

Abstract
Background
Pseudomonas aeruginosa ATCC 27853 was isolated from a hospital blood specimen in 1971 and has been widely used as a model strain to survey antibiotics susceptibilities, biofilm development, and metabolic activities of Pseudomonas spp.. Although four draft genomes of P. aeruginosa ATCC 27853 have been sequenced, the complete genome of this strain is still lacking, hindering a comprehensive understanding of its physiology and functional genome.
Results
Here we sequenced and assembled the complete genome of P. aeruginosa ATCC 27853 using the Pacific Biosciences SMRT (PacBio) technology and Illumina sequencing platform. We found that accessory genes of ATCC 27853 including prophages and genomic islands (GIs) mainly contribute to the difference between P. aeruginosa ATCC 27853 and other P. aeruginosa strains. Seven prophages were identified within the genome of P. aeruginosa ATCC 27853. Of the predicted 25 GIs, three contain genes that encode monoxoygenases, dioxygenases and hydrolases that could be involved in the metabolism of aromatic compounds. Surveying virulence-related genes revealed that a series of genes that encode the B-band O-antigen of LPS are lacking in ATCC 27853. Distinctive SNPs in genes of cellular adhesion proteins such as type IV pili and flagella biosynthesis were also observed in this strain. Colony morphology analysis confirmed an enhanced biofilm formation capability of ATCC 27853 on solid agar surface compared to Pseudomonas aeruginosa PAO1. We then performed transcriptome analysis of ATCC 27853 and PAO1 using RNA-seq and compared the expression of orthologous genes to understand the functional genome and the genomic details underlying the distinctive colony morphogenesis. These analyses revealed an increased expression of genes involved in cellular adhesion and biofilm maturation such as type IV pili, exopolysaccharide and electron transport chain components in ATCC 27853 compared with PAO1. In addition, distinctive expression profiles of the virulence genes lecA, lasB, quorum sensing regulators LasI/R, and the type I, III and VI secretion systems were observed in the two strains.
Conclusions
The complete genome sequence of P. aeruginosa ATCC 27853 reveals the comprehensive genetic background of the strain, and provides genetic basis for several interesting findings about the functions of surface associated proteins, prophages, and genomic islands. Comparative transcriptome analysis of P. aeruginosa ATCC 27853 and PAO1 revealed several classes of differentially expressed genes in the two strains, underlying the genetic and molecular details of several known and yet to be explored morphological and physiological potentials of P. aeruginosa ATCC 27853.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3842-z) contains supplementary material, which is available to authorized users.
Keywords: Pseudomonas Aeruginosa, Prophage, Genomic islands, Comparative transcriptome analysis, RNA-seq, Secretion system, Colony morphology
Background
Pseudomonas aeruginosa is a gram-negative, broad-host range, opportunistic pathogen found in diverse ecological niches. It is a frequent cause of many human infectious diseases including keratitis, burn infections, urinary tract infections (UTIs), sepsis, as well as acute and chronic infections of human airways. To understand the adaptation and pathogenesis of the bacterium, comprehensive investigations of the genomes and transcriptomes of P. aeruginosa strains from various sources are necessary.
Typical P. aeruginosa strains have a large genome size of 6–7 Mb encoding around 6000 genes contributing to the versatility of the species [1, 2]. The architecture of P. aeruginosa genomes exhibit a mosaic pattern composed of a core genome (5316 core genes) and a series of accessory genes inserted sporadically, including prophages, plasmids and islets [3]. Accessory genes could be acquired by horizontal gene transfer from various sources and they often contribute to the unique physiology, pathogenesis, or transmission capacity of the corresponding strains as has been demonstrated in several P. aeruginosa isolates [4, 5]. Although over one thousand genomes (deposited in NCBI GenBank) of P. aeruginosa have been sequenced, only 58 (as of May 2016) complete genomes are available, limiting a comprehensive understanding of this important group of opportunistic pathogens.
