)CD25(+) Treg cells in iodine-induced autoimmune thyroiditis in NOD.H-2(h4) mice[49]. γδT cells positively regulate contact sensitivity reactions by modulating INF-γ, IL-12, and TNF-α production[50]. Treg cells can inhibit the production of IFN-γ by antigen-specific memory γδT cells[51]. In addition, IL-10, TGF-β, and the transcription factor Foxp3 mediate immunoregulation[52]. The Treg cells restrain immune responses that are dependent upon expression of the IL-10 and the transcription factor Foxp3. Treg cell-derived IL-10 limits inflammation at environmental interfaces[53]. IL-10 may therefore play an important role in the suppressive function of Treg cells[54]. Other mechanisms, however, cannot be excluded, including direct cell-surface contact. These findings indicate a potential new mechanism by which CD4(+)CD25(+) Tregs can specifically suppress γδT cells and highlight the strategy of combining Treg inhibition with subsequent γδT-cell activation to enhance γδT-cell-mediated immunotherapy[55]. Tregs can inhibit γδT-cell proliferation in vitro via a cell-cell contact-independent mechanism[55]. Our data showed that IL-10 expression, as well as the absolute numbers and percentage of CD4(+)CD25(+) Treg cells in T-LPL, were significantly higher in the Se + chronic colitis group than in the chronic colitis group. Further understanding of the molecular mechanisms underlying γδT-cell-mediated exacerbation of chronic colitis may suggest better therapeutic strategies for human IBD.
NK cells are a subset of innate lymphocytes that contribute to host resistance and provide immune surveillance. The roles of NK cells in DSS-induced colitis, however, are less clear. NK cells produce cytokines, including TNF-α and IFN-γ[56], as well as protect mice from DSS-induced colitis by regulating neutrophil function via the NKG2A receptor[57]. Several proinflammatory cytokines, including IL-15, IL-21, and IL-23, may potently activate NK cells, inducing the secretion of high levels of the proinflammatory cytokines IFN-γ and TNF-α[58]. IL-21 enhances IBD NK cell cytotoxic responses, triggers T cells to produce proinflammatory cytokines, and induces IBD CD4(+) T cells to differentiate into Th17 cells, suggesting that IL-21 is involved in the pathogenesis of IBD and that blocking IL-21R signaling may have a therapeutic benefit in IBD[59]. NK T cells, a subset of T lymphocytes that express the TCR, play an important role in the pathogenesis of intestinal inflammation[60,61]. NK T cells elicit effector function by producing large amounts of IFN-γ, IL-4, and IL-10[62]. Activation of NK T cells has been shown to protect mice from DSS-induced colitis[60]. We found that the percentages and absolute numbers of NK and NKT cells in T-LPL did not differ significantly in the chronic colitis and Se + chronic colitis groups.
The Th17 pathway was shown to be very important in the pathogenesis of human IBD[63]. Th1 cell subsets are considered major factors in DSS-induced colitis[64]. The orphan nuclear receptor RORγt and T-bet direct Th17 and Th1 differentiation, respectively[65,66]. The amounts of Th1 and Th17 cells were increased and Treg cells were decreased in chronic colitis. In contrast, Treg cells may have protective effects in colitis. The expression of transcription factor Foxp3 has been shown to direct Treg differentiation[67]. However, Th1 and Th17 promote colitis whereas Tregs have protective effects[68,69]. The numbers of Th1 and Th17 cells were increased by the high expression of cytokines supporting Th17 cell differentiation, thus exacerbating the immunopathogenesis of IBD. These cytokines include IL-1, TGFβ, IL-6, IL-21, and IL-23. Our results showed that the percentages and absolute numbers of Th1 and Th17 cells in T-LPL were significantly lower in the Se + chronic colitis than in the chronic colitis group. In addition, IL-6, IFN-γ, IL-17A, IL-21, T-bet, and RORγt expression levels were lower. IL-10 is a negative regulator of inflammation and counters the activity of many proinflammatory cytokines. IL-10 has been shown to suppress intestinal inflammation. Foxp3 functionally inhibits RORγt[70]. Our results showed that IL-10 and Foxp3 expression levels were higher in the Se + chronic colitis group than in the chronic colitis group.Th17 cells are a subset of CD4+ T cells that produce IL-17A. IL-21 induces and maintains T-cell-dependent inflammatory responses, including both Th1 and Th17 responses[71]. IL-21 promotes Th17 cell differentiation by suppressing Foxp3 expression, with inhibition of IL-21 signaling reducing IL-17A and IFN-γ expression[72-74]. T-cell subsets are regarded as unstable and may convert into other subsets of T cells under certain circumstances[75].
