

Abstract
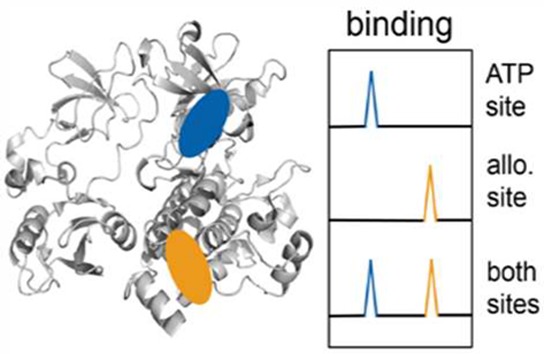
In modern kinase drug discovery, allosteric inhibitors have become a focus of attention due to their potential selectivity, but such compounds are difficult to identify. Here we describe an NMR-based competition assay using 19F-containing reporter molecules, which allows for rapid identification and discrimination between ATP-competitive and allosteric kinase inhibitors. We illustrate the principle of such a dual-site competition assay with the example of catalytic and allosteric ABL1 kinase inhibitors. The assay can also be used to identify and characterize mixed binding modes of well-known drugs, as shown for crizotinib and fingolimod.
Keywords: Reporter assay, 19F-NMR, Bcr-Abl, gatekeeper mutant, allosteric inhibition
Allosteric modulators of kinase activity are conceptually attractive as drugs for a number of reasons: They may be particularly selective because the allosteric site is less conserved than the ATP-binding site. They may cooperate with ATP-competitive inhibitors, to show synergy or additivity, so that they could be combined with ATP-site inhibitors. Allosteric modulators may also have different biological effects compared to ATP-site inhibitors, for example, providing opportunities for kinase activation, whereas orthosteric modulation inevitably leads to kinase inhibition.1,2 In spite of the great interest in such agents, it is difficult to identify and characterize allosteric kinase modulators, and hits from an unbiased screen cannot be easily classified as catalytic and allosteric modulators. NMR spectroscopy provides a robust method for the detection of weak ligands and characterization of their competitive behavior.3 Competition experiments with 19F-labeled reporter ligands are particularly attractive due to the lack of signal overlap with test compounds. We here report a study that simultaneously employed two orthogonal 19F-labeled ligands, one reporting on the catalytic site and one on the allosteric site, and we show that this dual-site assay can directly characterize test compounds as being orthosteric, allosteric, or having a mixed binding mode.
The method is illustrated for the Abelson (ABL1) kinase, which in chronic myelogenous leukemia (CML) patients is constitutively activated in the BCR-ABL1 fusion protein formed by a chromosome translocation in a hematopoietic stem cell. Tyrosine kinase inhibitors (TKIs) such as imatinib, nilotinib, and dasatinib are highly efficacious in treatment of CML and constitute current frontline therapies.4,5 These molecules all bind to the ATP-binding site within the catalytic domain of ABL1, thereby blocking kinase activity. Alternatively, small molecule inhibitors can bind to the myristate pocket of ABL1, thereby inducing a conformational change to block BCR-ABL1 by an allosteric mechanism.6,7 This class of inhibitors is exemplified by GNF-58 and ABL001 (manuscript in preparation), which is currently being evaluated in clinical trials. These allosteric BCR-ABL1 inhibitors are of clinical value because resistance mutations such as the “gatekeeper” T315I mutation9,10 can emerge, which renders current treatments with front-line orthosteric inhibitors inefficient, and the only available inhibitor of the T315I BCR-ABL1, ponatinib, has a narrow therapeutic index.11 Recently reported beneficial effects of a combination therapy with the two classes of BCR-ABL1 inhibitors12 further highlight the need for development of both catalytic and allosteric inhibitors in order to better manage the disease by controlling emergence of resistance mutations.