P. aeruginosa ATCC 27853 is commonly used in biomedical research and was initially isolated from a blood specimen in the Peter Bent Brigham Hospital in 1971 (Boston, USA) [6]. ATCC 27853 has been widely used as a model strain to survey antibiotics susceptibilities since 1978 [7, 8]. So far, four draft genomes of P. aeruginosa ATCC 27853 have been sequenced [9–12], but the complete genome of the strain is still lacking, hindering the understanding of its full physiological potentials.
In the present study, we sequenced and assembled the complete genome of P. aeruginosa ATCC 27853 using both PacBio’s SMRT and Illumina platforms. We then compared it with the complete genomes of two frequently used P. aeruginosa laboratory strains, P. aeruginosa PAO1 and P. aeruginosa PA14, to reveal distinct features of the ATCC 27853 genome. To advance our understanding of the physiology of the strain, specifically its morphogenesis, we performed comparative transcriptome analysis on ATCC 27853 and PAO1. These analyses revealed the presence of a large number (seven) of prophages in its genome and several unique physiological features of ATCC 27853, implying the striking ability of the strain to adapt to a variety of environmental niches and stresses.
Results
General features of the genome of P. aeruginosa ATCC27853
A total of 1.296 Gb raw data were produced by the PacBio platform. The error correction step produced 146,425 reads with an average length of 7564 bp and a maximum length of 39,699 bp. Corrected reads were assembled de novo, the contig was then polished and circularized using the SMRT Analysis pipeline to produce a single 6.833 Mb contig with 158× coverage. The assembly was also validated by mapping Illumina-generated reads. The GC content of the genome was 66.12%, which is comparable to other genomes within the P. aeruginosa species (Additional file 1: Table S1). A total of 6366 genes were predicted. Twelve rRNA genes, 66 tRNA genes and 215 tandem repeats were identified (Table 1).
Table 1.
List of the genomic features of P. aeruginosa ATCC 27853 revealed from the complete genome (this study) and those of previous publications
| Features | ATCC 27853 | LCT-PA102 | LCT-PA41 | Boston 41,501 | LCT-PA220 |
|---|---|---|---|---|---|
| Number of scaffolds greater than or equal to 500 bp | 1 | 124(296) | 48(191) | 1(10) | 45(194) |
| Total length (bp) | 6,833,187 | 6,887,913 | 6,887,679 | 6,819,384 | 6,746,593 |
| % GC Content | 66.12% | 66.15% | 66.16% | 66.1% | 66.17% |
| Genes | 6366 | 6474 | 6476 | 6295 | 6464 |
| rRNA | 12 | - | - | 31 | - |
| tRNA | 66 | 54 | 47 | 66 | 54 |
| Tandem repeats | 215 | - | 186 | - | 210 |
| Reference | This study | Fang et al. 2012 [9] | Liu et al. 2014 [10] | Minogue et al. 2014 [11] | Xu et al. 2014 [12] |
“-” indicates that data is not available
Phylogenetic relationship of the ATCC 27853 with other P. aeruginosa strains based on SNPs from all complete genomes
Since the 16S rRNA genes in the different strains of the P. aeruginosa species exhibit high similarity (>99%, data not shown) with low discriminating capability, single nucleotide polymorphisms (SNPs) were used to construct the phylogenetic relationship between ATCC 27853 and published strains. Using Harvest [13], we collected 269,561 SNPs from the complete genomes included. We generated the phylogenetic tree in MEGA [14] based on the maximum likelihood (ML) algorithm. It became apparent that P. aeruginosa ATCC 27853 is closely related to P. aeruginosa T38079, P. aeruginosa F9670 and P. aeruginosa S86968, all of which are clinical isolates (Fig. 1, Additional file 1: Table S1).
Fig. 1.

