

Abstract
A series of TRPA1 antagonists is described which has as its core structure an indazole moiety. The physical properties and in vitro DMPK profiles are discussed. Good in vivo exposure was obtained with several analogs, allowing efficacy to be assessed in rodent models of inflammatory pain. Two compounds showed significant activity in these models when administered either systemically or topically. Protein chimeras were constructed to indicate compounds from the series bound in the S5 region of the channel, and a computational docking model was used to propose a binding mode for example compounds.
Keywords: TRPA1, Transient receptor potential, Pain, Ion channel, Indazole, Cinnamaldehyde flare, AITC, Topical administration
The transient receptor potential (TRP) family of ion channels function as sensors of multiple chemical and physical stimuli (temperature, smell, taste, and noxious chemicals).1,2 The TRPA1 (transient receptor potential ankyrin-repeat 1) channel is a nonselective cation channel that is implicated in many aspects of sensation, including pain and thermosensation.3 TRPA1 is activated by a variety of ligands, including exogenous electrophiles, such as cinnamaldehyde, acrolein, allyl isothiocyanate (AITC), and the endogenous ligand 4-hydroxynonenal. Recombinant TRPA1 is activated by noxious cold (<17 °C). Electrophilic ligands have been shown to activate TRPA1 though covalent binding to cysteine residues present in the cytoplasmic N-terminus of TRPA1 in in vitro systems.4 When activated, TRPA1 permits the conduction of sodium and calcium ions from the extracellular environment into the cell, depolarizing the membrane and affecting calcium homeostasis in the primary afferents. Depolarization of the primary nerve terminals leads to action potential firing and consequently increased pain sensation and hyperalgesia in man.5
The TRPA1 channel has been directly linked to pain in humans by a gain-of-function mutation that causes familial episodic pain syndrome.6 TRPA1 antagonists have also been shown to reverse pain in various rodent models.7 These and related data have stimulated significant interest in the biomedical industry to seek potent and selective TRPA1 antagonists. A large number of disclosures have been made of TRPA1 chemotypes from across the industry. A selection of these, 1–7, are illustrated in Figure 1, taken from patent publications and the journal literature.2,3,5 Hydra Biosciences, in partnership with Cubist Pharmaceuticals, recently advanced a TRPA1 antagonist CB-189625 into a nociceptive pain Phase 1 clinical trial.8,9 Hydra have also advanced another compound, HX-100, into trials of painful diabetic neuropathy and allergic asthma.10 Glenmark Pharmaceuticals has reported positive data in a diabetic peripheral neuropathy Phase 2 study with GRC17536 in patients with neuropathic pain and asthma.11−13 The structures of these compounds have not been disclosed as yet, but they have been described in the literature as very potent and selective TRPA1 antagonists.
Figure 1.

Examples of chemotypes with reported TRPA1 activity.
Our program started with a high-throughput screen of the internal compound library using an antagonist mode FLIPR Ca2+ imaging assay in 384-well format. All hits were confirmed in a single rig electrophysiology (EP) assay. The hit rate from this screen was low but provided compound 8 as an initial hit (Figure 2). 8 was a quite lipophilic, weak base of moderate potency, which was confirmed in a manual patch (MP) clamp EP assay to have an IC50 of 0.55 μM and low LipE. Upon further investigation, 8 was found to have some affinity for other ion channels, most notably the KCNQ2/3 channel with an activation EC50 of 37 nM and the KCNQ1 channel with an inhibition IC50 of 2.6 μM,14 although no activity at the TRPV1 channel was observed.
Figure 2.

File screening hit.
It is notable that a recent report has described a series of compounds of similar structure (e.g., compound 3) from a Novartis group.15 At this stage, we wished to explore the structure activity relationships of this series more fully, to understand the structural basis of its TRPA1 potency. The initial FLIPR screen was valuable for hit identification and triage of focused libraries. However, EP was found to be more reliable, and the development of directed compound designs was driven by EP potency. Compounds were assayed in a medium throughput EP assay using the PatchXpress (PX) platform with a HEK293-T-rex human TRPA1-expressing cell line. Selected compounds were also tested in a rat cell line, and the most advanced tool compounds were further tested in MP assays.