In summary, our results suggest that sodium selenite has a protective role in DSS-induced chronic colitis. Se may alleviate colitis by inducing Tregs to suppress the secretion of proinflammatory cytokines and populations of Th1, Th17, and γδT cells. As sodium selenite has been widely used to treat human autoimmune diseases, it may be useful in treating IBD.
COMMENTS
Background
Inflammatory bowel disease (IBD) is a group of diseases responsible for chronic inflammation of the intestine. In a mouse model of dextran sulfate sodium (DSS)-induced colitis, Th17 cells promote, whereas regulatory T (Treg) cells protect against, intestinal inflammation. Selenium (Se) is an important natural antioxidant, with serum Se concentrations inversely correlated with IBD.
Research frontiers
CD4+ T cells have been divided into subsets, based on the cytokines they produce; these subsets include Th1, Th2, Th17, and Treg cells. Th1 and Th17 cells are involved in the pathogenesis of IBD, whereas Treg cells ameliorate intestinal inflammation by suppressing Th17 cells.
Innovations and breakthroughs
These results suggest that Se protects against DSS-induced chronic colitis perhaps by increasing the number of CD4(+)CD25(+) Tregs that suppress the secretion of proinflammatory cytokines and populations of Th1, Th17, and γδT cells.
Applications
The results suggest that sodium selenite has a protective role in DSS-induced chronic colitis. Se may alleviate colitis by inducing Tregs to suppress the secretion of proinflammatory cytokines and populations of Th1, Th17, and γδT cells. As sodium selenite has been widely used to treat human autoimmune diseases, it may be useful in treating IBD.
Peer-review
This is a well-designed and well-presented study for examining the antiinflammatory potential of sodium selenite in DSS colitis in mice.
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: China
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Institutional review board statement: All specimens from the mice were taken after ethical permission was obtained for participation in the study.
Institutional animal care and use committee statement: The experimental protocols were approved by Institutional Animal Care and Use Committee of China Medical University.
Conflict-of-interest statement: The authors disclose no potential competing interests.
Data sharing statement: No additional data are available.
Peer-review started: October 21, 2016
First decision: November 9, 2016
Article in press: March 15, 2017
P- Reviewer: Hayashi S, Howarth GS S- Editor: Gong ZM L- Editor: Wang TQ E- Editor: Zhang FF
References
- 1.Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, Albrecht M, Mayr G, De La Vega FM, Briggs J, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 2.Håkansson Å, Tormo-Badia N, Baridi A, Xu J, Molin G, Hagslätt ML, Karlsson C, Jeppsson B, Cilio CM, Ahrné S. Immunological alteration and changes of gut microbiota after dextran sulfate sodium (DSS) administration in mice. Clin Exp Med. 2015;15:107–120. doi: 10.1007/s10238-013-0270-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shaker ME, Ashamallah SA, Houssen ME. Celastrol ameliorates murine colitis via modulating oxidative stress, inflammatory cytokines and intestinal homeostasis. Chem Biol Interact. 2014;210:26–33. doi: 10.1016/j.cbi.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 4.Ananthakrishnan AN. Environmental risk factors for inflammatory bowel diseases: a review. Dig Dis Sci. 2015;60:290–298. doi: 10.1007/s10620-014-3350-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liang J, Sha SM, Wu KC. Role of the intestinal microbiota and fecal transplantation in inflammatory bowel diseases. J Dig Dis. 2014;15:641–646. doi: 10.1111/1751-2980.12211. [DOI] [PubMed] [Google Scholar]