Phylogenetic relationships of the currently available 59 complete genomes of Pseudomonas aeruginosa constructed based on the SNPs identified using Harvest with 100 bootstrap and maximum likelihood (ML) criterion in MEGA software. P. aeruginosa ATCC 27853 is highlighted in blue and italic style. The denotation of the strain is listed in the Additional file 1: Table S1
COG comparison
We compared Clusters of Orthologous Groups (COG) annotations of P. aeruginosa ATCC 27853 with those of P. aeruginosa PAO1, P. aeruginosa PA14 and P. aeruginosa LESB58 (an epidemic strain with known prophage functions) (Fig. 2 and Table 2). A total of 41 COGs are exclusively present in P. aeruginosa ATCC 27853 (Fig. 2 and Table 2), a much higher number if compared with the unique COGs in the other three genomes (Fig. 2). Most of these COGs are phage and plasmid proteins, consistent with the high number of prophages (seven) identified in P. aeruginosa ATCC 27853 (below). In addition, 58 COGs in P. aeruginosa ATCC 27853 are absent in P. aeruginosa PAO1. Nineteen of these genes have uncharacterized functions or with only hypothetical functions (Table 2). Several site-specific DNA methylase (COG0270 and COG0338) are also present in the list (Table 2).
Fig. 2.
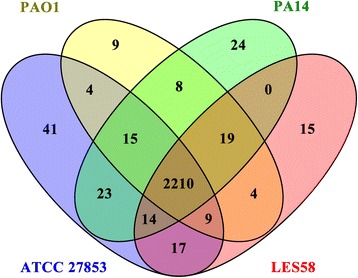
Venn diagram showing the number of shared and exclusive genes among four P. aeruginosa strains: P. aeruginosa ATCC 27853, P. aeruginosa PAO1, P. aeruginosa PA14, P. aeruginosa LESB58. The number of unique genes, those shared among two, three and all four strains of ATCC 27853, PAO1, PA14 and LESB58 strains based on the COG gene annotations are shown
Table 2.
COG identified in P. aeruginosa ATCC 27853 but absent in PAO1
| COG ID | Gene locus tag | Annotation |
|---|---|---|
| COG0213 | ACG06_12750 | Thymidine phosphorylase |
| COG0270 | ACG06_12115 ACG06_23945 |
Site-specific DNA methylase |
| COG0338 | ACG06_04040 ACG06_25705 |
Site-specific DNA methylase |
| COG0641 | ACG06_22850 | Arylsulfatase regulator (Fe-S oxidoreductase) |
| COG0798 | ACG06_13015 | Arsenite efflux pump ACR3 and related permeases |
| COG1205 | ACG06_26950 | Distinct helicase family with a unique C-terminal domain including a metal-binding cysteine cluster |
| COG1223 | ACG06_23485 | Predicted ATPase (AAA+ superfamily) |
| COG1479 | ACG06_19220 | Uncharacterized conserved protein |
| COG1541 | ACG06_14110 | Coenzyme F390 synthetase |
| COG1783 | ACG06_12260 | Phage terminase large subunit |
| COG2189 | ACG06_23500 | Adenine specific DNA methylase Mod |
| COG2253 | ACG06_23380 | Uncharacterized conserved protein |
| COG2372 | ACG06_12630 | Uncharacterized protein, homolog of Cu resistance protein CopC |
| COG2856 | ACG06_29550 | Predicted Zn peptidase |
| COG3383 | ACG06_20450 | Uncharacterized anaerobic dehydrogenase |
| COG3421 | ACG06_23505 | Uncharacterized protein conserved in bacteria |
| COG3440 | ACG06_26970 | Predicted restriction endonuclease |
| COG3464 | ACG06_12825 | Transposase and inactivated derivatives |
| COG3567 | ACG06_12270 | Uncharacterized protein conserved in bacteria |
| COG3575 | ACG06_27620 | Uncharacterized protein conserved in bacteria |
| COG3657 | ACG06_25795 | Uncharacterized protein conserved in bacteria |
| COG3723 | ACG06_12140 | Recombinational DNA repair protein (RecE pathway) |
| COG3762 | ACG06_12760 | Predicted membrane protein |
| COG4096 | ACG06_22820 | Type I site-specific restriction-modification system, R (restriction) subunit and related helicases |