The first analogues explored were variations of the amino substituent of 8 (Table 1). Larger substituents such as the CF3-pyridine 9 were substantially weaker than the starting Cl substituent, whereas the equivalent phenyl group in 10 was >10-fold more active than the pyridine derivative, albeit in a more lipophilic structure with similar LipE to 8. Capping of the hydrogen bond donating groups either individually (11 and 12) or together (13) lost potency, as did removing the indazole ring substituent (14). Alternative halogen substituents on the aniline group, such as the 3,4-diF group (15), were equivalent to Cl. Similarly, Cl (16) and OCF3 (17) were found to be equivalent substituents to a CF3 at the indazole 6-position, but larger groups, as in 18 and 19, were weaker.
Table 1. Amino-indazole Analogues of Screening Hit 8.

Insertion of a nitrogen atom was very well tolerated at the indazole 4-position (20), but less so at the 5-position (21).
We combined our findings from this phase of the project into a small number of targets, and found that the amino indazole 22 was now reaching very potent levels of TRPA1 inhibition, albeit of high LogP. Further attempts to reduce lipophilicity, for example through nitrogen insertion at the 2-position of the pyridine ring (23), were unsuccessful. It should be noted that, for those examples for which rat activity was tested, rat and human potencies were typically within 10-fold of each other.
In a parallel effort, we also explored replacing the amino substituent (Table 2).
Table 2. Aryl-indazole Analogues of Screening Hit 8.

Simply removing the amino group produced a significant drop in potency (24), but inserting a nitrogen atom at the indazole 4-position allowed a modest recovery in activity (25). It was found that OCF3 versions such as 26 were of very similar TRPA1 activity and the tolerance of 6-substituents on the indazole ring was broadly similar to the amino series, with large (or more polar/charged) groups such as those in 27 and 28 less well tolerated.
An extended aryl group at the 3-position (29) was somewhat tolerated, particularly when combined with a 4-aza substitution (30). Alternative aryl groups such as a thiophene (31) were potent, but once again, polarity as in the pyridine 32 was weaker than the phenyl equivalents (33 and 34).
We therefore had a range of compounds that were either highly potent TRPA1 antagonists but were lipophilic, or examples of more polar compounds that had much weaker primary pharmacology. Examples of two compounds that possessed the former more lipophilic profile are illustrated in Table 3, in which we evaluated physicochemical and biopharmaceutic properties more fully.
Table 3. Physicochemical Data of Selected Analoguesa.
| Comp | Human TRPA1 PX est. IC50 (nM) | HLM/RLM Clint (μL/min/mg) | cLogP (LogD) | MWt | MDR1 (RRCK) (10–6 cm/s) | Aq sol (μg/mL) |
|---|---|---|---|---|---|---|
| 22 | 20 | 73/99 | 5.3 (4.6) | 330 | 2/7 (4) | 1.6 |
| 33 | 686 | <8/58 | 4.6 (4.6) | 303 | 4/8 (<0.1) | 4.8 |
The metabolism assays are as described.16 Metabolism data is reported in units of intrinsic clearance. Aqueous solubility was measured from a DMSO stock solution using a kinetic miniaturized shake flask method.
Both compounds were of low aqueous solubility and modest passive permeability, but we noted that the aryl-indazole 33 was considerably more stable in human hepatic microsomes than the amino derivative 22 (Figure 3).
Figure 3.

Example tool compounds.
We further profiled both compounds for activity against other ion channels, in particular against the KCNQ channels that the original hit 8 displayed. 22 retained KCNQ activity, while the aryl-indazole 33 had ablated this off-target activity completely. Interestingly, the latter compound was very weak in the FLIPR assay, highlighting the importance of using EP platforms for SAR development within this chemotype.
The series had therefore provided good TRPA1 potency and in the aryl-indazole series selective inhibition, but physicochemical properties were not ideal for an oral indication. We next explored the likely binding site for the series, and explored ways to evaluate the in vivo efficacy of the tool compounds.
To establish the binding site of compounds such as 22 and 33, TRPA1 species ortholog chimeras using opossum TRPA1 were generated to identify the critical regions of interaction, as described previously.1722 was >1000× more potent against hTRPA1 (IC50 = 20 nM) compared to oTRPA1 (IC50 > 30 μM). A 27 amino acid sequence within the S5 helix was swapped and 22 potency tested. The potency of the compound positively correlated with the species source of this 27 amino acid S5 region. Gain of potency was observed when the S5 region of human TRPA1 was swapped into oTRPA1 (hS5, IC50 = 132 nM). Loss of function was observed in the counterpart chimera (oS5, IC50 = 16 μM). Taken together, these data suggest the activity of 22 at TRPA1 is dependent on the S5 helix.