- 6.Fiocchi C. Inflammatory bowel disease: etiology and pathogenesis. Gastroenterology. 1998;115:182–205. doi: 10.1016/s0016-5085(98)70381-6. [DOI] [PubMed] [Google Scholar]
- 7.Wang S, Zhang Y, Saas P, Wang H, Xu Y, Chen K, Zhong J, Yuan Y, Wang Y, Sun Y. Oridonin’s therapeutic effect: suppressing Th1/Th17 simultaneously in a mouse model of Crohn’s disease. J Gastroenterol Hepatol. 2015;30:504–512. doi: 10.1111/jgh.12710. [DOI] [PubMed] [Google Scholar]
- 8.Steinbach EC, Kobayashi T, Russo SM, Sheikh SZ, Gipson GR, Kennedy ST, Uno JK, Mishima Y, Borst LB, Liu B, et al. Innate PI3K p110δ regulates Th1/Th17 development and microbiota-dependent colitis. J Immunol. 2014;192:3958–3968. doi: 10.4049/jimmunol.1301533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li L, Liu S, Xu Y, Zhang A, Jiang J, Tan W, Xing J, Feng G, Liu H, Huo F, et al. Human umbilical cord-derived mesenchymal stem cells downregulate inflammatory responses by shifting the Treg/Th17 profile in experimental colitis. Pharmacology. 2013;92:257–264. doi: 10.1159/000354883. [DOI] [PubMed] [Google Scholar]
- 10.Rutgeerts P, Sandborn WJ, Feagan BG, Reinisch W, Olson A, Johanns J, Travers S, Rachmilewitz D, Hanauer SB, Lichtenstein GR, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2005;353:2462–2476. doi: 10.1056/NEJMoa050516. [DOI] [PubMed] [Google Scholar]
- 11.Sands BE, Anderson FH, Bernstein CN, Chey WY, Feagan BG, Fedorak RN, Kamm MA, Korzenik JR, Lashner BA, Onken JE, et al. Infliximab maintenance therapy for fistulizing Crohn’s disease. N Engl J Med. 2004;350:876–885. doi: 10.1056/NEJMoa030815. [DOI] [PubMed] [Google Scholar]
- 12.Rayman MP. The importance of selenium to human health. Lancet. 2000;356:233–241. doi: 10.1016/S0140-6736(00)02490-9. [DOI] [PubMed] [Google Scholar]
- 13.Kuroki F, Matsumoto T, Iida M. Selenium is depleted in Crohn’s disease on enteral nutrition. Dig Dis. 2003;21:266–270. doi: 10.1159/000073346. [DOI] [PubMed] [Google Scholar]
- 14.Hoffenberg EJ, Deutsch J, Smith S, Sokol RJ. Circulating antioxidant concentrations in children with inflammatory bowel disease. Am J Clin Nutr. 1997;65:1482–1488. doi: 10.1093/ajcn/65.5.1482. [DOI] [PubMed] [Google Scholar]
- 15.Ishida T, Himeno K, Torigoe Y, Inoue M, Wakisaka O, Tabuki T, Ono H, Honda K, Mori T, Seike M, et al. Selenium deficiency in a patient with Crohn’s disease receiving long-term total parenteral nutrition. Intern Med. 2003;42:154–157. doi: 10.2169/internalmedicine.42.154. [DOI] [PubMed] [Google Scholar]
- 16.Andoh A, Hirashima M, Maeda H, Hata K, Inatomi O, Tsujikawa T, Sasaki M, Takahashi K, Fujiyama Y. Serum selenoprotein-P levels in patients with inflammatory bowel disease. Nutrition. 2005;21:574–579. doi: 10.1016/j.nut.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 17.Bogatov NV. [Selenium deficiency and its dietary correction in patients with irritable bowel syndrome and chronic catarrhal colitis] Vopr Pitan. 2007;76:35–39. [PubMed] [Google Scholar]
- 18.Tirosh O, Levy E, Reifen R. High selenium diet protects against TNBS-induced acute inflammation, mitochondrial dysfunction, and secondary necrosis in rat colon. Nutrition. 2007;23:878–886. doi: 10.1016/j.nut.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 19.Takada Y, Ray N, Ikeda E, Kawaguchi T, Kuwahara M, Wagner EF, Matsuo K. Fos proteins suppress dextran sulfate sodium-induced colitis through inhibition of NF-kappaB. J Immunol. 2010;184:1014–1021. doi: 10.4049/jimmunol.0901196. [DOI] [PubMed] [Google Scholar]