Characterization of kinase inhibitors according to their binding site by NMR-based competition experiments requires the availability of suitable reporter ligands. Each such ligand must report specifically on one site. In our effort to find ABL1 reporter ligands for the orthosteric and allosteric sites, we have focused on compounds containing a trifluoromethyl (CF3) group, as these can be observed in 19F-detected NMR experiments with high sensitivity, low protein consumption, and lack of signal overlap.13
The Novartis CF3 fragment library comprising 540 compounds was screened as mixtures of 30 compounds using the SH3-SH2-SH1 ABL1(83–534) construct at 4 μM protein and 25 μM ligand concentration (per fragment) in the following manner. First, binding to unliganded protein was detected by loss of the signal intensity of the respective compounds upon interaction (Supplementary Figure S1A,B). Then, the highly potent allosteric inhibitor ABL001 was added to displace myristate-pocket binders, resulting in recovery of their signal (Supplementary Figure S1C). Finally, imatinib was added (Supplementary Figure S1D) to detect ATP-site binders and compounds, which were not fully displaced by ABL001 (i.e., dual site binders). Based on the results obtained for mixtures, compounds were selected for validation as singles. From this analysis, two ATP-site and four myristate pocket reporters were found. Ligands CAT-1 and ALLO-1 (Figure 1A) were found to be optimal reporters for their respective binding site, as they bound exclusively to their respective site, were fully displaced by imatinib and ABL001, respectively, and showed no cross-affinity to the secondary site (Supplementary Figure 2). Furthermore, both reporters are commercially available.
Figure 1.
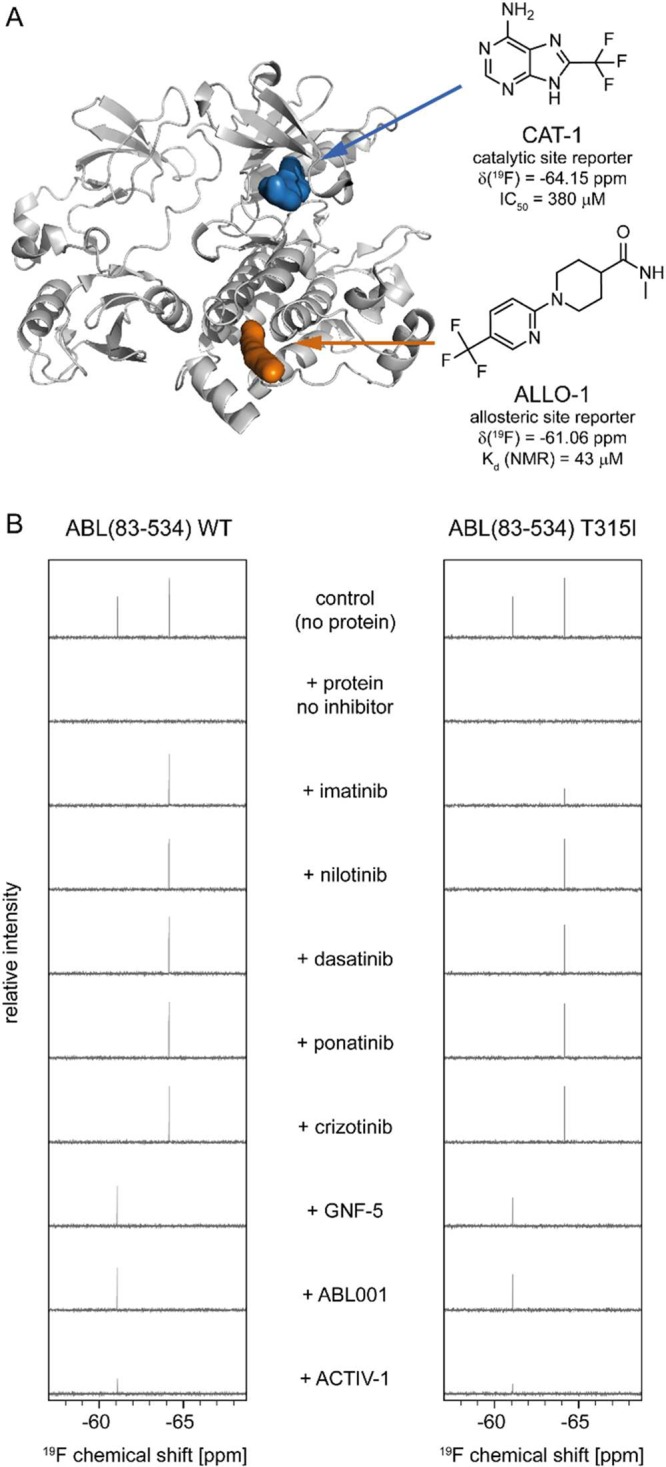
(A) Cartoon representation of the structure of Abelson kinase (pdb code: 2FO0). The binding sites for the catalytic- and allosteric-pocket reporters are indicated in blue and orange, respectively. (B) Dual-site reporter assay allows for distinction of orthosteric and allosteric binders in a panel of kinase activity modulators, for both wild-type and mutant ABL1. The protein concentration was 4 μM, and the concentrations of reporter ligand and competitor were 25 μM each. The 19F relaxation period was 320 ms.