We then sought to provide more granularity of critical residues of interaction using computational docking. Specific residues in the S5 region have been suggested to be involved in interactions with small molecule TRPA1 modulators. Xiao et al.18 has suggested that Ser873 and Thr874 in human TRPA1 are important for small molecule binding.19 We docked 33 into the cryo-EM structure of human TRPA1 described by Paulsen et al.20 and focused on the region around Ser873 and Thr874.
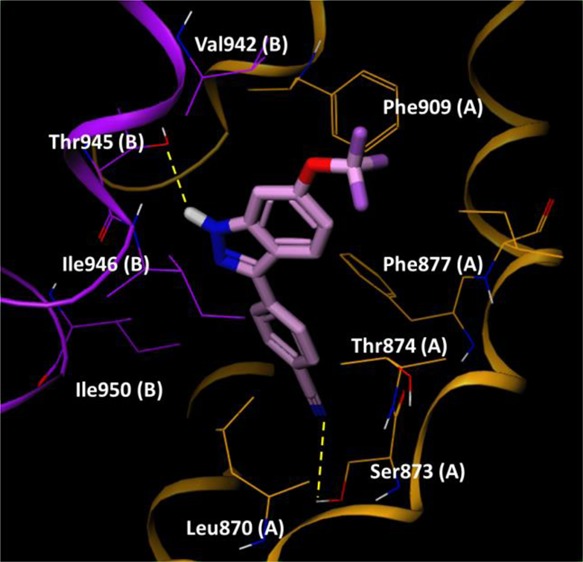
Figure 4 shows an example binding mode of 33. Two H-bond interactions stabilize the complex, formed between the OH group of Thr945 and the NH of the indazole, and the other between the CN group and the OH group of Ser873. Thr874 lies close to the indazole 4-position, which could explain the quite specific SAR in this location. 4-Aza substituents resulted in enhancement of human TRPA1 potency, potentially through the OH group of Thr874 forming an H-bond to this N. The 6-position of the indazole was located at the entrance of the binding site. We propose that Val942 in human TRPA1 when mutated to Ile945 in rat TRPA1 supports stronger hydrophobic interactions to extended 6-substituents as in 27 and 28, which show greater rat than human potency. Similar docking and binding site analysis for compound 22 also highlighted the same S5 binding region (see Supporting Information), which is very close to the region described by Grandl and co-workers for their indazole series.15
Figure 4.
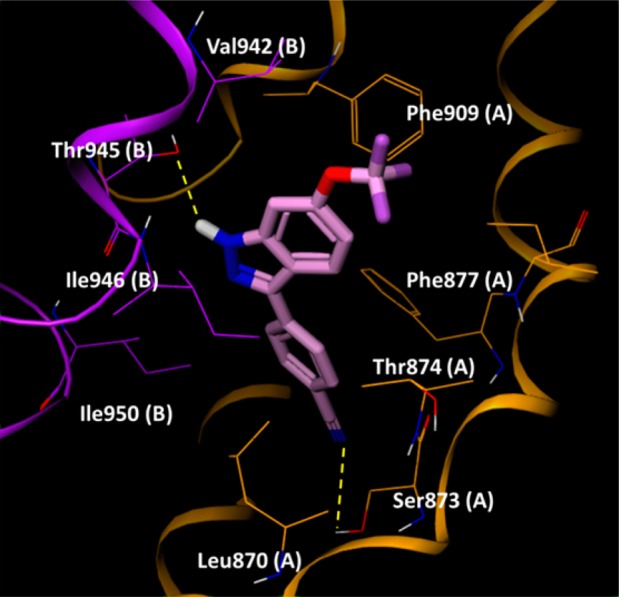
33 docked into a human TRPA1 construct. Residues from chain A and B are colored orange and purple, respectively, and highlighted residues are suffixed by chain ID for clarity.
The two tool compounds were next evaluated in two models of inflammatory pain. In these studies, we were cognizant of their lipophilicity and low solubility, and therefore that they may be unsuitable as systemic agents. We therefore also wanted to explore the potential of the indazole series to provide a topical agent.