- 20.El-Salhy M, Umezawa K, Gilja OH, Hatlebakk JG, Gundersen D, Hausken T. Amelioration of severe TNBS induced colitis by novel AP-1 and NF- κ B inhibitors in rats. ScientificWorldJournal. 2014;2014:813804. doi: 10.1155/2014/813804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pillai SS, Sugathan JK, Indira M. Selenium downregulates RAGE and NFκB expression in diabetic rats. Biol Trace Elem Res. 2012;149:71–77. doi: 10.1007/s12011-012-9401-1. [DOI] [PubMed] [Google Scholar]
- 22.József L, Filep JG. Selenium-containing compounds attenuate peroxynitrite-mediated NF-kappaB and AP-1 activation and interleukin-8 gene and protein expression in human leukocytes. Free Radic Biol Med. 2003;35:1018–1027. doi: 10.1016/s0891-5849(03)00439-8. [DOI] [PubMed] [Google Scholar]
- 23.Shalini S, Bansal MP. Alterations in selenium status influences reproductive potential of male mice by modulation of transcription factor NFkappaB. Biometals. 2007;20:49–59. doi: 10.1007/s10534-006-9014-2. [DOI] [PubMed] [Google Scholar]
- 24.Tyszka-Czochara M, Pasko P, Zagrodzki P, Gajdzik E, Wietecha-Posluszny R, Gorinstein S. Selenium Supplementation of Amaranth Sprouts Influences Betacyanin Content and Improves Anti-Inflammatory Properties via NFκB in Murine RAW 264.7 Macrophages. Biol Trace Elem Res. 2016;169:320–330. doi: 10.1007/s12011-015-0429-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun X, Yamada H, Yoshihara K, Awaya A, Yoshikai Y. In vivo treatment with a nonapeptide thymic hormone, facteur thymique serique (FTS), ameliorates chronic colitis induced by dextran sulphate sodium in mice. Int Immunopharmacol. 2007;7:928–936. doi: 10.1016/j.intimp.2007.02.014. [DOI] [PubMed] [Google Scholar]
- 26.Carneiro MF, Rhoden CR, Amantéa SL, Barbosa F. Low concentrations of selenium and zinc in nails are associated with childhood asthma. Biol Trace Elem Res. 2011;144:244–252. doi: 10.1007/s12011-011-9080-3. [DOI] [PubMed] [Google Scholar]
- 27.Stone J, Doube A, Dudson D, Wallace J. Inadequate calcium, folic acid, vitamin E, zinc, and selenium intake in rheumatoid arthritis patients: results of a dietary survey. Semin Arthritis Rheum. 1997;27:180–185. doi: 10.1016/s0049-0172(97)80018-2. [DOI] [PubMed] [Google Scholar]
- 28.Barrett CW, Singh K, Motley AK, Lintel MK, Matafonova E, Bradley AM, Ning W, Poindexter SV, Parang B, Reddy VK, et al. Dietary selenium deficiency exacerbates DSS-induced epithelial injury and AOM/DSS-induced tumorigenesis. PLoS One. 2013;8:e67845. doi: 10.1371/journal.pone.0067845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bitiren M, Karakilcik AZ, Zerin M, Ozardali I, Selek S, Nazligül Y, Ozgonul A, Musa D, Uzunkoy A. Protective effects of selenium and vitamin E combination on experimental colitis in blood plasma and colon of rats. Biol Trace Elem Res. 2010;136:87–95. doi: 10.1007/s12011-009-8518-3. [DOI] [PubMed] [Google Scholar]
- 30.Qualls JE, Kaplan AM, van Rooijen N, Cohen DA. Suppression of experimental colitis by intestinal mononuclear phagocytes. J Leukoc Biol. 2006;80:802–815. doi: 10.1189/jlb.1205734. [DOI] [PubMed] [Google Scholar]
- 31.Buell MG, Berin MC. Neutrophil-independence of the initiation of colonic injury. Comparison of results from three models of experimental colitis in the rat. Dig Dis Sci. 1994;39:2575–2588. doi: 10.1007/BF02087693. [DOI] [PubMed] [Google Scholar]
- 32.Yamada T, Zimmerman BJ, Specian RD, Grisham MB. Role of neutrophils in acetic acid-induced colitis in rats. Inflammation. 1991;15:399–411. doi: 10.1007/BF00917356. [DOI] [PubMed] [Google Scholar]
