

Abstract

A series of new 8-nitro-6-(trifluoromethyl)-1,3-benzothiazin-4-one(BTZ) derivatives containing a C-2 nitrogen spiro-heterocycle moiety based on the structures of BTZ candidates BTZ043 and PBTZ169 were designed and synthesized as new antitubercular agents. Many of them were found to have excellent in vitro activity (MIC < 0.15 μM) against the drug susceptive Mycobacterium tuberculosis H37Rv strain and two clinically isolated multidrug-resistant strains. Compounds 11l and 11m display acceptable safety, greater aqueous solubility, and better pharmacokinetic profiles than PBTZ169, suggesting their promising potential to be lead compounds for future antitubercular drug discovery.
Keywords: Antitubercular agents, benzothiazinones, spiro-heterocycles, structure−activity relationships, pharmacokinetics
Tuberculosis (TB), one of the leading causes of death from an infectious disease ranking above HIV/AIDS, is caused mainly by Mycobacterium tuberculosis (MTB).1 The World Health Organization (WHO) estimated that approximately 10.4 million people were infected and 1.4 million died from TB worldwide in 2015.2 Recently, the spread of multidrug-resistant (MDR) TB and the emergence of extensively drug-resistant (XDR) TB have revitalized drug discovery efforts in search of new drugs.3−5 Although Bedaquiline (inhibition of mitochondrial ATP synthase) and Delamanid (inhibition of mycolic acid biosynthesis) were approved for the treatment of MDR-TB, over a huge gap of over 40 years,6,7 both of them have pronounced issues, including hERG toxicity concerns, as well as multiple ADME issues due to their high lipophilicity.3 Moreover, there are only three new chemical entities TBA-354 (nitroimidazole), PBTZ169 (benzothiazinone), and Q203 (imidazopyridine)8 currently in Phase 1 clinical trials. Therefore, there is a significant unmet medical need for safer, more effective TB drugs with novel mechanisms.
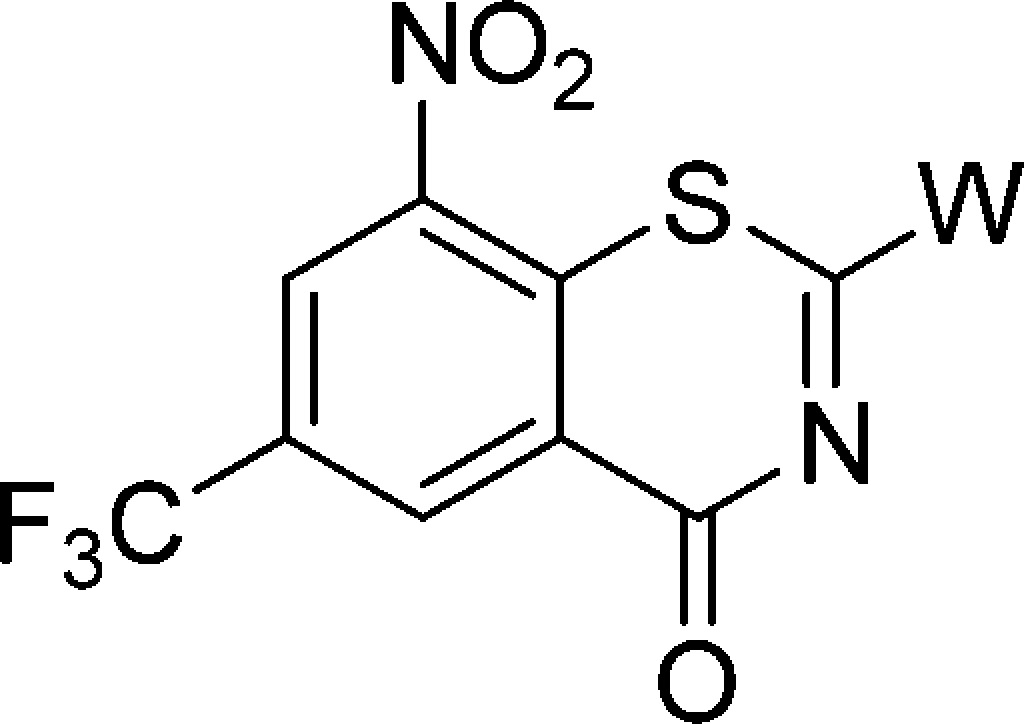
Decaprenyl phosphoryl-β-d-ribose 2′-epimerase (DprE1), an essential aspect of MTB survival and a novel mechanism of antitubercular activity, has been identified as a potential target for developing potent and safer anti-TB agents.9−11 Some new chemical entities (NCE) were found to have excellent activity against MDR/XDR-MTB as covalent or noncovalent inhibitors of the DprE1 enzyme.12−17 As the most advanced scaffold among these NCEs, nitrobenzothiazinones (BTZs) have garnered great interest recently, and many series of BTZ derivatives were reported.18−26 The structure–activity relationship (SAR) and mechanistic studies suggest that the −NO2 group at position 8 and the sulfur atom at position 1 are critical for activity, that the −CF3 at position 6 plays an important role in maintaining activity,18,19,27,28 and that structural modifications at position 2 are the most successful and efficient. For example, BTZ043 (Figure 1), a 2-spiroketal BTZ nearing phase I clinical trials, displays nanomolar bactericidal activity both in vitro and in ex vivo models of TB.18,24 PBTZ169(Figure 1), a 2-piperazino BTZ with slightly worse pharmacokinetic (PK) properties relative to BTZ043, has improved in vivo potency because of its greater solubility through protonation of the tertiary amino nitrogen on the piperazine.20 According to the latest report, a Phase I trial of PBTZ169 was completed in Russia, and a second Phase I trial will be undertaken in Switzerland in 2017.2
Figure 1.

