

Abstract
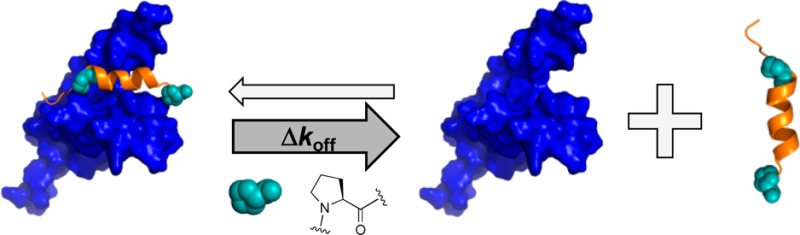
Appropriate integration of cellular signals requires a delicate balance of ligand–target binding affinities. Increasing the level of residual structure in intrinsically disordered proteins (IDPs), which are overrepresented in these cellular processes, has been shown previously to enhance binding affinities and alter cellular function. Conserved proline residues are commonly found flanking regions of IDPs that become helical upon interacting with a partner protein. Here, we mutate these helix-flanking prolines in p53 and MLL and find opposite effects on binding affinity upon an increase in free IDP helicity. In both cases, changes in affinity were due to alterations in dissociation, not association, rate constants, which is inconsistent with conformational selection mechanisms. We conclude that, contrary to previous suggestions, helix-flanking prolines do not regulate affinity by modulating the rate of complex formation. Instead, they influence binding affinities by controlling the lifetime of the bound complex.
Intrinsically disordered proteins (IDPs) or intrinsically disordered regions of proteins (IDRs) lack a well-defined three-dimensional fold and exist as ensembles of conformations with variable levels of transient structure.1 A subset of IDRs fold upon binding to their partner macromolecule.2 For IDRs that form α-helices upon coupled folding and binding, increasing the residual helicity of the free IDR has been shown to increase the affinity for the partner protein.3−5 Conserved proline residues are commonly found flanking transiently helical binding segments of IDRs6,7 (Figure 1). The abundance and conservation of helix-flanking prolines in IDPs point toward an important biological function. Proline residues are known helix breakers, and these conserved helix-flanking prolines may act to restrict the transient residual helicity of the free IDR6,7 and consequently the affinity for the partner protein. We have previously shown that this is the case for the disordered transactivation domain of p53, where mutation of conserved helix-flanking prolines increases the peak residual helicity of the free state by approximately 40%.3 When the proline 27 to alanine (P27A) mutation is present, this increase in residual helicity is accompanied by a 10-fold increase in affinity for its ordered binding partner, MDM2.
Figure 1.

Position and conservation of helix-flanking prolines in IDRs. Bound structures of IDPs/IDRs that undergo coupled folding and binding to form α-helices that are flanked by prolines. From top to bottom: p53:MDM2 [Protein Data Bank (PDB) entry 1YCR], MLL:CBP KIX (PDB entry 2LXS), c-Myb:CBP KIX (PDB entry 1SB0), CREB:CBP KIX (PDB entry 1KDX), SHP-1:SF-1 (PDB entry 1YMT), and PGC-1 α:PPARγ (PDB entry 3CS8), respectively. Folded partner proteins are colored gray and IDPs and IDRs red. Helix-flanking prolines are represented as cyan spheres. Cyan circles indicate helix-flanking prolines that are not present in the structure. N denotes the N-terminus of the IDP/IDR. For each IDP/IDR, the sequences from Homo sapiens, Mus musculus, Gallus gallus, and Danio rerio were aligned using Clutsal Omega.11,12 The p53 alignment is from ref (3). The peptide region used in the structure is highlighted in gray, with residues that form α-helices upon binding highlighted in red. Helix-flanking prolines are highlighted. As determined with Clustal Omega, positions of full residue conservation are indicated with an asterisk, a colon indicates conservation of strongly similar amino acid properties, and weakly similar properties are specified with a period.