- 33.Kühl AA, Kakirman H, Janotta M, Dreher S, Cremer P, Pawlowski NN, Loddenkemper C, Heimesaat MM, Grollich K, Zeitz M, et al. Aggravation of different types of experimental colitis by depletion or adhesion blockade of neutrophils. Gastroenterology. 2007;133:1882–1892. doi: 10.1053/j.gastro.2007.08.073. [DOI] [PubMed] [Google Scholar]
- 34.Zindl CL, Lai JF, Lee YK, Maynard CL, Harbour SN, Ouyang W, Chaplin DD, Weaver CT. IL-22-producing neutrophils contribute to antimicrobial defense and restitution of colonic epithelial integrity during colitis. Proc Natl Acad Sci USA. 2013;110:12768–12773. doi: 10.1073/pnas.1300318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamamoto-Furusho JK, Miranda-Pérez E, Fonseca-Camarillo G, Sánchez-Muñoz F, Dominguez-Lopez A, Barreto-Zuñiga R. Colonic epithelial upregulation of interleukin 22 (IL-22) in patients with ulcerative colitis. Inflamm Bowel Dis. 2010;16:1823. doi: 10.1002/ibd.21235. [DOI] [PubMed] [Google Scholar]
- 36.Brand S, Beigel F, Olszak T, Zitzmann K, Eichhorst ST, Otte JM, Diepolder H, Marquardt A, Jagla W, Popp A, et al. IL-22 is increased in active Crohn’s disease and promotes proinflammatory gene expression and intestinal epithelial cell migration. Am J Physiol Gastrointest Liver Physiol. 2006;290:G827–G838. doi: 10.1152/ajpgi.00513.2005. [DOI] [PubMed] [Google Scholar]
- 37.Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, Blumberg RS, Xavier RJ, Mizoguchi A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest. 2008;118:534–544. doi: 10.1172/JCI33194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fina D, Sarra M, Fantini MC, Rizzo A, Caruso R, Caprioli F, Stolfi C, Cardolini I, Dottori M, Boirivant M, et al. Regulation of gut inflammation and th17 cell response by interleukin-21. Gastroenterology. 2008;134:1038–1048. doi: 10.1053/j.gastro.2008.01.041. [DOI] [PubMed] [Google Scholar]
- 39.Murai M, Turovskaya O, Kim G, Madan R, Karp CL, Cheroutre H, Kronenberg M. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat Immunol. 2009;10:1178–1184. doi: 10.1038/ni.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li B, Alli R, Vogel P, Geiger TL. IL-10 modulates DSS-induced colitis through a macrophage-ROS-NO axis. Mucosal Immunol. 2014;7:869–878. doi: 10.1038/mi.2013.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Watanabe N, Ikuta K, Okazaki K, Nakase H, Tabata Y, Matsuura M, Tamaki H, Kawanami C, Honjo T, Chiba T. Elimination of local macrophages in intestine prevents chronic colitis in interleukin-10-deficient mice. Dig Dis Sci. 2003;48:408–414. doi: 10.1023/a:1021960401290. [DOI] [PubMed] [Google Scholar]
- 42.Pull SL, Doherty JM, Mills JC, Gordon JI, Stappenbeck TS. Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury. Proc Natl Acad Sci USA. 2005;102:99–104. doi: 10.1073/pnas.0405979102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaushal N, Kudva AK, Patterson AD, Chiaro C, Kennett MJ, Desai D, Amin S, Carlson BA, Cantorna MT, Prabhu KS. Crucial role of macrophage selenoproteins in experimental colitis. J Immunol. 2014;193:3683–3692. doi: 10.4049/jimmunol.1400347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gandhi UH, Kaushal N, Ravindra KC, Hegde S, Nelson SM, Narayan V, Vunta H, Paulson RF, Prabhu KS. Selenoprotein-dependent up-regulation of hematopoietic prostaglandin D2 synthase in macrophages is mediated through the activation of peroxisome proliferator-activated receptor (PPAR) gamma. J Biol Chem. 2011;286:27471–27482. doi: 10.1074/jbc.M111.260547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsuchiya T, Fukuda S, Hamada H, Nakamura A, Kohama Y, Ishikawa H, Tsujikawa K, Yamamoto H. Role of gamma delta T cells in the inflammatory response of experimental colitis mice. J Immunol. 2003;171:5507–5513. doi: 10.4049/jimmunol.171.10.5507. [DOI] [PubMed] [Google Scholar]