| COG4245 | ACG06_22780 | Uncharacterized protein encoded in toxicity protection region of plasmid R478, contains von Willebrand factor (vWF) domain |
| COG4248 | ACG06_22770 | Uncharacterized protein with protein kinase and helix-hairpin-helix DNA-binding domains |
| COG4371 | ACG06_15075 ACG06_15120 ACG06_15175 ACG06_15230 |
Predicted membrane protein |
| COG4373 | ACG06_03925 | Mu-like prophage FluMu protein gp28 |
| COG4387 | ACG06_03965 | Mu-like prophage protein gp36 |
| COG4570 | ACG06_12235 | Holliday junction resolvase |
| COG4938 | ACG06_22795 | Uncharacterized conserved protein |
| COG5005 | ACG06_03940 | Mu-like prophage protein gpG |
| COG5268 | ACG06_18730 | Type IV secretory pathway, TrbD component |
| COG5283 | ACG06_23880 | Phage-related tail protein |
| COG5484 | ACG06_23765 | Uncharacterized conserved protein |
| COG5492 | ACG06_12325 | Bacterial surface proteins containing Ig-like domains |
| COG5518 | ACG06_23760 | Bacteriophage capsid portal protein |
| COG5569 | ACG06_12595 | Uncharacterized conserved protein |
| COG5639 | ACG06_18690 | Uncharacterized conserved small protein |
Genomic islands
A total of 25 genomic islands (GIs) were identified in the genome of P. aeruginosa ATCC 27853 by IslandViewer [15] using SIGI-HMM [16] and IslandPath-DIMOB [17] algorithms. The lengths of these GIs range from 4055 bp to 36,677 bp with four GIs associated with prophages (Table 3, and below). Some genes in the remaining GIs were assigned to functional groups including metal resistance, virulence, regulatory proteins etc. (Table 3). Knowledge of the exact functions of these genes would require further investigations. Compared with PAO1, three GIs that are unique to P. aeruginosa ATCC 27853 contain a number of genes encoding monoxoygenase, dioxygenase and hydrolase, which are likely responsible for catabolism of aromatic compounds. Genes in these GIs were not annotated as they only displayed high similarity to certain genes present in a handful of draft genomes of P. aeruginosa strains that lack functional annotation.
Table 3.
List of genomic islands identified in P. aeruginosa ATCC 27853
| Start | End | Size (bp) | Gene locus tag range | Mainly annotated genes |
|---|---|---|---|---|
| 1,208,749 | 1,213,145 | 4396 | ACG06_05790- ACG06_05800 | - |
| 1,357,527 | 1,363,441 | 5914 | ACG06_06510- ACG06_06545 | Prophage 3 |
| 1,369,295 | 1,374,450 | 5155 | ACG06_06585 ACG06_06595 | Prophage 3 |
| 1,375,007 | 1,379,950 | 4943 | ACG06_06610- ACG06_06645 | Prophage 3 |
| 2,433,632 | 2,438,325 | 4693 | ACG06_11700- ACG06_11730 | General secretion pathway protein |
| 2,550,405 | 2,554,460 | 4055 | ACG06_12335- ACG06_12355 | Prophage 4 |
| 2,635,133 | 2,641,374 | 6241 | ACG06_12645- ACG06_12670 | Heavy metal, cooper response |
| 2,668,513 | 2,677,091 | 8578 | ACG06_12800- ACG06_12845 | Mercuric resistance |
| 2,690,535 | 2,695,956 | 5421 | ACG06_12920- ACG06_12960 | - |
| 2,736,058 | 2,742,733 | 6675 | ACG06_13180- ACG06_13200 | Virulence |
| 3,210,856 | 3,216,077 | 5221 | ACG06_14975- ACG06_15000 | Hydrolase |
| 3,217,225 | 3,221,802 | 4577 | ACG06_15010- ACG06_15040 | Hypothetical protein |
| 3,260,302 | 3,265,059 | 4757 | ACG06_15285- ACG06_15305 | Antibiotics biosynthesis |
| 3,271,888 | 3,278,016 | 6128 | ACG06_15330- ACG06_15350 | Monooxygenase and hydrolase |
| 4,118,826 | 4,132,842 | 14,016 | ACG06_19215 ACG06_19245 | Integrase and dehydrogenase |
| 4,345,306 | 4,358,424 | 13,118 | ACG06_20250- ACG06_20300 | - |
| 4,855,376 | 4,892,053 | 36,677 | ACG06_22680- ACG06_22805 | TetR family Transcriptional regulator |