Design of the BTZ derivatives.
In our previous study, the spiroketal moiety of BTZ043 was replaced by various cyclic ketoximes or the terminal nitrogen on the piperazine ring of BPTZ169 was shifted outside the ring, and some of the resulting compounds were found to have improved activity and aqueous solubility, or acceptable PK profiles.26 In this study, we intended to integrate the structural features of BTZ043 and PBTZ169, and construct structurally unique compounds, nitrogen spiro-heterocycles as side chains at the 2-position of BTZs (Figure 1). Clearly, these side chains combine the characteristics of both the spiro-cycle of BTZ043 and the alkaline tertiary-amine of BTZ169. The antimycobacterial activity, solubility, toxicity, and PK properties of these target compounds were evaluated, aiming to identify alternative groups at position 2 of BTZs and find optimized potent anti-TB drug candidates with improved drug-like properties through SAR study.
Detailed synthetic pathways to target compounds 11–13 are outlined in Scheme 1. Introduction of various R groups to Boc-protected spiro-heterocycles 1–3 by reductive amination with aldehydes,29 nucleophilic substitution with bromides, or amidation with carboxylic acid in the presence of EDC and HOBt30 gave compounds 4–6, which upon removal of the Boc protecting group afforded the desired side chain compounds 7–9 by TFA in DCM in good yields. Additionally, the Boc group of 3 was also reduced by LiALH4 to produce the methide 9.31 The target compounds 11–13 were conveniently obtained from 10(20) by nucleophilic substitution with spiro-heterocycle derivatives 7–9 or 1–3.
Scheme 1. Synthesis of Compounds 11–13.

Regents and conditions: (a) (i) RCHO, MeOH, AcOH, NaCNBH3, rt–50 °C, 60–85%; or (ii) RBr, MeCN, K2CO3, rt, 55–65%; or (iii) RCOOH, EDC, HOBt, Et3N, DCM, rt, 67%; (b) TFA, DCM, rt, 80–95%; (c) LiAlH4, THF, 60 °C, 65%; (d) MeOH, rt–50 °C, 43–70%.
The target compounds 11a–i, 12a–d, and 13a–c bearing different kinds of substituent groups to ensure side chain flexibility and structure diversity were first synthesized (Table 1). They were preliminarily screened for in vitro activity against MTB H37Rv ATCC27294 strain, using the Microplate Alamar Blue Assay (MABA).32 The minimum inhibitory concentration (MIC) is defined as the lowest concentration effecting a reduction in fluorescence of >90% relative to the mean of replicate bacterium-only controls. The MIC values of the compounds along with PBTZ169, isoniazid (INH), and rifampicin (RFP) for comparison were obtained from three independent experiments and presented in μM in Table 1.
Table 1. Structures and Activities of 11a–i, 12a–d, and 13a–c against MTB H37Rv.

The data reveal that with a few exceptions (11e–h, 12d, 13c), all the BTZ derivatives show considerable activity against this strain (MIC < 1 μM). Among them, compounds 12b and 12c (MIC = 0.062 and 0.060 μM) are more active than INH (MIC = 0.284 μM) and RFP (MIC = 0.092 μM), and slightly less active than PBTZ169 (MIC < 0.035 μM). The potency of the BTZ derivatives is related to the sizes of both cycles A and B in the spiro-heterocycle moiety, as well as the nature of substituent groups. When ring A is piperidine (11–13), the contribution of ring B to the activity seems to be as follows: pyrrolidine > azetidine > piperidine (11a vs 12a vs 13a; 11b vs 12b vs 13b); whereas, when ring B is piperidine, the contribution of ring A is in this order: piperidine > azetidine > pyrrolidine (11i vs 12d vs 13c). However, replacement of the cyclohexylmethyl group derived from PBTZ169 with benzyl or acetate moieties leads to increased activity (11a vs 11b–d; 12a vs 12b–c; 13a vs 13b). Removal of the cyclohexyl group (11e) or replacement of the cyclohexylmethyl with benzoyl (11h) abolishes the potency against this strain. Moreover, N-acetamide was also found to be much less active than the corresponding N-acetate (11b vs 11g; 11d vs 11f). Consequently, this set of modifications suggests that the spiro-heterocycle (piperidine as ring A) with N-benzylic group might be important for antimycobacterial activity and worthy of further optimizations, considering the probable instability in vivo of the N-acetate moiety.