- 46.McVay LD, Li B, Biancaniello R, Creighton MA, Bachwich D, Lichtenstein G, Rombeau JL, Carding SR. Changes in human mucosal gamma delta T cell repertoire and function associated with the disease process in inflammatory bowel disease. Mol Med. 1997;3:183–203. [PMC free article] [PubMed] [Google Scholar]
- 47.Kühl AA, Pawlowski NN, Grollich K, Blessenohl M, Westermann J, Zeitz M, Loddenkemper C, Hoffmann JC. Human peripheral gammadelta T cells possess regulatory potential. Immunology. 2009;128:580–588. doi: 10.1111/j.1365-2567.2009.03162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nanno M, Kanari Y, Naito T, Inoue N, Hisamatsu T, Chinen H, Sugimoto K, Shimomura Y, Yamagishi H, Shiohara T, et al. Exacerbating role of gammadelta T cells in chronic colitis of T-cell receptor alpha mutant mice. Gastroenterology. 2008;134:481–490. doi: 10.1053/j.gastro.2007.11.056. [DOI] [PubMed] [Google Scholar]
- 49.Xue H, Wang W, Li Y, Shan Z, Li Y, Teng X, Gao Y, Fan C, Teng W. Selenium upregulates CD4(+)CD25(+) regulatory T cells in iodine-induced autoimmune thyroiditis model of NOD.H-2(h4) mice. Endocr J. 2010;57:595–601. doi: 10.1507/endocrj.k10e-063. [DOI] [PubMed] [Google Scholar]
- 50.Strzepa A, Majewska-Szczepanik M, Szczepanik M. GammadeltaT cells positively regulate contact sensitivity (CS) reaction via modulation of INF-gamma, IL-12 and TNF-alpha production. Folia Biol (Krakow) 2013;61:205–210. doi: 10.3409/fb61_3-4.205. [DOI] [PubMed] [Google Scholar]
- 51.Li L, Wu CY. CD4+ CD25+ Treg cells inhibit human memory gammadelta T cells to produce IFN-gamma in response to M tuberculosis antigen ESAT-6. Blood. 2008;111:5629–5636. doi: 10.1182/blood-2008-02-139899. [DOI] [PubMed] [Google Scholar]
- 52.Pereira S, Teixeira L, Aguilar E, Oliveira M, Savassi-Rocha A, Pelaez JN, Capettini L, Diniz MT, Ferreira A, Alvarez-Leite J. Modulation of adipose tissue inflammation by FOXP3+ Treg cells, IL-10, and TGF-β in metabolically healthy class III obese individuals. Nutrition. 2014;30:784–790. doi: 10.1016/j.nut.2013.11.023. [DOI] [PubMed] [Google Scholar]
- 53.Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, Treuting P, Siewe L, Roers A, Henderson WR, et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 2008;28:546–558. doi: 10.1016/j.immuni.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 54.Unutmaz D, Pulendran B. The gut feeling of Treg cells: IL-10 is the silver lining during colitis. Nat Immunol. 2009;10:1141–1143. doi: 10.1038/ni1109-1141. [DOI] [PubMed] [Google Scholar]
- 55.Kunzmann V, Kimmel B, Herrmann T, Einsele H, Wilhelm M. Inhibition of phosphoantigen-mediated gammadelta T-cell proliferation by CD4+ CD25+ FoxP3+ regulatory T cells. Immunology. 2009;126:256–267. doi: 10.1111/j.1365-2567.2008.02894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, Yokoyama WM, Ugolini S. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331:44–49. doi: 10.1126/science.1198687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hall LJ, Murphy CT, Quinlan A, Hurley G, Shanahan F, Nally K, Melgar S. Natural killer cells protect mice from DSS-induced colitis by regulating neutrophil function via the NKG2A receptor. Mucosal Immunol. 2013;6:1016–1026. doi: 10.1038/mi.2012.140. [DOI] [PubMed] [Google Scholar]
- 58.Yadav PK, Chen C, Liu Z. Potential role of NK cells in the pathogenesis of inflammatory bowel disease. J Biomed Biotechnol. 2011;2011:348530. doi: 10.1155/2011/348530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu Z, Yang L, Cui Y, Wang X, Guo C, Huang Z, Kan Q, Liu Z, Liu Y. Il-21 enhances NK cell activation and cytolytic activity and induces Th17 cell differentiation in inflammatory bowel disease. Inflamm Bowel Dis. 2009;15:1133–1144. doi: 10.1002/ibd.20923. [DOI] [PubMed] [Google Scholar]