The more metabolically stable 33 was investigated in an allyl isothiocyanate (AITC) model of inflammatory pain (see Supporting Information). After demonstrating similar hTRPA1 and rTRPA1 EP potency, we more rigorously tested rTRPA1 potency in MP using 5 pt concentration–response curves. The PX and MP IC50s were within 2-fold (PX est IC50 = 0.246 μM, MP IC50 = 0.195 μM), and thus, the MP value was used for subsequent PK/PD calculations. We also extended the selectivity testing of compound 33 to other gene family pain targets in MP. Compound 33 was >100× selective over TRPM8, TRPV3, Nav1.8, and Cav2.2. Upon oral dosing of 33 to rats treated with AITC subcutaneously in the right hind paw, a significant dose-dependent reduction in flinching was observed. 33 showed a minimum effective dose (MED) of 100 mg/kg in this model, which equated to an approximately IC50 level of free plasma exposure based on rat MP data.
We also examined the efficacy of 33 in ablating a cinnamaldehyde-induced flare response in the rat using laser Doppler sonography (Figure 5).
Figure 5.
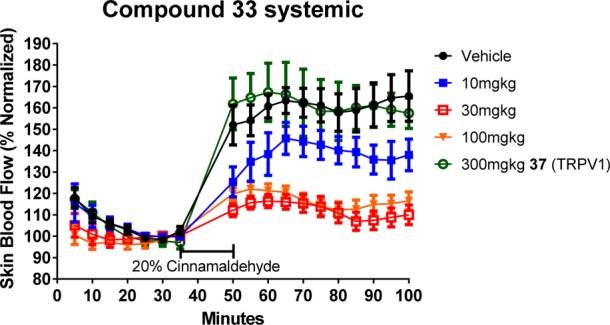
Cinnamaldehyde flare response of systemically applied 33.
Three oral doses of 33 were investigated: 10, 30, and 100 mg/kg, which achieved mean systemic concentrations of between 11 and 52 nM, below the rat MP in vitro IC50 (195 nM). An example TRPV1 antagonist 37,21−23 was used as a comparative compound of disparate mechanism in the studies, and was not expected to show efficacy in a flare study at a dose that resulted in measured plasma-free exposure of >100× the reported TRPV1 IC50 (approximately 3 nM). The laser Doppler data indicated a response at all doses of 33, which achieved a suppression of the cinnamaldehyde-induced flare response compared to both vehicle and the TRPV1 antagonist. The two higher doses showed a greater response, and overall, the level of response for these relatively low concentrations was very encouraging.
These data provided support for next looking at a topical flare study with the more potent, but less stable 22. In preparation for this study, we examined the skin flux properties of 22, which showed low skin permeability (1.2 × 10–5 cm/h) and moderate concentrations both in the epidermal layer (304 μg/g) and in the dermal layer (20 μg/g) following topical administration, confirming the compound had some skin exposure. We also confirmed that 22 had similar MP potency at the rat TRPA1 channel (IC50 11 nM). We designed a topical flare study in the rat, in which 22 was formulated in vehicle, applied to the skin, and left for a defined period of time.21 A 20% v/v formulation of cinnamaldehyde in the study vehicle was applied to the same location and blood flow then measured in that region by laser Doppler sonography.
The free concentrations achieved with a 50 mg/mL dose in all animals were in the range 7–59 nM (see Supporting Information), similar values to the in vitro IC50 in both PX and MP (rat MP IC50 11 nM), although presumably the skin concentrations of compound were significantly higher. The suppression of the flare response was very similar to that observed with the systemic application of 33, clearly showing that a topically administered TRPA1 antagonist can ablate an inflammatory flare response.
In conclusion, we have identified potent and selective inhibitors of the TRPA1 channel based on an indazole template. Efforts to enhance the in vitro ADME, off-target pharmacology, and physicochemical properties of this series met with mixed success, but allowed us to identify tool compounds that were used to confirm their region of engagement with the TRPA1 protein. An example compound from the series showed good efficacy in a rat AITC study at approximately IC50 levels of compound, and efficacy in a cinnamaldehyde flare study at sub-IC50 levels. A further example of the series was examined in a topical flare study and showed efficacy at approximately IC50 levels measured systemically. The skin is a major neurosensory organ where the symptoms of pain and itch in chronic diseases are most often first perceived; therefore, we hypothesize that indazole based inhibitors of TRPA1 could offer potential topical treatments for pain or itch.