| 4,899,363 | 4,906,116 | 6753 | ACG06_22830- ACG06_22850 | Hypothetical protein |
| 5,064,552 | 5,079,564 | 15,012 | ACG06_23610- ACG06_23680 | PFGI-1-like_cluster_1 |
| 5,089,064 | 5,097,821 | 8757 | ACG06_23720- ACG06_23755 | Peptidase and Thioredoxin |
| 5,506,547 | 5,511,513 | 4966 | ACG06_25640- ACG06_25670 | Prophage 6 |
| 5,573,404 | 5,577,851 | 4447 | ACG06_26025- ACG06_26055 | Mercuric resistance |
| 5,597,700 | 5,606,740 | 9040 | ACG06_26180- ACG06_26220 | Transcriptional regulator |
| 5,757,464 | 5,780,636 | 23,172 | ACG06_26905- ACG06_26970 | Bacterial regulatory proteins, AsnC family |
| 6,493,061 | 6,497,532 | 4471 | ACG06_30145- ACG06_30150 | Hypothetical protein |
Prophages
Prophage prediction using Prophinder [18] and PHAST [19] revealed seven prophages in the genome of P. aeruginosa ATCC 27853. All these prophages were assigned as accessory genes and are designated as Prophage 1–7 (Table 4, Fig. 3). Prophage 1 which is closely related to phi CTX is located between genes encoding anthranilate synthase component I and component II. It is noteworthy that this prophage is observed in all available genomes of P. aeruginosa and its genomic location (between trpE and trpG genes) is also highly conserved, based on the PHASTER database [20]. The specific location of Prophage1 and its effect on the physiology of the P. aeruginosa host, particularly the antranilate biosynthesis, remain to be explored. Prophage 2 is 38,604 bp and harbors 50 open reading frames (ORFs). It is located between 797,729–836,333, upstream of the first phenazine biosynthesis gene cluster phz1 (see below) (Fig. 4). This prophage does not interrupt any genes involved in phenazine biosynthesis (Fig. 4). Most ORFs in this prophage encode phage components such as phage head and tail, transposases and integrases (Fig. 3 and Additional file 1: Table S2). Besides these structural genes, one transcription factor which belongs to the DNA-binding IclR family could be annotated. A previous study showed that this prophage shares high similarity with prophage B3, a Mu-Like bacteriophage identified by Braid et al. (2004) [21]. Interestingly, prophage prediction in the complete genomes of P. aeruginosa revealed that this prophage exists in a few other P. aeruginosa strains such as NCGM2.S1, VRFPA04 and Carb01_63, but in different genome locations and with distinctive flanking regions (Additional file 1: Figure S1).
Table 4.
List of prophages identified in P. aeruginosa ATCC 27853
| Prophage ID | Score | Normalized | Coordinate range | Length | Win size | Close phage |
|---|---|---|---|---|---|---|
| Prophage 1 | 15.3525 | 0.9031 | 683,173–696,044 | 12,871 | 20 | phi CTX |
| Prophage 2 | 63.7992 | 1.3292 | 797,729–836,333 | 38,604 | 50 | B3 |
| Prophage 3 | 44.6107 | 0.6759 | 1,337,276–1,379,950 | 42,674 | 100 | F10/lambda |
| Prophage 4 | 53.0771 | 0.8042 | 2,508,898–2,560,402 | 51,504 | 100 | D3/ES18 |
| Prophage 5 | 47.7643 | 0.7582 | 5,093,820–5,142,761 | 48,941 | 100 | phi CTX |
| Prophage 6 | 1.6659 | 0.0490 | 5,504,457–5,531,885 | 27,428 | 50 | B3 |
| Prophage 7 | 13.3825 | 0.8364 | 5,783,641–5,794,256 | 10,615 | 20 | Pf1 |
Score. It is a significance score from Prophinder, more reliable prediction with higher score
Normalised. Each significant score is normalized based on its number of CDS
Win size. Prophinder runs with different window sizes to screen the genome. Here is reported with which window size the prophage was detected
Fig. 3.