To further characterize these molecules and their interaction with ABL1, we determined the IC50 value of CAT-1 as 380 μM in a biochemical enzyme inhibition assay. The IC50 value of ALLO-1 in this assay is not meaningful since inhibitory activity of allosteric ABL1 binders depends upon their ability to induce a conformational change in helix I within the C-lobe of the kinase domain, and the IC50 therefore does not necessarily correlate with the binding affinity.7 For ALLO-1, we have therefore measured the Kd value by NMR titration experiments and determined it to be 43 μM. This means that affinities of test compounds in the double-digit micromolar range or higher can be detected in competition experiments with ALLO-1.
CAT-1 and ALLO-1 were then employed as reporters in a dual-site competition assay to characterize the binding specificity of known ABL1 inhibitors. In this dual-site assay, the 19F-signals of both reporters disappear in the presence of SH3-SH2-SH1 ABL1(83–534), due to binding of the reporters. If an added inhibitor leads to reappearance of the signal of CAT-1, which is reporting on the catalytic (orthosteric) site, the inhibitor binds to the catalytic site. Analogously, reappearance of the ALLO-1 signal indicates binding of the inhibitor to the allosteric site. Mixed binding modes can be identified by partial reappearance of either of the two reporters.
Figure 1B shows results of the dual-site competition assay. The left panel shows results with wild-type ABL1 kinase. The left 19F-signal at −61 ppm comes from ALLO-1 and reports on the allosteric site, whereas the right signal at −64 ppm comes from CAT-1 and reports on the catalytic site. The clinically used ABL1 inhibitors imatinib, nilotinib, dasatinib, and ponatinib all fully displace CAT-1, the reporter for the catalytic site. This is expected since all of these inhibitors are known to bind to the ATP-site. It is, however, noteworthy that imatinib does not show any displacement of ALLO-1. This might have been expected based on a crystal structure of the Abelson-related kinase ABL2, which shares 94% sequence homology with ABL1 (residues 46–534 of ABL1b), in complex with imatinib (pdb code: 3GVU). In this structure, imatinib is bound within the ATP-site, and a second molecule of the inhibitor occupies the myristate pocket. The fact that imatinib does not compete with ALLO-1 binding to ABL1 indicates that its binding affinity to the myristate pocket of ABL1 has only double-digit micromolar or weaker affinity.
Since both reporters, CAT-1 and ALLO-1, bind to wild-type as well as to T315I ABL1 kinase, the inhibitors can also be tested for binding to the T315I mutant form of ABL1 kinase. The right panel of Figure 1B shows the results of analogous experiments with SH3-SH2-SH1 ABL1 T315I, for which imatinib, nilotinib, and dasatinib have greatly reduced affinity and are clinically inactive. Figure 1B shows that imatinib, the first and weakest of the three inhibitors, indeed leads to only partial displacement of CAT-1, consistent with a strongly reduced binding affinity to T315I ABL1 in the micromolar range. At the concentrations used for the experiments (25 μM), however, both nilotinib and dasatinib fully displace CAT-1 from T315I ABL1, indicating an affinity in the double-digit micromolar range or stronger. In fact, at these concentrations (which are clinically not relevant), nilotinib and dasatinib are indistinguishable from ponatinib, an inhibitor that has been specifically developed for T315I ABL1. Lowering the concentrations of reporter ligand and protein could increase the dynamic range for compound ranking to low single-digit micromolar.
Recently, crizotinib, a pan-kinase inhibitor with strong inhibition of ALK, MET, ABL1, and several other kinases,14 which is in clinical use for the treatment of nonsmall cell lung carcinoma, was suggested to be capable of allosterically inhibiting BCR-ABL1 by binding to the myristate pocket.15 Our data from the dual-site competition assay clearly show displacement of CAT-1 by crizotinib, proving binding to the catalytic site, but no displacement at all of ALLO-1 (Figure 1). This demonstrates that crizotinib has no or only very weak (triple-digit micromolar) affinity to the myristate pocket of ABL1 kinase, and that only the crizotinib action through the ATP-site is biologically relevant. Even if crizotinib bound to the ABL1 myristate pocket with relevant affinity, it would still have to induce helix I bending in order to act as an allosteric inhibitor.7
The multiple sclerosis drug fingolimod (Gilenya, FTY720) is another molecule in clinical use that has been reported to act as an allosteric ABL1 inhibitor.16−18 When tested in our assay, FTY720 was indeed found to interact with ABL1 and bind to the allosteric pocket (Figure 2), as could be expected considering structural similarities between FTY720, sphingosine, and myristic acid. However, the inhibition of BCR-ABL1 dependent proliferation of murine 32D or Ba/F3 cells is too weak (IC50 > 3 μM) for these effects to be clinically relevant.