- 60.Saubermann LJ, Beck P, De Jong YP, Pitman RS, Ryan MS, Kim HS, Exley M, Snapper S, Balk SP, Hagen SJ, et al. Activation of natural killer T cells by alpha-galactosylceramide in the presence of CD1d provides protection against colitis in mice. Gastroenterology. 2000;119:119–128. doi: 10.1053/gast.2000.9114. [DOI] [PubMed] [Google Scholar]
- 61.Shibolet O, Kalish Y, Klein A, Alper R, Zolotarov L, Thalenfeld B, Engelhardt D, Rabbani E, Ilan Y. Adoptive transfer of ex vivo immune-programmed NKT lymphocytes alleviates immune-mediated colitis. J Leukoc Biol. 2004;75:76–86. doi: 10.1189/jlb.0703351. [DOI] [PubMed] [Google Scholar]
- 62.Taniguchi M, Harada M, Kojo S, Nakayama T, Wakao H. The regulatory role of Valpha14 NKT cells in innate and acquired immune response. Annu Rev Immunol. 2003;21:483–513. doi: 10.1146/annurev.immunol.21.120601.141057. [DOI] [PubMed] [Google Scholar]
- 63.Zhang H, Wu H, Liu L, Li H, Shih DQ, Zhang X. 1,25-dihydroxyvitamin D3 regulates the development of chronic colitis by modulating both T helper (Th)1 and Th17 activation. APMIS. 2015;123:490–501. doi: 10.1111/apm.12378. [DOI] [PubMed] [Google Scholar]
- 64.Yue M, Shen Z, Yu CH, Ye H, Li YM. The therapeutic role of oral tolerance in dextran sulfate sodium-induced colitis via Th1-Th2 balance and γδ T cells. J Dig Dis. 2013;14:543–551. doi: 10.1111/1751-2980.12068. [DOI] [PubMed] [Google Scholar]
- 65.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 66.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 67.Tang Q, Bluestone JA. The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nat Immunol. 2008;9:239–244. doi: 10.1038/ni1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Park JW, Bae H, Lee G, Hong BG, Yoo HH, Lim SJ, Lee K, Kim J, Ryu B, Lee BJ, et al. Prophylactic effects of Lonicera japonica extract on dextran sulphate sodium-induced colitis in a mouse model by the inhibition of the Th1/Th17 response. Br J Nutr. 2013;109:283–292. doi: 10.1017/S0007114512001122. [DOI] [PubMed] [Google Scholar]
- 69.Yao J, Wei C, Wang JY, Zhang R, Li YX, Wang LS. Effect of resveratrol on Treg/Th17 signaling and ulcerative colitis treatment in mice. World J Gastroenterol. 2015;21:6572–6581. doi: 10.3748/wjg.v21.i21.6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang F, Meng G, Strober W. Interactions among the transcription factors Runx1, RORgammat and Foxp3 regulate the differentiation of interleukin 17-producing T cells. Nat Immunol. 2008;9:1297–1306. doi: 10.1038/ni.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sarra M, Pallone F, Macdonald TT, Monteleone G. IL-23/IL-17 axis in IBD. Inflamm Bowel Dis. 2010;16:1808–1813. doi: 10.1002/ibd.21248. [DOI] [PubMed] [Google Scholar]
- 72.Korn T, Bettelli E, Gao W, Awasthi A, Jäger A, Strom TB, Oukka M, Kuchroo VK. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, Tian Q, Watowich SS, Jetten AM, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 74.Monteleone G, Monteleone I, Fina D, Vavassori P, Del Vecchio Blanco G, Caruso R, Tersigni R, Alessandroni L, Biancone L, Naccari GC, et al. Interleukin-21 enhances T-helper cell type I signaling and interferon-gamma production in Crohn’s disease. Gastroenterology. 2005;128:687–694. doi: 10.1053/j.gastro.2004.12.042. [DOI] [PubMed] [Google Scholar]
- 75.Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. 2009;30:646–655. doi: 10.1016/j.immuni.2009.05.001. [DOI] [PubMed] [Google Scholar]