Acknowledgments
The authors would like to thank Neil Flanagan and Thean Yeoh for providing the skin flux and skin retention data.
Glossary
Abbreviations
- TRPA1
transient receptor potential channel A1
- AITC
allyl isothiocyanate
- EP
electrophysiology
- FLIPR
fluorescent imaging plate reader
- KCNQ
potassium voltage-gated channel subfamily KQT
- MP
manual patch
- cryo-EM
cryo-electron microscopy
- PX
PatchXpress
- ADME
absorption, distribution, metabolism, elimination
- MED
minimum effective dose
- RRCK
Madin–Darby canine kidney cell line low efflux
- MDR1
multidrug resistance gene
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00140.
Detailed experimental methods, data, synthetic chemistry procedures and additional references are provided as Supporting Information in pdf format. The Supporting Information is available free of charge on the ACS Publications Web site. (PDF)
Author Present Address
∥ (D.C.P.) Curadev UK, Innovation House, Discovery Park, Sandwich Kent, CT13 9ND, UK.
All authors are or were employed by Pfizer, and all experiments described herein were carried out in Pfizer laboratories.
The authors declare the following competing financial interest(s): All authors are, or were at the time of the work being undertaken, employees of Pfizer.
Supplementary Material
References
- Kaneko Y.; Szallasi A. Transient Receptor Potential (TRP) Channels: a Clinical Perspective. Br. J. Pharmacol. 2014, 171 (10), 2474–2507. 10.1111/bph.12414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B.; Szallasi A. Transient Receptor Potential Channels as Drug Targets: From the Science of Basic Research to the Art of Medicine. Pharmacol. Rev. 2014, 66 (3), 676–814. 10.1124/pr.113.008268. [DOI] [PubMed] [Google Scholar]
- Chen J.; Hackos D. H. TRPA1 as a Drug Target – Promise and Challenges. Naunyn-Schmiedeberg's Arch. Pharmacol. 2015, 388 (4), 451–463. 10.1007/s00210-015-1088-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordt S. E.; Bautista D. M.; Chuang H. H.; McKemy D. D.; Zygmunt P. M.; Hogestatt E. D.; Meng I. D.; Julius D. Mustard Oils and Cannabinoids Excite Sensory Nerve Fibres Through the TRP channel ANKTM1. Nature 2004, 427 (6971), 260–265. 10.1038/nature02282. [DOI] [PubMed] [Google Scholar]
- Kaneko Y.; Szallasi A. Transient Receptor Potential (TRP) Channels: a Clinical Perspective. Br. J. Pharmacol. 2014, 171 (10), 2474–2507. 10.1111/bph.12414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremeyer B.; Lopera F.; Cox J. J.; Momin A.; Rugiero F.; Marsh S.; Woods C. G.; Jones N. G.; Paterson K. J.; Fricker F. R.; Villegas A.; Acosta N.; Tineda-Trujilo N. G.; Ramirez J. D.; Zea J.; Burley M.-W.; Bedoya G.; Bennett D. L. H.; Wood J. N.; Ruiz-Linares A. A Gain-of-Function Mutation in TRPA1 Causes Familial Episodic Pain Syndrome. Neuron 2010, 66 (5), 671–680. 10.1016/j.neuron.2010.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Joshi S. K.; DiDomenico S.; Perner R. J.; Mikusa J. P.; Gauvin D. M.; Segreti J. A.; Han P.; Zhang X.-F.; Niforatos W.; Bianchi B. R.; Baker S. J.; Zhong C.; Simler G. H.; McDonald H. A.; Schmidt R. G.; McGaraughty S. P.; Chu K. L.; Faltynek C. L.; Kort M. E.; Reilly R. M.; Kym P. R. Selective Blockade of TRPA1 Channel Attenuates Pathogical Pain Without Altering Noxious Cold Sensation or Body Temperature. Pain 2011, 152 (5), 1165–1172. 10.1016/j.pain.2011.01.049. [DOI] [PubMed] [Google Scholar]
- Cubist Pharmaceuticals and Hydra Biosciences Announce Plans to Begin Phase 1 Clinical Trial for Novel TRPA1 Modulator to Treat Acute Pain. http://hydrabiosciences.com/pdf/press_releases/2012_01_10.pdf.