Figure 2.
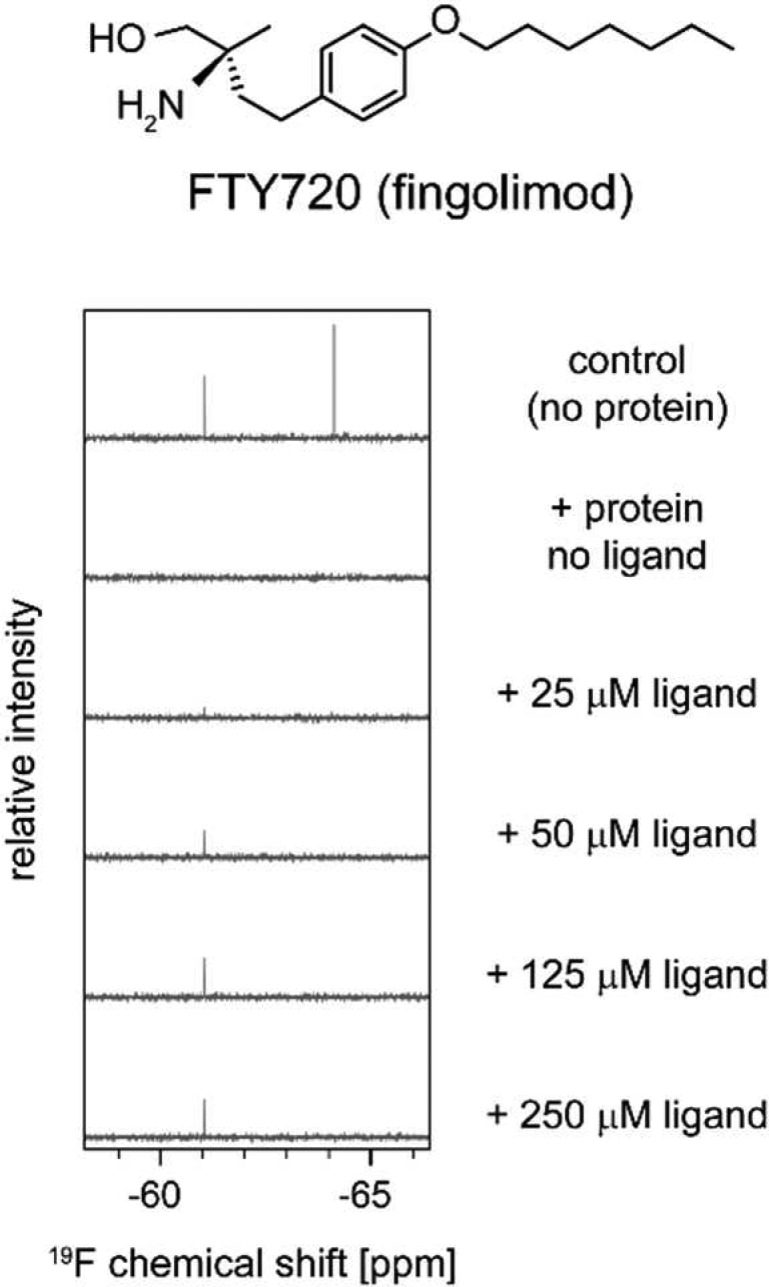
Multiple sclerosis drug fingolimod (FTY720) binds to the myristate pocket of ABL1, as indicated by displacement of the allosteric-site reporter ALLO-1.
As noted above, the reporter assay only detects binding to ABL1 but, in case of interaction with the myristate pocket, does not discriminate between allosteric activators and allosteric inhibitors. Therefore, follow-up biochemical or cellular experiments are required to investigate the effects of binding on enzymatic activity of ABL1.