It has been suggested that observing a concomitant enhancement in IDR residual helicity and IDR:target partner protein affinity, will lead to an increase in the proportion of binding competent species within the conformational ensemble and, thus, an increase in the rate of complex formation.4,8 However, kinetic information is required to justify such mechanistic conclusions.9,10 Here we determine the mechanism behind the increase in the affinity of p53 for MDM2 upon proline to alanine mutation (PtoA). We then investigate helix-flanking prolines in the transactivation domain of MLL, another IDR, to determine the commonality of this mechanism. We demonstrate that the principal effect is on complex lifetime, rather than rate of formation, but that conserved helix-flanking prolines have more subtlety in the way that they can control affinity than a simple model might predict.
Materials and Methods
Methods for protein expression, purification and labeling, circular dichroism, and equilibrium binding experiments are described in the Supporting Information.
Peptides
MLL peptides were synthesized by Biomatik and purchased as trifluoroacetate salts with a fluorescein isothiocyanate (FITC)-labeled or free N-terminus. Wild-type (WT) MLL consisted of 31 amino acids: residues 2838–2869 (Uniprot entry Q03164) with residue C2841 removed to avoid disulfide bond formation. p53 peptides were produced recombinantly and labeled with Alexa Fluor dyes, as described in the Supporting Information.
Biophysical Buffer
Biophysical buffer for p53 and MDM2 consisted of 50 mM sodium phosphate, 100 mM NaCl, 1 mM EDTA, 2 mM DTT, and 0.02% sodium azide (pH 6.8). Biophysical buffer for MLL and KIX consisted of 100 mM sodium phosphate and 0.05% Tween 20 (pH 7.4).
Nuclear Magnetic Resonance (NMR) Data Collection and Analysis
NMR experiments for WT MLL and the P9/21A mutant were performed using uniformly 15N- and 13C-labeled samples at 160 and 155 μM, respectively, at 10 °C on a Varian VNMRS 800 MHz spectrometer equipped with a triple-resonance pulse field Z-axis gradient cold probe. To make the amide 1H and 15N as well as 13Cα, 13Cβ, and 13CO resonance assignments, sensitivity-enhanced 1H–15N HSQC and three-dimensional HNCACB and HNCO experiments were performed on the uniformly 15N- and 13C-labeled samples in 90% H2O/10% D2O, 50 mM phosphate buffer with 50 mM NaCl, 1 mM EDTA, and 0.02% NaN3 (pH 6.8). For the HNCACB experiment, data were acquired in the 1H, 13C, and 15N dimensions using 9689.9 (t3) Hz × 14074.1 (t2) Hz × 1944.5 (t1) Hz sweep widths and 1024 (t3) × 128 (t2) × 32 (t1) complex data points. For the HNCO, the sweep widths were 9689.9 (t3) Hz × 3770.1 (t2) Hz × 1944.5 (t1) Hz, and complex data points were identical to those of the HNCACB.13−15 The sweep widths and complex points of the HSQC were 9689.9 (t2) Hz × 1944.5 (t1) Hz and 1024 (t2) × 128 (t1), respectively. For WT MLL, processing and analysis of the HNCACB data resulted in 28 non-proline, amide 1H, 15N, 13Cα, and 26 13Cβ resonance assignments and three proline 13Cα and 13Cβ resonance assignments. Twenty-seven 13CO resonance assignments were made using the HNCO data. For mutant MLL, processing and analysis of the HNCACB data resulted in 30 non-proline, amide 1H, 15N, 13Cα, and 28 13Cβ resonance assignments and one proline 13Cα and 13Cβ resonance assignment. Twenty-nine 13CO resonance assignments were made using the HNCO data.
All NMR spectra were processed with NVFx and analyzed using nmrViewJ.16,17 Apodization was achieved in the 1H, 13C, and 15N dimensions using a squared sine bell function shifted by 70°. Apodization was followed by zero filling to double the number of real data points, and linear prediction was used in the 15N dimension. The 1H carrier frequency was set on 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS was used as the reference frequency).18 Secondary chemical shift values were calculated by subtracting the residue specific random coil chemical shifts in the neighbor-corrected IDP chemical shift library (ncIDP) from the measured chemical shifts.19 Secondary structure populations were calculated with d2D using the measured proton, nitrogen, and α, β, and carbonyl carbon chemical shifts.20 The overall helicity was calculated as the mean of the per residue d2D helical population estimates.