Next, the optimizations were focused on R group (Table 2) of the benzene ring. An additional set of N-benzylated analogues listed in Table 2 were designed and synthesized. Considering the commercial availability of the spiro-heterocycles, compounds with piperidine as B ring synthesized from the cheapest starting material 1 were selected for the preliminary SAR study. Our results indicate that most of them show potent activity against MTB H37Rv strain (MIC < 0.15 μM) and that the most active compounds, 11m, 11o and 12f, were found to have the same MIC values of <0.035 μM as PBTZ169. Exploration of SAR at the para-position of phenyl group with piperidine as ring B was first conducted by substitution of various functional groups (11j–m). Compared with the lead compound 11c, introduction of an electron-withdrawing group (F, Br, CF3) offers more potency (11k–m), whereas the presence of an electron-donating one (CH3O) results in slightly decreased activity (11j). Moreover, a meta-fluoro atom leads to increased activity (11k vs 11n), and an additional meta-fluoro one is even more favorable (11n vs 11o). The SAR applies equally to the analogues with pyrrolidine ring as ring B. For example, introduction of an electron-donating group (CH3O) is detrimental to the potency (12c vs 12e; 12c vs 12g). Conversely, an electron-withdrawing one (F) causes improved activity (12c vs 12f). Additionally, a compound with azetidine ring as ring B (13c) was also synthesized to investigate the potential impact of the size of ring B on activity. It is shown that azetidine ≈ pyrrolidine ≫ piperidine (11j vs 12e vs 13d), suggesting that BTZs with azetidine or pyrrolidine as ring B appears to be more active than piperidine. Inspired by this, more N-benzylated analogues with azetidine or pyrrolidine as B ring are now being synthesized in our lab, and the results will be reported in due course.
Table 2. Structures and Activity of N-Benzylated Analogues against MTB H37Rv.
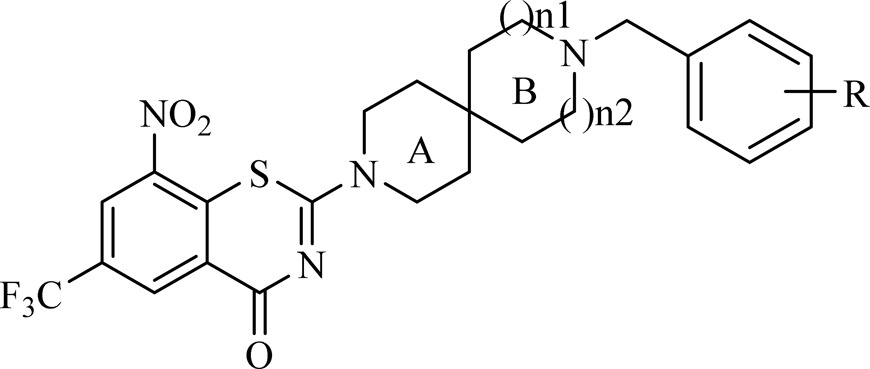
| compd. | n1 | n2 | R | LogP | MIC (μM) |
|---|---|---|---|---|---|
| 11c | 1 | 1 | H | 5.78 | 0.847 |
| 11j | 1 | 1 | 4-methoxyl | 5.91 | 1.005 |
| 11k | 1 | 1 | 4-fluoro | 5.92 | 0.210 |
| 11l | 1 | 1 | 4-bromo | 6.67 | 0.057 |
| 11m | 1 | 1 | 4-trifluoromethyl | 6.76 | <0.035 |
| 11n | 1 | 1 | 3-fluoro | 5.92 | 0.054 |
| 11o | 1 | 1 | 3,5-difluoro | 6.06 | <0.035 |
| 11p | 1 | 1 | 3,5-dimethoxyl | 6.05 | 0.825 |
| 12c | 0 | 1 | H | 5.33 | 0.059 |
| 12e | 0 | 1 | 4-methoxyl | 5.46 | 0.112 |
| 12f | 0 | 1 | 4-fluoro | 5.46 | <0.035 |
| 12g | 0 | 1 | 2-methoxyl | 5.46 | 1.676 |
| 13d | 0 | 0 | 4-methoxyl | 5.00 | 0.113 |
| PBTZ 169 | <0.035 |
It was reported there is a strong correlation between MIC and lipophilicity of N-alkylpiperazyl-BTZs.20 The logP values of the N-benzylated spiro-heterocycle series were calculated by Chemdraw 15.0 and listed in Table 2. However, no clear correlation between logP and antimycobacterial activity was found in this study.
Encouraged by their strong potency against the drug sensitive MTB H37Rv strain (MIC < 0.15 μM), compounds 11l–o, 12b-c, 12e–f, and 13d were further evaluated against two clinical isolated MTB-MDR (12525 and14231) strains resistant to both INH and RFP. As shown in Table 3, all of them exhibit excellent activity (MIC < 0.035–0.123 μM) comparable to or slightly less potent than PBTZ169, suggesting their promising potential for both drug-sensitive and resistant MTB strains (Tables 1–3).