The reporter assay can also be utilized to identify and characterize true dual-site binders. For example, DUAL-1, a fragment of GNF-2, was found to bind to both the allosteric pocket, and with weaker affinity also to the ATP-site. DUAL-1 is based on an aminopyrimidine scaffold, which is a classical hinge-binding motif. However, in GNF-2 this scaffold is 4,6-disubstituted rather than 2,4-disubstituted as in most classical inhibitors.19Figure 3 shows titration experiments with DUAL-1. Almost full displacement of ALLO-1, the reporter for the myristate pocket, but no displacement of CAT-1 are seen at 25 μM DUAL-1. The addition of 50 μM DUAL-1 leads to full displacement of ALLO-1 without significant effects on CAT-1. Only at 125 μM, and more pronounced at 250 μM, does CAT-1 get displaced by DUAL-1. Considering the 43 and 380 μM affinities for ALLO-1 and CAT-1, respectively, this indicates a preferential binding of DUAL-1 to the allosteric site, and corroborates that the 4,6-disubstituted aminopyrimidine is indeed not a preferred scaffold for the catalytic site.19
Figure 3.

DUAL-1 can bind to both sites on ABL1, but preferentially interacts with the myristate pocket.
In conclusion, we have developed a dual-site competition binding assay to simultaneously assess compound binding to the catalytic and allosteric pocket of ABL1 kinase in a robust and sensitive manner. In screening mode, the dual-site competition assay can immediately answer the question whether newly identified hits bind to the catalytic or allosteric pocket. In characterization mode, the mechanism of action of known inhibitors can be deconvoluted by assessing the relative affinities to the catalytic and allosteric pockets, as shown here for crizotinib and fingolimod. Even weak affinities to alternative binding sites are detectable. Ranking of ligands is possible with a dynamic range from millimolar to micromolar affinities. This approach should be applicable to other kinases, if reporters for each binding site can be identified.
Acknowledgments
The authors would like to thank Patrik Roethlisberger for performing the kinase inhibition assay and Dr. Paul Manley for critical reading of the manuscript.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00084.
Supporting Figures S1 and S2, details of experimental methods (PDF)
Author Contributions
The manuscript was written through contributions of all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Rauch J.; Volinsky N.; Romano D.; Kolch W. The secret life of kinases: functions beyond catalysis. Cell Commun. Signaling 2011, 9, 23. 10.1186/1478-811X-9-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan-Jacob S. W.; Jahnke W.; Knapp S. Novel approaches for targeting kinases: allosteric inhibition, allosteric activation and pseudokinases. Future Med. Chem. 2014, 6, 541–561. 10.4155/fmc.13.216. [DOI] [PubMed] [Google Scholar]
- Skora L.; Jahnke W. Contributions of Biomolecular NMR to Allosteric Drug Discovery. Chimia 2015, 69, 421–424. 10.2533/chimia.2015.421. [DOI] [PubMed] [Google Scholar]
- Baccarani M.; Castagnetti F.; Gugliotta G.; Rosti G. A review of the European LeukemiaNet recommendations for the management of CML. Ann. Hematol. 2015, 94, S141–S147. 10.1007/s00277-015-2322-2. [DOI] [PubMed] [Google Scholar]
- Fava C.; Rege-Cambrin G.; Saglio G. The choice of first-line chronic myelogenous leukemia treatment. Ann. Hematol. 2015, 94, S123–S131. 10.1007/s00277-015-2321-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adrián F. J.; Ding Q.; Sim T.; Velentza A.; Sloan C.; Liu Y.; Zhang G.; Hur W.; Ding S.; Manley P.; Mestan J.; Fabbro D.; Gray N. S. Allosteric inhibitors of Bcr-abl-dependent cell proliferation. Nat. Chem. Biol. 2006, 2, 95–102. 10.1038/nchembio760. [DOI] [PubMed] [Google Scholar]