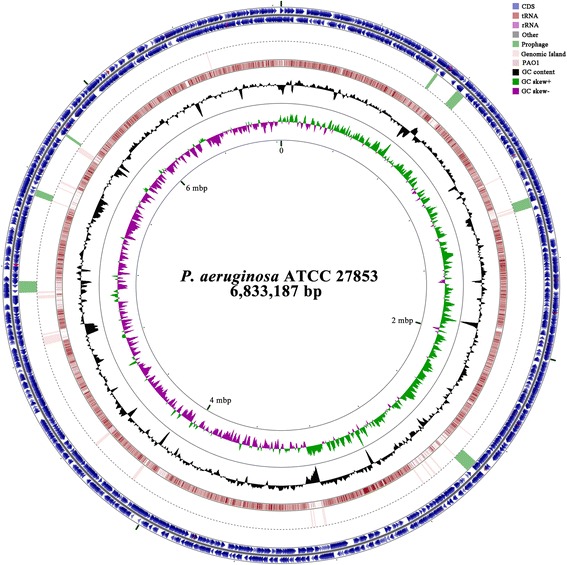
Circular genome map of P. aeruginosa ATCC 27853 showing the Genomic Islands (GIs) predicted by IslandViewer and prophages. From the outside: circles 1 and 2 (clockwise and counterclockwise) genes on the + and - strands, respectively; circles 3, prophages; 4, Genomic Islands; 5, PAO1 genes; 6, GC content; 7, GC skew. The scale in mbp is indicated on the innermost of the map
Fig. 4.
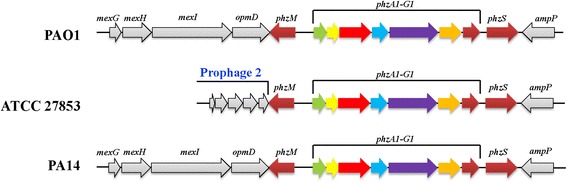
Comparison of the gene cluster of phenazine biosynthesis (Phz1) and its flanking regions in three strains of P. aeruginosa: ATCC 27853, PAO1 and PA14. Genomic location of the prophage 2 upstream of phz1 gene cluster in ATCC 27853 is highlighted
Prophage 3 is located at the genomic site of 1,337,276–1,379,950 with a size of 42,674 bp. Several genes that encode virulence associated proteins and transcriptional regulators were also identified, such as ACG06_06430 (gene locus tag in the genome of ATCC 27853 annotated by NCBI) which belongs to the LuxR family transcription factor that modulate quorum sensing [22]. Prophage 4 is the largest predicted prophages in ATCC 27853 genome and is composed of genes from different prophages such as phages ES18 and D3, indicating a complicated evolutionary history. In addition to typical phage components, other genes contained in the predicted prophages in the genome of P. aeruginosa ATCC 27853 include those of virulence factors and other functional genes, e.g. an adenylate kinase in Prophage 5.