Table 3. Activity, Solubility, Cytotoxicity and Acute Toxicity of Selected Compounds.
| MIC
(μM) |
|||||
|---|---|---|---|---|---|
| compd. | MDR-MTB 12525 | MDR-MTB 14231 | water solubilitya (mg/mL) | CC50b Vero cells (μM) | acute toxicityc |
| 11l | 0.092 | 0.086 | 1.17 | 1677.85 | 5/5 |
| 11m | <0.035 | <0.035 | 1.24 | 145.35 | 5/5 |
| 11n | <0.035 | <0.035 | 1.13 | 1398.13 | 5/5 |
| 11o | <0.035 | <0.035 | 0.67 | 1169.75 | 5/5 |
| 12b | 0.080 | 0.060 | 2.04 | 266.34 | 5/5 |
| 12c | <0.035 | <0.035 | 1.56 | 289.26 | 3/5 |
| 12e | 0.056 | 0.095 | 1.40 | 86.20 | NT |
| 12f | <0.035 | 0.034 | 1.12 | 810.61 | 4/5 |
| 13d | 0.119 | 0.123 | 1.20 | 78.86 | NT |
| PBTZ-169 | <0.035 | <0.035 | 0.90 | 1402.82 | NT |
| INH | >291.7 | 34.48 | |||
| RFP | 27.24 | >48.60 | |||
The water solubility was tested in 0.01 M HCl solution (approximate pH 2.0).
The 50% cytotoxic concentration.
No. of animals that survived/total no. of animals; NT, not tested.
Considering acidic gastrointestinal environments, compounds 11l–o, 12b–c, 12e–f, and 13d were evaluated for their water solubility at pH 2 (0.01 M HCl solution) by using an HPLC-UV method.33 As expected, the compounds containing a basic nitrogen spiro-heterocycle moiety display good solubility (0.67–2.04 mg/mL). All of them are, with the exception of 11o, more water-soluble than PBTZ169 (0.90 mg/mL). Compound 12b with an additional acetate moiety was found to have the highest solubility (2.04 mg/mL), which is more than two times that of PBTZ169 (Table 3).
Compounds 11l–o, 12b-c, 12e-f, and 13d were examined for cytotoxicity (CC50) in a mammalian Vero cell line by MTT assay,34 and the results are reported in Table 3. A comparison demonstrates that only three compounds 11l, 11n, and 11o (CC50: 1169.75–1677 μM) display comparable cytotoxic to PBTZ169 (CC50: 1402.82 μM), and compounds 12e and 13d containing a N-(4-methoxyl)benzyl group are the most cytotoxic (CC50: 86.20 and 78.86 μM). Moreover, all but one (11m) of the derivatives with piperidine as ring B have higher CC50 than those with pyrrolidine or azetidine as ring B.
Based on the measured activity levels against all of the tested strains and cytotoxicity, compounds 11l–o, 12b–c, and 12f were further tested for in vivo tolerability by recording the number of survivors after a single oral dose in mice of 500 mg/kg, followed by a 7-day observation. As shown in Table 3, all of the compounds with piperidine as ring B (11l–o) and compound 12b with an acetate moiety display the lowest oral acute lethal toxicity, followed by pyrrolidine (ring B)-containing derivatives 12f and then 12c.
The in vivo PK profiles of compounds 11l–o, 12b, and 12f were evaluated in mice after a single oral administration of 50 mg/kg. As shown in Table 4, all the tested compounds display similar or significantly longer T1/2 (3.11–10.66 h) and MRT (4.00–15.9 h) than PBTZ169 (2.87 and 3.73 h). Among the compounds with piperidine as ring B (11l–o), compounds 11l and 11m with a substituent at the para-position on benzene ring display the best PK profiles, their T1/2, Cmax, AUC0-inf, and MRT are comparable to or significantly longer/higher than PBTZ169, whereas the Cmax and AUC0-inf of compounds 11n–o with a substituent at the meta-position are similar to or significantly lower than PBTZ169. The results suggest that the presence of a substituent at the para-position on benzene ring is more favorable for the PK profiles than the meta-position. In addition, compound 12f with a para-fluoro atom on benzene ring has similar PK profiles to PBTZ169. However, compound 12b has very poor drug exposures as expected; its Cmax (17.6 ng/mL) and AUC0-inf (41.8 ng/mL) are significantly lower than PBTZ169, and the hydrolysis of the acetate moiety in acidic conditions may account for this.
Table 4. PK Profiles of 11k–n and 12b, 12e Dosed Orally in Mice at 50 mg/kg (n = 3)a.