Binding Kinetics
Binding kinetics were followed using SX18 or SX20 stopped-flow spectrometers (Applied Photophysics). Temperatures were maintained at 25 or 5 °C for p53:MDM2 or MLL:KIX, respectively. The lower temperatures were required to follow the fast observed rates of MLL:KIX. Excitation wavelengths of 493 and 593 nm were used in conjunction with 515 and 610 nm long pass filters, respectively. Data within the dead time of mixing (the first 1 ms) were removed before fitting.
Association experiments were performed under pseudo-first-order conditions, such that the concentration of the partner protein was at least 10-fold higher than the concentration of the peptide. For each concentration of excess protein, 30–70 traces for p53 were collected and averaged. Kinetic traces were fit to a single-exponential decay function to extract observed association rate constants (kobs). Association rate constants (kon) were obtained from the gradient of the straight line fit of the observed rate versus the concentration of excess protein (Figures S1a and S3b).
For dissociation experiments, 0.25–0.5 μM labeled peptide was pre-equilibrated with 1–2.5 μM partner protein and mixed with various concentrations (0–50 μM) of unlabeled ligand. Kinetic traces were fit to a single-exponential decay function to extract kobs. p53 P27A (Alexa 594-labeled) fit poorly to a single exponential and was instead fit to a double-exponential function. Two dissociation rates were also observed with p53 P27A labeled with another dye, Alexa 488, suggesting dye-specific causes were not responsible for the observed biphasic dissociation kinetics. The most likely explanation is therefore that p53 P27A follows a three-state reaction, as discussed in Figure 2. We note that WT p53 may also follow this pathway, but with rates or amplitudes that cannot be detected by stopped-flow methods. We note that this does not affect our conclusions. Dissociation rate constants (koff) were obtained either from the fit of dissociation kobs, as a function of the unlabeled ligand concentration, as described previously,21 or from the mean of the kobs obtained at concentrations of competing peptide at which kobs was essentially concentration-independent, as described previously.22
Figure 2.

Observed rate constants for the interaction between p53 and MDM2. (A) Observed rate constants (kobs) from pseudo-first-order association binding experiments, with MDM2 in excess. The association rate constant (kon) is given by the gradient of the straight line fit. (B) Dissociation kobs from competition dissociation studies. Biphasic dissociation kinetics for p53 P27A may be due to either the presence of an intermediate on the dissociation pathway or a secondary binding event. The dissociation rate constant (koff) is given by the asymptote. Error bars, representing the error of the fit, are smaller than the data points.
Fitting MLL P9/20A and L8A Binding Kinetics
Between 60 and 120 traces were collected for each concentration of excess partner protein/peptide. Each trace was individually fit to a single-exponential decay function. The individual rate constants obtained for single traces (at each concentration of excess partner protein) were plotted as histograms and fit to a Gaussian function (eq 1) to extract an average rate:
| 1 |
where a is a scaling constant, μ is the mean, and σ is the standard deviation.
To assist Gaussian fitting, only individual fitted rates between 0 and 1000 s–1 were included: rates below 0 s–1 have no physical meaning, and rates above 1000 s–1 cannot be feasibly detected with the stopped-flow setup used to collect these data.
To uncover and minimize any potential dependence of the extracted rate on the chosen bin size, 10 bin sizes (from 5 to 50 in increments of 5) were used to generate histograms. kobs was determined as the mean μ over all bin sizes. kon was obtained from the gradient of the straight line fit of kobs versus partner protein concentration. Dissociation kobs values at each concentration of excess peptide were determined as described above. koff was then obtained by taking the mean of dissociation kobs at different concentrations of out-competitor.
These described data analyses for MLL P9/20A and L8 were performed using a bespoke script created in Mathematica (Wolfram).