- Preti D.; Szallasi A.; Patacchini R. TRP channels as therapeutic targets in airway disorders: a patent review. Expert Opin. Ther. Pat. 2012, 22 (6), 663–695. 10.1517/13543776.2012.696099. [DOI] [PubMed] [Google Scholar]
- Hydra Biosciences Receives Approval from Health Canada to Begin Phase 1 Trial for HX-100. http://hydrabiosciences.com/pdf/press_releases/2015_04_14.pdf.
- http://glenmarkpharma.com/sites/default/files/GlenmarksTRPA1antagonistGRC_17536.pdf.
- Mukhopadhyay I.; Kulkarni A.; Aranake S.; Karnik P.; Shetty M.; et al. Transient Receptor Potential Ankyrin 1 Receptor Activation In Vitro and In Vivo by Pro-tussive Agents: GRC 17536 as a Promising Anti-Tussive Therapeutic. PLoS One 2014, 9 (5), e97005. 10.1371/journal.pone.0097005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- http://adisinsight.springer.com/drugs/800032789.
- WO2002074388. Bisarylamines as potassium channel openers. Icagen Inc.; 11th March 2002.
- Moldenhauer H.; Latorre R.; Grandl J.. The Pore-Domain of TRPA1Mediates the Inhibitory Effect of the Antagonist 6-Methyl-5-(2-(trifluoromethyl)phenyl)-1H-indazole. PLoS One, September 2, 2014; 9, e106776. 10.1371/journal.pone.0106776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di L.; Whitney-Pickett C.; Umland J. P.; Zhang H.; Zhang X.; Gebhard D. F.; Lai Y.; Federico J. J.; Davidson R. E.; Smith R.; Reyner E. L.; Lee C.; Feng B.; Rotter C.; Varma M. V.; Kempshall S.; Fenner K.; El-Kattan A. F.; Liston T. E.; Troutman M. D. Development of a New Permeability Assay Using Low-efflux MDCKII Cells. J. Pharm. Sci. 2011, 100 (11), 4974–4985. 10.1002/jps.22674. [DOI] [PubMed] [Google Scholar]
- Pryde D. C.; Marron B.; West C. G.; Reister S.; Amato G.; Yoger K.; Padilla K.; Turner J.; Swain N. A.; Cox P. J.; Skerratt S. E.; Ryckmans T.; Blakemore D. C.; Warmus J.; Gerlach A. C. The Discovery of a Potent Series of Carboxamide TRPA1 Antagonists. MedChemComm 2016, 7, 2145–2158. 10.1039/C6MD00387G. [DOI] [Google Scholar]
- Xiao B.; Dubin A. E.; Bursulaya B.; Viswanath V.; Jegla T. J.; Patapoutian A. Identification of Transmembrane Domain 5 as a Critical Molecular Determinant of Menthol Sensitivity in Mammalian TRPA1 Channels. J. Neurosci. 2008, 28 (39), 9640–9651. 10.1523/JNEUROSCI.2772-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsuka K.; Gupta R.; Saito S.; Banzawa N.; Takahashi K.; Tominaga M.; Ohta T. Identification of Molecular Determinants for a Potent Mammalian TRPA1 Antagonist by Utilizing Species Differences. J. Mol. Neurosci. 2013, 51 (3), 754–762. 10.1007/s12031-013-0060-2. [DOI] [PubMed] [Google Scholar]
- Paulsen C. E.; Armache J.-P.; Gao Y.; Cheng Y.; Julius D. Structure of the TRPA1 Ion Channel Suggests Regulatory Mechanisms. Nature 2015, 520, 511–517. 10.1038/nature14367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WO2007100758, Amide Derivatives as Ion-Channel Ligands and Pharmaceutical Compositions and Methods of Using the Same, Renovis Inc., 7th September 2007.
- WO2006093832, Amide Derivatives as Ion-Channel Ligands and Pharmaceutical Compositions and Methods of Using the Same, Renovis Inc., 8th September 2006.
- US20060194801, Amide Derivatives as Ion-Channel Ligands and Pharmaceutical Compositions and Methods of Using the Same, Renovis Inc., 31st August 2006.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.