| compd. | T1/2 (h) | Tmax (h) | Cmax (ng/mL) | AUC0-inf (h·ng/mL) | MRT (h) |
|---|---|---|---|---|---|
| 11l | 10.66 ± 5.62* | 1.00 ± 0 | 1682 ± 307 | 24619 ± 3478** | 15.9 ± 8.66* |
| 11m | 3.59 ± 0.64 | 1.33 ± 0.58 | 2700 ± 933* | 28716 ± 9878* | 6.07 ± 0.35** |
| 11n | 3.53 ± 1.63 | 0.67 ± 0.29 | 901 ± 110 | 4871 ± 935 | 4.83 ± 0.66 |
| 11o | 4.68 ± 0.92* | 0.50 ± 0 | 426 ± 243* | 2599 ± 1220 | 5.80 ± 0.84* |
| 12b | 3.46 ± 3.23 | 0.67 ± 0.29 | 17.6 ± 5.41** | 41.8 ± 33.0** | 5.20 ± 4.50 |
| 12f | 3.11 ± 0.35 | 1.08 ± 0.88 | 1234 ± 159 | 6024 ± 379 | 4.00 ± 0.85 |
| PBTZ169 | 2.87 ± 1.03 | 0.83 ± 0.29 | 1300 ± 422 | 5478 ± 1730 | 3.73 ± 0.94 |
Note: noncompartmental calculations gave these parameter estimates. *P < 0.05. **P < 0.01, one-tail t test compared with PBTZ169.
In summary, a series of new BTZ derivatives with a nitrogen spiro-heterocycle moiety at position 2 were designed as new TB agents through combining the structural features of both BTZ043 and PBTZ169. Many of them exhibit excellent in vitro inhibitory activity against both drug-sensitive MTB strain H37Rv and drug-resistant clinical isolates (MIC< 0.15 μM). Compounds 11l and 11m, with N-(4-bromobenzyl)- and N-(4-trifuorobenzyl)-3,9-diazaspiro[5,5]undecane moieties, respectively, display acceptable safety, higher aqueous solubility and better PK properties than PBTZ169, and both of them may serve as new and promising lead compounds for further antitubercular drug discovery. Studies to determine the in vivo efficacy of 11l and 11m are currently underway.
Glossary
ABBREVIATIONS
- ADME
absorption, distribution, metabolism, excretion, toxicity
- hERG
human ether-a-go-go related gene
- PK
pharmacokinetic
- TB
tuberculosis
- HIV
human immunodeficiency virus
- AIDS
acquired immune deficiency syndrome
- MTB
Mycobacterium tuberculosis
- MDR
multidrug-resistant
- XDR
extensively drug-resistant
- WHO
World Health Organization
- ATP
adenosine triphosphate
- DprE1
decaprenyl phosphoryl-β-d-ribose 2′-epimerase
- NCE
new chemical entities
- BTZs
nitrobenzothiazinones
- SAR
structure–activity relationship
- EDC
1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide
- THF
tetrahydrofuran
- HOBt
hydroxybenzotriazole
- TFA
trifluoroacetic acid
- DCM
methylene chloride
- MIC
minimum inhibitory concentration
- MABA
microplate alamar blue assay
- INH
isoniazid
- RFP
rifampicin
- logP
lipophilicity
- HPLC
high-performance liquid chromatography
- UV
ultraviolet
- CC
cytotoxicity
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- T1/2
half-life
- Cmax
maximum serum concentration
- Tmax
time at which the Cmax is observed
- AUC
area under the curve
- MRT
mean residence time
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00106.
Experimental procedures, analytical data, 1H NMR and 13C NMR copies for compounds 11–13, and HPLC copies of compound 11o for solubility determination (PDF)
Author Contributions
M.L.L., Y.L., X.F.Y., and H.Y.G. conceived and designed the project. K.L., B.W., Z.W., Y.C., B.W., and A.W. conducted the experiments, K.L. and M.L. analyzed the data and prepared the manuscript. All authors read and approved the manuscript. All authors have given approval to the final version of the manuscript.
We gratefully acknowledge the financial support by the National S&T Major Special Project on Major New Drug Innovations (2015ZX09102007-008), NSFC (81502923, 81373267, 21502237), CAMS Innovation Fund for Medical Sciences (CIFMS2016-I2M-1-010), and Beijing Municipal Administration of Hospitals Clinical Medicine Development of Special Funding Support (ZYLX201304).
All animal experiments were carried out in accordance with the guidelines of the Chinese Association for Laboratory Animal Sciences and approved by the institutional ethical committee (IEC) of Peking Union Medical College.
The authors declare no competing financial interest.
This paper was published ASAP on May 12, 2017 with errors in the abstract graphic, figure 1, and table 2. The corrected version was reposted on May 15, 2017.
Supplementary Material
References
- Nusrath Unissa A.; Hanna L. E.; Swaminathan S. A Note on Derivatives of Isoniazid, Rifampicin, and Pyrazinamide Showing Activity Against Resistant Mycobacterium tuberculosis. Chem. Biol. Drug Des. 2016, 87 (4), 537–50. 10.1111/cbdd.12684. [DOI] [PubMed] [Google Scholar]
- World Health Organization. Global Tuberculosis Report 2016. http://www.who.int/tb/publications/global_report/en/.