Results and Discussion
Using stopped-flow fluorescence, we investigated the kinetic basis for the increase in affinity of p53 for MDM2, upon mutation of helix-flanking prolines to alanine (PtoA). If the increased level of residual structure increased the proportion of binding competent p53, i.e., a conformational selection mechanism, the PtoA mutations would be expected to increase the association rate constant (kon), yet despite increasing the residual helicity within the region that becomes helical upon binding by 2.5-fold,3 an only 1.23 ± 0.06-fold increase in kon (Figure 2A) was observed for the P27A mutant. Clearly, an enhanced rate of complex formation does not therefore explain the 10-fold increase in affinity for p53 P27A.3 All other p53 proline to alanine (PtoA) mutants displayed similarly small changes in kon (Table 1). Two dissociation rate constants (koff) were observed for the P27A p53 mutant compared to one for WT p53 (Figure 2B). An approximate 12-fold reduction in koff was observed for the faster of the two rates, while the slower rate was around 60-fold lower than that of the WT (Figure 2B). Thus, the PtoA mutation increases the stability of p53:MDM2 by reducing the dissociation rate constant, suggesting that the influence of conserved helix-flanking prolines is on the stability of the bound complex.
Table 1. Peptide Helicities and Equilibrium and Rate Constantsa.
| peptide | helicity (NMR) (%) | helicity (CD) (%) | equilibrium Kd (nM) | kinetic Kd (nM) | kon (×106 M–1 s–1) | koff (s–1) |
|---|---|---|---|---|---|---|
| p53 WT | 3 | ndb | 240 ± 60 | 176 ± 8 | 25 ± 1 | 4.40 ± 0.03 |
| p53 P27A | 6 | ndb | 25 ± 3 | ndb | 30.7 ± 0.7 | fast, 0.37 ± 0.01 |
| slow, 0.072 ± 0.005 | ||||||
| p53 P12/13A | 3 | ndb | 220 ± 30 | ndb | 22 ± 2 | ndb |
| p53 P12/13/27A | 6 | ndb | 17 ± 6 | ndb | 32.4 ± 0.9 | ndb |
| MLL WT | 2 | 13 | 660 ± 60 | 530 ± 30 | 24 ± 1 | 12.4 ± 0.3 |
| MLL P21A | ndb | 14 | 700 ± 120 | 560 ± 15 | 22.0 ± 0.6 | 12.22 ± 0.08 |
| MLL P9/21A | 3 | 16 | 17000 ± 2000 | 9000 ± 6000 | 20 ± 14 | 200 ± 16 |
| MLL L8A | ndb | 12 | 35000 ± 4000 | ndb | ndb | 170 ± 14 |
The percent helicity values reported in this table were calculated as described in Materials and Methods and represent the change across the entire peptide sequence. Overall changes in helicity are predominantly due to increased helicity within the binding region (Figure 3). Errors for equilibrium Kd measurements represent the standard error of the mean (SEM; n = 3). Errors for rate constants represent the error of the fit, except for MLL P9/21A and MLL L8A, where the error represents the SEM (n = 7 and 5, respectively). Kinetic Kd was calculated as koff/kon, with the errors propagated using standard methods. Data for p53 equilibrium Kd and helicity (NMR) were taken from ref (3). A recent mutational analysis of MLL showed that the equivalent leucine to alanine mutation (L8A) was stabilizing at pH 4,23 although the authors could not measure the effects at pH 7.2 and noted that several residues displayed different behaviors with a change in pH, which might explain the differing result.
Not determined.
To determine whether this is a common function of conserved proline residues in IDPs, we investigated the interaction between the disordered transactivation domain of MLL and the folded KIX domain of CBP (Figure 1). Single (P2858A) and double (P2846/2858A) PtoA mutants were made for MLL (Figure S1). Hereafter, these MLL mutations are referenced to the peptide investigated in this study (P9A and P9/21A).
Using circular dichroism spectroscopy (CD), an increase in helicity upon PtoA mutation was observed (Figure S2A), consistent with both the previous p53 data3 and the idea that helix-flanking prolines control the level of residual helical structure.7 Such an observation might be due to a simple extension of the helix or, as observed in p53, to an increase in the stability of the helical region itself (Figure 3A). To confirm and locate the changes in helicity, MLL WT and P9/21A were investigated using NMR spectroscopy. As expected, increases in helicity were observed at and around the site of the mutation (Figure 3B and Figure S2B), with a 1.4-fold increase in helicity observed within the region that becomes helical upon binding. For MLL, mutation of the N-terminal helix-flanking proline increased the helicity to an extent greater than that seen when the C-terminal proline was mutated (Table 1).