- Jahnke W.; Grotzfeld R. M.; Pellé X.; Strauss A.; Fendrich G.; Cowan-Jacob S. W.; Cotesta S.; Fabbro D.; Furet P.; Mestan J.; Marzinzik A. L. Binding or bending: distinction of allosteric Abl kinase agonists from antagonists by an NMR-based conformational assay. J. Am. Chem. Soc. 2010, 132, 7043–7048. 10.1021/ja101837n. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Adrián F. J.; Jahnke W.; Cowan-Jacob S. W.; Li A. G.; Iacob R. E.; Sim T.; Powers J.; Dierks C.; Sun F.; Guo G. R.; Ding Q.; Okram B.; Choi Y.; Wojciechowski A.; Deng X.; Liu G.; Fendrich G.; Strauss A.; Vajpai N.; Grzesiek S.; Tuntland T.; Liu Y.; Bursulaya B.; Azam M.; Manley P. W.; Engen J. R.; Daley G. Q.; Warmuth M.; Gray N. S. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature 2010, 463, 501–506. 10.1038/nature08675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorre M. E.; Mohammed M.; Ellwood K.; Hsu N.; Paquette R.; Rao P. N.; Sawyers C. L. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001, 293, 876–880. 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- O’Hare T.; Eide C. A.; Deininger M. W. N. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood 2007, 110, 2242–2249. 10.1182/blood-2007-03-066936. [DOI] [PubMed] [Google Scholar]
- Fava C.; Morotti A.; Dogliotti I.; Saglio G.; Rege-Cambrin G. Update on emerging treatments for chronic myeloid leukemia. Expert Opin. Emerging Drugs 2015, 20, 183–196. 10.1517/14728214.2015.1031217. [DOI] [PubMed] [Google Scholar]
- Wylie A.; Schoepfer J.; Berellini G.; Cai H. B.; Caravatti G.; Cotesta S.; Dodd S.; Donovan J.; Erb B.; Furet P.; Gangal G.; Grotzfeld R.; Hassan Q.; Hood T.; Iyer V.; Jacob S.; Jahnke W.; Lombardo F.; Loo A.; Manley P. W.; Marzinzik A.; Palmer M.; Pelle X.; Salem B.; Sharma S.; Thohan S.; Zhu S.; Keen N.; Petruzzelli L.; Vanasse K. G.; Sellers W. R. ABL001, a Potent Allosteric Inhibitor of BCR-ABL, Prevents Emergence of Resistant Disease When Administered in Combination with Nilotinib in an in Vivo Murine Model of Chronic Myeloid Leukemia. Blood 2014, 124, 398. [Google Scholar]
- Dalvit C. Ligand- and substrate-based 19F NMR screening: Principles and applications to drug discovery. Prog. Nucl. Magn. Reson. Spectrosc. 2007, 51, 243–271. 10.1016/j.pnmrs.2007.07.002. [DOI] [Google Scholar]
- Cui J. J.; Tran-Dubé M.; Shen H.; Nambu M.; Kung P. P.; Pairish M.; Jia L.; Meng J.; Funk L.; Botrous I.; McTigue M.; Grodsky N.; Ryan K.; Padrique E.; Alton G.; Timofeevski S.; Yamazaki S.; Li Q.; Zou H.; Christensen J.; Mroczkowski B.; Bender S.; Kania R. S.; Edwards M. P. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. 10.1021/jm2007613. [DOI] [PubMed] [Google Scholar]
- Mahajna J.; Khamaisie H.; Ruimi N.; Agbarya A.; Eshel E.; Dally N.; Ottmann O.; Biondi R.; Mian A. A.; Ruthardt M. Crizotinib is an allosteric ABL-inhibitor targeting both native BCR/ABL and BCR/ABL-T315I in vitro and in vivo models of PH+ leukemia. Eur. J. Cancer 2014, 50, S191–S192. 10.1016/S0959-8049(14)50699-5. [DOI] [Google Scholar]
- Neviani P.; Santhanam R.; Oaks J. J.; Eiring A. M.; Notari M.; Blaser B. W.; Liu S.; Trotta R.; Muthusamy N.; Gambacorti-Passerini C.; Druker B. J.; Cortes J.; Marcucci G.; Chen C. S.; Verrills N. M.; Roy D. C.; Caligiuri M. A.; Bloomfield C. D.; Byrd J. C.; Perrotti D. FTY720, a new alternative for treating blast crisis chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphocytic leukemia. J. Clin. Invest. 2007, 117, 2408–2421. 10.1172/JCI31095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Chiang E. T.; Simmons J. T.; Garcia J. G.; Dudek S. M. FTY720-induced human pulmonary endothelial barrier enhancement is mediated by c-Abl. Eur. Respir. J. 2011, 38, 78–88. 10.1183/09031936.00047810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley P. W.; Cowan-Jacob S. W.; Cozens R.; Fendrich G.; Jahnke W.; Roesel J. L.; Fabbro D. Fingolimod (FTY720) Inhibits BCR-ABL Signaling Allosterically by Binding to the Myristate Binding Site. Blood 2011, 118, 2746. [Google Scholar]
- Deng X.; Okram B.; Ding Q.; Zhang J.; Choi Y.; Adrián F. J.; Wojciechowski A.; Zhang G.; Che J.; Bursulaya B.; Cowan-Jacob S. W.; Rummel G.; Sim T.; Gray N. S. Expanding the diversity of allosteric bcr-abl inhibitors. J. Med. Chem. 2010, 53, 6934–6946. 10.1021/jm100555f. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.