- Hoagland D. T.; Liu J.; Lee R. B.; Lee R. E. New agents for the treatment of drug-resistant Mycobacterium tuberculosis. Adv. Drug Delivery Rev. 2016, 102, 55–72. 10.1016/j.addr.2016.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D. Tuberculosis success. Nat. Rev. Drug Discovery 2013, 12 (3), 175–6. 10.1038/nrd3957. [DOI] [PubMed] [Google Scholar]
- Schraufnagel D.; Abubaker J. Global action against multidrug-resistant tuberculosis. JAMA, J. Am. Med. Assoc. 2000, 283 (1), 54–5. 10.1001/jama.283.1.54. [DOI] [PubMed] [Google Scholar]
- Cohen J. Infectious disease. Approval of novel TB drug celebrated--with restraint. Science 2013, 339 (6116), 130. 10.1126/science.339.6116.130. [DOI] [PubMed] [Google Scholar]
- Ryan N. J.; Lo J. H. Delamanid: first global approval. Drugs 2014, 74 (9), 1041–5. 10.1007/s40265-014-0241-5. [DOI] [PubMed] [Google Scholar]
- Pethe K.; Bifani P.; Jang J.; Kang S.; Park S.; Ahn S.; Jiricek J.; Jung J.; Jeon H. K.; Cechetto J.; Christophe T.; Lee H.; Kempf M.; Jackson M.; Lenaerts A. J.; Pham H.; Jones V.; Seo M. J.; Kim Y. M.; Seo M.; Seo J. J.; Park D.; Ko Y.; Choi I.; Kim R.; Kim S. Y.; Lim S.; Yim S. A.; Nam J.; Kang H.; Kwon H.; Oh C. T.; Cho Y.; Jang Y.; Kim J.; Chua A.; Tan B. H.; Nanjundappa M. B.; Rao S. P.; Barnes W. S.; Wintjens R.; Walker J. R.; Alonso S.; Lee S.; Kim J.; Oh S.; Oh T.; Nehrbass U.; Han S. J.; No Z.; Lee J.; Brodin P.; Cho S. N.; Nam K.; Kim J. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat. Med. 2013, 19 (9), 1157–60. 10.1038/nm.3262. [DOI] [PubMed] [Google Scholar]
- Manina G.; Pasca M. R.; Buroni S.; De Rossi E.; Riccardi G. Decaprenylphosphoryl-beta-D-ribose 2′-epimerase from Mycobacterium tuberculosis is a magic drug target. Curr. Med. Chem. 2010, 17 (27), 3099–108. 10.2174/092986710791959693. [DOI] [PubMed] [Google Scholar]
- Riccardi G.; Pasca M. R.; Chiarelli L. R.; Manina G.; Mattevi A.; Binda C. The DprE1 enzyme, one of the most vulnerable targets of Mycobacterium tuberculosis. Appl. Microbiol. Biotechnol. 2013, 97 (20), 8841–48. 10.1007/s00253-013-5218-x. [DOI] [PubMed] [Google Scholar]
- Brecik M.; Centarova I.; Mukherjee R.; Kolly G. S.; Huszar S.; Bobovska A.; Kilacskova E.; Mokosova V.; Svetlikova Z.; Sarkan M.; Neres J.; Kordulakova J.; Cole S. T.; Mikusova K. DprE1 Is a Vulnerable Tuberculosis Drug Target Due to Its Cell Wall Localization. ACS Chem. Biol. 2015, 10 (7), 1631–36. 10.1021/acschembio.5b00237. [DOI] [PubMed] [Google Scholar]
- Crellin P. K.; Brammananth R.; Coppel R. L. Decaprenylphosphoryl-beta-D-Ribose 2 ′-Epimerase, the Target of Benzothiazinones and Dinitrobenzamides, Is an Essential Enzyme in Mycobacterium smegmatis. PLoS One 2011, 6 (2), e16869. 10.1371/journal.pone.0016869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley S. A.; Grant S. S.; Kawate T.; Iwase N.; Shimizu M.; Wivagg C.; Silvis M.; Kazyanskaya E.; Aquadro J.; Golas A.; Fitzgerald M.; Dai H. Q.; Zhang L. X.; Hung D. T. Identification of Novel Inhibitors of M. tuberculosis Growth Using Whole Cell Based High-Throughput Screening. ACS Chem. Biol. 2012, 7 (8), 1377–84. 10.1021/cb300151m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F.; Sambandan D.; Halder R.; Wang J. N.; Batt S. M.; Weinrick B.; Ahmad I.; Yang P. Y.; Zhang Y.; Kim J.; Hassani M.; Huszar S.; Trefzer C.; Ma Z. K.; Kaneko T.; Mdluli K. E.; Franzblau S.; Chatterjee A. K.; Johnson K.; Mikusova K.; Besra G. S.; Futterer K.; Jacobs W. R.; Schultz P. G. Identification of a small molecule with activity against drug-resistant and persistent tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (27), 2510–17. 