Figure 3.
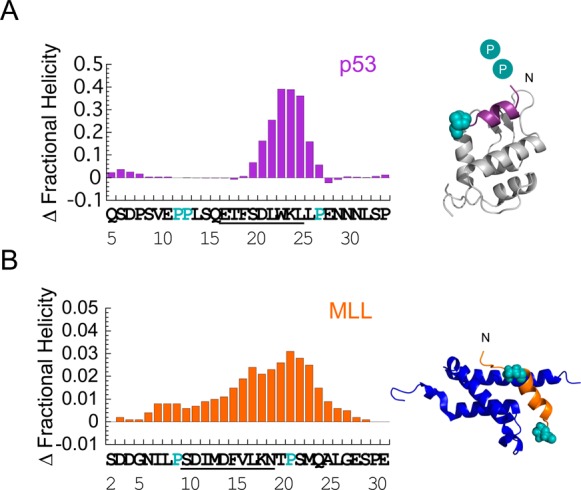
Helix-flanking prolines modulate residual structure in p53 and MLL. Change in per residue fractional helicity estimates upon PtoA mutation for (A) p53 P27A and (B) MLL P9/21A. WT residues that become helical upon binding are underlined. Conserved helix-flanking prolines are highlighted. p53 NMR helicity data are from ref (3). Individual fractional helicity plots for MLL WT and P9/21A are shown in Figure S2B. Estimates from NMR and CD gave similar changes in overall MLL helicity (Table 1). Note that the mean helicity within the region that becomes helical upon binding was increased by 2.5-fold in p53 P27A and 1.4-fold in MLL P9/21A, compared to that of the WT. Helix-flanking prolines are shown as cyan spheres in the structures of (A) p53:MDM2 (PDB entry 1YCR) and (B) MLL:KIX (PDB entry 2LXS). Cyan circles represent helix-flanking prolines that are not present in the structure.
Surprisingly, given the 1.4-fold increase in residual helicity, equilibrium binding experiments demonstrated a large, approximate 25-fold, reduction in affinity for MLL P9/21A, compared to that of the WT or P21A (Figure 4 and Figure S3A). MLL, therefore, does not fit the prevailing view that an increase in the level of residual structure enhances binding affinity.3−5 To determine the cause of these unexpected changes, we turned again to the reaction kinetics. As for the p53:MDM2 interaction, the association rate constant was almost unaffected in our PtoA mutants [a <1.2 ± 0.6-fold decrease in kon was observed for both MLL mutants (Table 1, Figure 4, and Figure S3B)]. Again, the change in affinity for MLL P9/21A was almost entirely due to changes in koff (Table 1, Figure 4, and Figure S3C). This observation has been made previously in mutagenesis studies of other peptides that form simple helices upon binding,24 and is consistent with an induced fit mechanism of binding. We note that the error in kon is large because of the difficulty in collecting the data;a however, determining kon by dividing koff by Kd gave a similar 2 ± 0.3-fold decrease in kon for MLL P9/21A.
Figure 4.
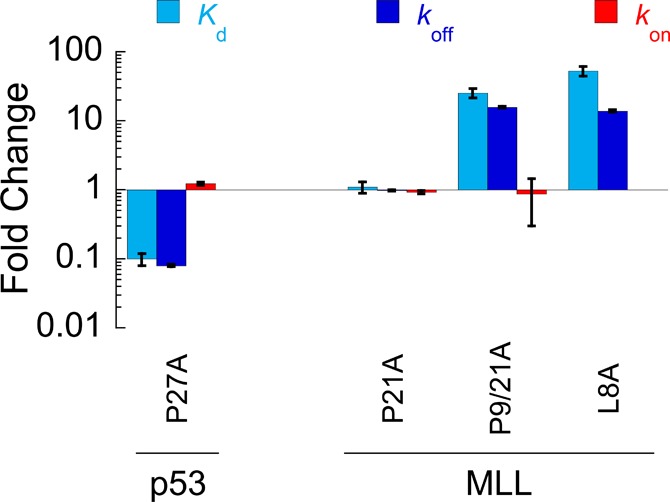
Fold change in thermodynamic and kinetic parameters upon mutation. kon not determined for MLL L8A. Kd error bars represent the standard error of the mean (SEM; n = 3). Error bars for kon and koff represent the error of the fit, except for MLL P9/21A and MLL L8A, where error bars represent the SEM (n = 7 and 5, respectively).
What causes the unexpected 25-fold reduction in affinity of MLL for KIX upon mutation of Pro9 to Ala? Like any other mutation, the PtoA mutation has the potential to remove interactions of the MLL proline side chain with KIX, which could explain the decrease in affinity. However, compared to those of WT and P21A:KIX, we had noticed a change in the maximal anisotropy of the MLL P9/21A:KIX complex (Figure S3A), and CD indicated that there was an increase in helicity within the bound complex (Figure S4A). We thus examined the bound structure. Immediately N-terminal of P9, a leucine (L8) of MLL packs into a hydrophobic pocket of KIX25 (Figure S4B). Proline residues are known to influence the conformation of preceding residues,26 so perhaps mutation of P9A could be indirectly disrupting the interaction of L8A with KIX. To test our hypothesis, we mutated L8, which we reasoned should lead to similar changes in affinity. Accordingly, mutation of L8 to Ala decreased the affinity for KIX approximately 53-fold (Table 1, Figure 4, and Figure S3A). Again, the decrease was predominantly explained by an increase in koff (Table 1, Figure 4, and Figure S3C). We note that the 13.9 ± 0.6-fold shift in koff for the MLL L8A mutants accounts for the majority of the 15.8 ± 0.5-fold change in koff observed for MLL P9A. This indicates that the disruption of the interaction of L8 with KIX is the predominant reason for the decrease in affinity upon P9A mutation, rather than a loss of proline side chain interactions.
Conclusions
In contrast to the suggestion that their role is to determine the population of structures in a binding competent state,6,7 we find that conserved IDR helix-flanking prolines control affinity by modulating the lifetime of the bound complex. Therefore, while helix-flanking prolines may reduce the level of residual free IDP/IDR helical structure, any influence on affinity occurs predominantly through effects in the bound state of the IDP/IDR:target complex. Remarkably, mutation of helix-flanking prolines to alanines was not always associated with an increase in affinity, as a significant decrease was observed for MLL:KIX. Control of IDP/IDR:target binding affinity is crucial for cellular function.3 Clearly, evolution can result in helix-flanking prolines being conserved to allow subtle, system specific, ways of controlling the affinity and lifetime of important regulatory complexes.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.7b00179.
Materials and methods used for protein expression, purification and labeling, circular dichroism, and equilibrium binding experiments; peptide and protein sequences used in this investigation (Figure S1); CD and NMR data for MLL mutants (Figure S2); thermodynamic and kinetic characterization of MLL mutants (Figure S3); and structural comparison of MLL WT:KIX and MLL P9/21A:KIX complexes (Figure S4) (PDF)
Author Present Address
∥ S.L.S.: Department of Biochemistry, University of Oxford, Oxford OX1 3QU, U.K.
This work was supported by the Wellcome Trust (WT 095195MA). J.C. is a Wellcome Trust Senior Research Fellow. M.D.C. is supported by a Biotechnology and Biological Sciences Research Council (BBSRC) studentship. G.W.D. is supported by the National Institutes of Health (2R01CA14124406-A1 and 1R01GM115556-01A1).
The authors declare no competing financial interest.
Footnotes
The small amount of complex formation (high Kd), combined with the approximate 25-fold increase in koff for MLL P9/21A, meant the kinetic traces for this mutant were short, with only small signal changes. It is challenging to extract consistent rates under these conditions, which are at the limit of operation of our stopped-flow apparatus. To address this issue, we developed a new fitting strategy, which is described in detail in Materials and Methods.