10.1073/pnas.1309171110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik M.; Humnabadkar V.; Tantry S. J.; Panda M.; Narayan A.; Guptha S.; Panduga V.; Manjrekar P.; Jena L. K.; Koushik K.; Shanbhag G.; Jatheendranath S.; Manjunatha M. R.; Gorai G.; Bathula C.; Rudrapatna S.; Achar V.; Sharma S.; Ambady A.; Hegde N.; Mahadevaswamy J.; Kaur P.; Sambandamurthy V. K.; Awasthy D.; Narayan C.; Ravishankar S.; Madhavapeddi P.; Reddy J.; Prabhakar K. R.; Saralaya R.; Chatterji M.; Whiteaker J.; McLaughlin B.; Chiarelli L. R.; Riccardi G.; Pasca M. R.; Binda C.; Neres J.; Dhar N.; Signorino-Gelo F.; McKinney J. D.; Ramachandran V.; Shandil R.; Tommasi R.; Iyer P. S.; Narayanan S.; Hosagrahara V.; Kavanagh S.; Dinesh N.; Ghorpade S. R. 4-Aminoquinolone Piperidine Amides: Noncovalent Inhibitors of DprE1 with Long Residence Time and Potent Antimycobacterial Activity. J. Med. Chem. 2014, 57 (12), 5419–34. 10.1021/jm5005978. [DOI] [PubMed] [Google Scholar]
- Panda M.; Ramachandran S.; Ramachandran V.; Shirude P. S.; Humnabadkar V.; Nagalapur K.; Sharma S.; Kaur P.; Guptha S.; Narayan A.; Mahadevaswamy J.; Ambady A.; Hegde N.; Rudrapatna S. S.; Hosagrahara V. P.; Sambandamurthy V. K.; Raichurkar A. Discovery of Pyrazolopyridones as a Novel Class of Noncovalent DprE1 Inhibitor with Potent Anti-Mycobacterial Activity. J. Med. Chem. 2014, 57 (11), 4761–71. 10.1021/jm5002937. [DOI] [PubMed] [Google Scholar]
- Shirude P. S.; Shandil R.; Sadler C.; Naik M.; Hosagrahara V.; Hameed S.; Shinde V.; Bathula C.; Humnabadkar V.; Kumar N.; Reddy J.; Panduga V.; Sharma S.; Ambady A.; Hegde N.; Whiteaker J.; McLaughlin R. E.; Gardner H.; Madhavapeddi P.; Ramachandran V.; Kaur P.; Narayan A.; Guptha S.; Awasthy D.; Narayan C.; Mahadevaswamy J.; Vishwas K. G.; Ahuja V.; Srivastava A.; Prabhakar K. R.; Bharath S.; Kale R.; Ramaiah M.; Choudhury N. R.; Sambandamurthy V. K.; Solapure S.; Iyer P. S.; Narayanan S.; Chatterji M. Azaindoles: Noncovalent DprE1 Inhibitors from Scaffold Morphing Efforts, Kill Mycobacterium tuberculosis and Are Efficacious in Vivo. J. Med. Chem. 2013, 56 (23), 9701–8. 10.1021/jm401382v. [DOI] [PubMed] [Google Scholar]
- Neres J.; Pojer F.; Molteni E.; Chiarelli L. R.; Dhar N.; Boy-Rottger S.; Buroni S.; Fullam E.; Degiacomi G.; Lucarelli A. P.; Read R. J.; Zanoni G.; Edmondson D. E.; De Rossi E.; Pasca M. R.; McKinney J. D.; Dyson P. J.; Riccardi G.; Mattevi A.; Cole S. T.; Binda C. Structural basis for benzothiazinone-mediated killing of Mycobacterium tuberculosis. Sci. Transl. Med. 2012, 4 (150), 150ra121. 10.1126/scitranslmed.3004395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarov V.; Manina G.; Mikusova K.; Mollmann U.; Ryabova O.; Saint-Joanis B.; Dhar N.; Pasca M. R.; Buroni S.; Lucarelli A. P.; Milano A.; De Rossi E.; Belanova M.; Bobovska A.; Dianiskova P.; Kordulakova J.; Sala C.; Fullam E.; Schneider P.; McKinney J. D.; Brodin P.; Christophe T.; Waddell S.; Butcher P.; Albrethsen J.; Rosenkrands I.; Brosch R.; Nandi V.; Bharath S.; Gaonkar S.; Shandil R. K.; Balasubramanian V.; Balganesh T.; Tyagi S.; Grosset J.; Riccardi G.; Cole S. T. Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science 2009, 324 (5928), 801–4. 10.1126/science.1171583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarov V.; Lechartier B.; Zhang M.; Neres J.; van der Sar A. M.; Raadsen S. A.; Hartkoorn R. C.; Ryabova O. B.; Vocat A.; Decosterd L. A.; Widmer N.; Buclin T.; Bitter W.; Andries K.; Pojer F.; Dyson P. J.; Cole S. T. Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol. Med. 2014, 6 (3), 372–83. 10.1002/emmm.201303575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trefzer C.; Rengifo-Gonzalez M.; Hinner M. J.; Schneider P.; Makarov V.; Cole S. T.; Johnsson K. Benzothiazinones: Prodrugs That Covalently Modify the Decaprenylphosphoryl-beta-D-ribose 2 ′-epimerase DprE1 of Mycobacterium tuberculosis. J. Am. Chem. Soc. 2010, 132 (39), 13663–65. 10.1021/ja106357w. [DOI] [PubMed] [Google Scholar]