Supplementary Material
References
- Dyson H. J.; Wright P. E. (2005) Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 6, 197–208. 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- Dyson H. J.; Wright P. E. (2002) Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 12, 54–60. 10.1016/S0959-440X(02)00289-0. [DOI] [PubMed] [Google Scholar]
- Borcherds W.; Theillet F.-X.; Katzer A.; Finzel A.; Mishall K. M.; Powell A. T.; Wu H.; Manieri W.; Dieterich C.; Selenko P.; Loewer A.; Daughdrill G. W. (2014) Disorder and residual helicity alter p53-Mdm2 binding affinity and signaling in cells. Nat. Chem. Biol. 10, 1000–1002. 10.1038/nchembio.1668. [DOI] [PubMed] [Google Scholar]
- Krieger J. M.; Fusco G.; Lewitzky M.; Simister P. C.; Marchant J.; Camilloni C.; Feller S. M.; De Simone A. (2014) Conformational recognition of an intrinsically disordered protein. Biophys. J. 106, 1771–1779. 10.1016/j.bpj.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iešmantavičius V.; Dogan J.; Jemth P.; Teilum K.; Kjaergaard M. (2014) Helical propensity in an intrinsically disordered protein accelerates ligand binding. Angew. Chem., Int. Ed. 53, 1548–51. 10.1002/anie.201307712. [DOI] [PubMed] [Google Scholar]
- Theillet F.-X.; Kalmar L.; Tompa P.; Han K.-H.; Selenko P.; Dunker A. K.; Daughdrill G. W.; Uversky V. N. (2013) The alphabet of intrinsic disorder. Intrinsically Disord. Proteins 1, e24360–e24373. 10.4161/idp.24360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.; Kalmar L.; Xue B.; Tompa P.; Daughdrill G. W.; Uversky V. N.; Han K.-H. (2014) Contribution of proline to the pre-structuring tendency of transient helical secondary structure elements in intrinsically disordered proteins. Biochim. Biophys. Acta, Gen. Subj. 1840, 993–1003. 10.1016/j.bbagen.2013.10.042. [DOI] [PubMed] [Google Scholar]
- Brown C. J.; Johnson A. K.; Dunker A. K.; Daughdrill G. W. (2011) Evolution and disorder. Curr. Opin. Struct. Biol. 21, 441–446. 10.1016/j.sbi.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shammas S. L.; Crabtree M. D.; Dahal L.; Wicky B. I. M.; Clarke J. (2016) Insights into coupled folding and binding mechanisms from kinetic studies. J. Biol. Chem. 291, 6689–6695. 10.1074/jbc.R115.692715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianni S.; Dogan J.; Jemth P. (2014) Distinguishing induced fit from conformational selection. Biophys. Chem. 189, 33–39. 10.1016/j.bpc.2014.03.003. [DOI] [PubMed] [Google Scholar]
- Goujon M.; McWilliam H.; Li W.; Valentin F.; Squizzato S.; Paern J.; Lopez R. (2010) A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 38, W695–W699. 10.1093/nar/gkq313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F.; Wilm A.; Dineen D.; Gibson T. J.; Karplus K.; Li W.; Lopez R.; McWilliam H.; Remmert M.; Soding J.; Thompson J. D.; Higgins D. G.; Aniba M.; Poch O.; Thompson J.; Blackshields G.; Sievers F.; Shi W.; Wilm A.; Higgins D.; Bradley R.; Roberts A.; Smoot M.; Juvekar S.; Do J.; Dewey C.; Holmes I.; Pachter L.; Clamp M.; Cuff J.; Searle S.; Barton G.; Do C.; Mahabhashyam M.; Brudno M.; Batzoglou S.; Eddy S.; Edgar R.; Edgar R.; Finn R.; Mistry J.; Tate J.; Coggill P.; Heger A.; Pollington J.; Gavin O.; Gunasekaran P.; Ceric G.; Forslund K.; Holm L.; Sonnhammer E.; Eddy S.; Bateman A.; Gouy M.; Guindon S.; Gascuel O.; Hogeweg P.; Hesper B.; Katoh K.; Misawa K.; Kuma K.; Miyata T.; Katoh K.; Toh H.; Larkin M.; Blackshields G.; Brown N.; Chenna R.; McGettigan P.; McWilliam H.; Valentin F.; Wallace I.; Wilm A.; Lopez R.; Thompson J.; Gibson T.; Higgins D.; Lassmann T.; Sonnhammer E.; Liu Y.; Schmidt B.; Maskell D.; Löytynoja A.; Goldman N.; Mizuguchi K.; Deane C.; Blundell T.; Overington J.; Morgenstern B.; Frech K.; Dress A.; Werner T.; Notredame C.; Higgins D.; Heringa J.; Pirovano W.; Feenstra K.; Heringa J.; Söding J.; Thompson J.; Koehl P.; Ripp R.; Poch O.; Wilm A.; Higgins D.; Notredame C. (2014) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay L. E.; Keifer P.; Saarinen T. (1992) Pure Absorption Gradient Enhanced Heteronuclear Single Quantum Correlation Spectroscopy with Improved Sensitivity. J. Am. Chem. Soc. 114, 10663–10665. 10.1021/ja00052a088. [DOI] [Google Scholar]
- Muhandiram D. R.; Kay L. E. (1994) Gradient-enhanced triple-resonance three-dimensional NMR experiments with improved sensitivity. J. Magn. Reson., Ser. B 103, 203–216. 10.1006/jmrb.1994.1032. [DOI] [Google Scholar]
- Wittekind M.; Mueller L. (1993) HNCACB, a high-sensitivity 3D NMR experiment to correlate amide-proton and nitrogen resonances with the alpha- and beta-carbon resonances in proteins. J. Magn. Reson., Ser. B 101, 201–205. 10.1006/jmrb.1993.1033. [DOI] [Google Scholar]
- Johnson B. A. (2004) Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol. Biol. 278, 313–52. 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
- Johnson B. A.; Blevins R. A. (1994) NMR View: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4, 603–14. 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- Markley J. L.; Bax a; Arata Y.; Hilbers C. W.; Kaptein R.; Sykes B. D.; Wright P. E.; Wüthrich K. (1998) Recommendations for the presentation of NMR structures of proteins and nucleic acids--IUPAC-IUBMB-IUPAB Inter-Union Task Group on the standardization of data bases of protein and nucleic acid structures determined by NMR spectroscopy. Eur. J. Biochem. 256, 1–15. 10.1046/j.1432-1327.1998.2560001.x. [DOI] [PubMed] [Google Scholar]
- Tamiola K.; Acar B.; Mulder F. A. A. (2010) Sequence-specific random coil chemical shifts of intrinsically disordered proteins. J. Am. Chem. Soc. 132, 18000–18003. 10.1021/ja105656t. [DOI] [PubMed] [Google Scholar]
- Camilloni C.; De Simone A.; Vranken W. F.; Vendruscolo M. (2012) Determination of secondary structure populations in disordered states of proteins using nuclear magnetic resonance chemical shifts. Biochemistry 51, 2224–2231. 10.1021/bi3001825. [DOI] [PubMed] [Google Scholar]
- Shammas S. L.; Travis A. J.; Clarke J. (2014) Allostery within a transcription coactivator is predominantly mediated through dissociation rate constants. Proc. Natl. Acad. Sci. U. S. A. 111, 12055–12060. 10.1073/pnas.1405815111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers J. M.; Oleinikovas V.; Shammas S. L.; Wong C. T.; De Sancho D.; Baker C. M.; Clarke J. (2014) Interplay between partner and ligand facilitates the folding and binding of an intrinsically disordered protein. Proc. Natl. Acad. Sci. U. S. A. 111, 15420–15425. 10.1073/pnas.1409122111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toto A.; Gianni S. (2016) Mutational analysis of the binding-induced folding reaction of the mixed-lineage leukemia protein to the KIX domain. Biochemistry 55, 3957–3962. 10.1021/acs.biochem.6b00505. [DOI] [PubMed] [Google Scholar]
- Rogers J. M.; Wong C. T.; Clarke J. (2014) Coupled folding and binding of the disordered protein PUMA does not require particular residual structure. J. Am. Chem. Soc. 136, 5197–5200. 10.1021/ja4125065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brüschweiler S.; Konrat R.; Tollinger M. (2013) Allosteric communication in the KIX domain proceeds through dynamic repacking of the hydrophobic core. ACS Chem. Biol. 8, 1600–1610. 10.1021/cb4002188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur M. W.; Thornton J. M. (1991) Influence of proline residues on protein conformation. J. Mol. Biol. 218, 397–412. 10.1016/0022-2836(91)90721-H. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.