- de Jesus Lopes Ribeiro A. L.; Degiacomi G.; Ewann F.; Buroni S.; Incandela M. L.; Chiarelli L. R.; Mori G.; Kim J.; Contreras-Dominguez M.; Park Y. S.; Han S. J.; Brodin P.; Valentini G.; Rizzi M.; Riccardi G.; Pasca M. R. Analogous Mechanisms of Resistance to Benzothiazinones and Dinitrobenzamides in Mycobacterium smegmatis. PLoS One 2011, 6 (11), e26675. 10.1371/journal.pone.0026675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karoli T.; Becker B.; Zuegg J.; Mollmann U.; Ramu S.; Huang J. X.; Cooper M. A. Identification of Antitubercular Benzothiazinone Compounds by Ligand-Based Design. J. Med. Chem. 2012, 55 (17), 7940–7944. 10.1021/jm3008882. [DOI] [PubMed] [Google Scholar]
- Pasca M. R.; Degiacomi G.; Ribeiro A. L.; Zara F.; De Mori P.; Heym B.; Mirrione M.; Brerra R.; Pagani L.; Pucillo L.; Troupioti P.; Makarov V.; Cole S. T.; Riccardi G. Clinical isolates of Mycobacterium tuberculosis in four European hospitals are uniformly susceptible to benzothiazinones. Antimicrob. Agents Chemother. 2010, 54 (4), 1616–8. 10.1128/AAC.01676-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batt S. M.; Jabeen T.; Bhowruth V.; Quill L.; Lund P. A.; Eggeling L.; Alderwick L. J.; Futterer K.; Besra G. S. Structural basis of inhibition of Mycobacterium tuberculosis DprE1 by benzothiazinone inhibitors. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (28), 11354–59. 10.1073/pnas.1205735109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R.; Lv K.; Wang B.; Li L. H.; Wang B.; Liu M. L.; Guo H. Y.; Wang A. P.; Lu Y. Design, synthesis and antitubercular evaluation of benzothiazinones containing an oximido or amino nitrogen heterocycle moiety. RSC Adv. 2017, 7 (3), 1480–83. 10.1039/C6RA25712G. [DOI] [Google Scholar]
- Kloss F.; Krchnak V.; Krchnakova A.; Schieferdecker S.; Dreisbach J.; Krone V.; Mollmann U.; Hoelscher M.; Miller M. J. In Vivo Dearomatization of the Potent Antituberculosis Agent BTZ043 via Meisenheimer Complex Formation. Angew. Chem., Int. Ed. 2017, 56 (8), 2187–91. 10.1002/anie.201609737. [DOI] [PubMed] [Google Scholar]
- Tiwari R.; Miller P. A.; Chiarelli L. R.; Mori G.; Sarkan M.; Centarova I.; Cho S. H.; Mikusova K.; Franzblau S. G.; Oliver A. G.; Miller M. J. Design, Syntheses, and Anti-TB Activity of 1,3-Benzothiazinone Azide and Click Chemistry Products Inspired by BTZ043. ACS Med. Chem. Lett. 2016, 7 (3), 266–70. 10.1021/acsmedchemlett.5b00424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter E. W. Reductive aminations of carbonyl compounds with borohydride and borane reducing agents. Organic Reactions 2001, 59, 1–714. 10.1002/0471264180.or059.01. [DOI] [Google Scholar]
- Mahmoud K. A.; Long Y. T.; Schatte G.; Kraatz H. B. Rearrangement of the active ester intermediate during HOBt/EDC amide coupling. Eur. J. Inorg. Chem. 2005, 1, 173–180. 10.1002/ejic.200400504. [DOI] [Google Scholar]
- Corruble A.; Davoust D.; Desjardins S.; Fressigne C.; Giessner-Prettre C.; Harrison-Marchand A.; Houte H.; Lasne M. C.; Maddaluno J.; Oulyadi H.; Valnot J. Y. A NMR and theoretical study of the aggregates between alkyllithium and chiral lithium amides: control of the topology through a single asymmetric center. J. Am. Chem. Soc. 2002, 124 (51), 15267–79. 10.1021/ja016945d. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Zheng M.; Wang B.; Fu L.; Zhao W.; Li P.; Xu J.; Zhu H.; Jin H.; Yin D.; Huang H.; Upton A. M.; Ma Z. Clofazimine analogs with efficacy against experimental tuberculosis and reduced potential for accumulation. Antimicrob. Agents Chemother. 2011, 55 (11), 5185–93. 10.1128/AAC.00699-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L. Q.; Zhu L.; Qian K.; Qin B.; Huang L.; Chen C. H.; Lee K. H.; Xie L. Design, synthesis, and preclinical evaluations of novel 4-substituted 1,5-diarylanilines as potent HIV-1 non-nucleoside reverse transcriptase inhibitor (NNRTI) drug candidates. J. Med. Chem. 2012, 55 (16), 7219–29. 10.1021/jm3007678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y.; Wan Z. L.; Wang B.; Guo H. Y.; Liu M. L. Synthesis and in vitro antibacterial activity of 7-(4-alkoxyimino-3-amino-3-methylpiperidin-1-yl)fluoroquinolone derivatives. Eur. J. Med. Chem. 2009, 44 (10), 4063–9. 10.1016/j.ejmech.2009.04.041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.