Phenazine biosynthesis
Phenazine compounds comprise an important class of secondary metabolites and virulence factors in Pseudomonas species. All phenazines contain a dibenzol annulated pyrazine ring represented by several structurally related compounds [23]. In most of the annotated P. aeruginosa genomes, two clusters of genes that encode phenazine biosynthetic pathways (Phz1 and Phz2) are present. The genes in the phenazine biosynthesis in ATCC 27853 and PAO1are highly similar (98.98 to 99.70% at nucleotide level). However, the phz1 gene cluster in ATCC 27853 is preceded by Prophage 2 island (see above, ORFs with gene locus tags: ACG06_03785-ACG06_04040) (Fig. 4). On the other hand, the orthologous gene cluster of phz1 in PAO1 (genes: PA4209-PA4217, gene locus tag in PAO1 from annotations by NCBI), is precededby a large fragment encompassing opmD, mexI, mexH and mexG genes (genes: PA4120-PA4208, Fig. 4) which are components of a Resistance Nodulation Division (RND) type efflux system and is proposed to pump the phenazine derivate 5-methylphenazine-1-carboxylate (5-Me-PCA) out of the cell [24]. These genes were absent in P. aeruginosa ATCC 27853. To examine whether this genomic difference affects phenazine production pattern, we measured the production of a major phenazine compound in P. aeruginosa, pyocyanin (PYO), in the two strains cultured in LB medium at room temperature. We observed a higher level of PYO in ATCC 27853 than in PAO1 at all time points examined (Fig. 5), suggesting that the different genomic architecture flanking the phz1 gene cluster may indeed affect the PYO production in P. aeruginosa strains [25, 26].
Fig. 5.
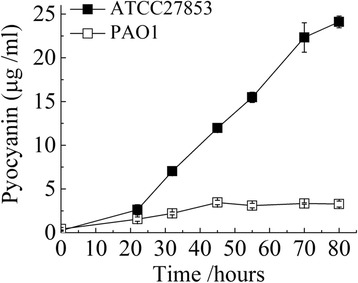
Measurements of pyocyanin in P. aeruginosa ATCC 27853 and PAO1 cultured in LB media
Virulence, surface-associated, and motility proteins
We compiled a database of 369 virulence genes based on a list of conserved virulence factors of Pseudomonas species with a primary focus on P. aeruginosa PAO1 and P. aeruginosa PA14 using the Virulence Factor Database (VFDB) [27] and the Victors Virulence Factors (PHIDIAS) (http://www.phidias.us/victors/index.php). Comparing ATCC 27853 genome against this database revealed that 254 of these virulence genes are also present in the genome of P. aeruginosa ATCC 27853 (Table 5). A class of virulence genes that are absent in P. aeruginosa ATCC 27853 include the wbp genes which encode the B-band lipopolysaccharide O antigen, with the exception of wbpX. B-band O-antigen of the lipopolysaccharide serotype O5 (such as that in PAO1) is important in conferring serum resistance in host pathogen interactions. Its presence or absence has also been shown to influence biofilm formation of the corresponding strain due to its capability to influence the hydrophilicity of cell surfaces and consequently the interaction of the cell with different surface materials and neighboring environment [28]. Absence of this system in P. aeruginosa ATCC 27853 probably indicates a defect in its defense mechanism against the host serum system and an altered biofilm formation capacity from that of the B+ strains such as PAO1.
Table 5.
List of virulence genes present in PAO1 but is absent or with low identity in P. aeruginosa ATCC 27853
| PAO1 | ATCC 27853 | Identity on protein level (%) | Annotation |
|---|---|---|---|
| PA1092 | ACG06_21330 | 63.04 | Flagellin type B |
| PA1093 | ACG06_21325 | 41.48 | Hypothetical protein/flaG protein |
| PA1094 | ACG06_21320 | 43.07 | Flagellar capping protein FliD |
| PA1095 | ACG06_21310/ACG06_21315 | 66.40/39.67 | Flagellar protein FliS |
| PA1096 | ACG06_21305 | 46.94 | Hypothetical protein |
| PA2397 | ACG06_14125 | 64.55 | PvdE, pyoverdine biosynthesis protein |
| PA2398 | ACG06_14120 | 28.37 | Ferripyoverdine receptor |
| PA2399 | N/A | N/A | PvdD, pyoverdine biosynthetase D |
| PA2400 | N/A | N/A | PvdJ, pyoverdine biosynthetic process |
| PA2402 | ACG06_14105 | 69.40 | Probable non-ribosomal peptide synthetase |
| PA2424 | ACG06_13995/ACG06_14110 | 99.61/49.19 | Probable non-ribosomal peptide synthetase/PvdL |
| PA2427 | ACG06_13980 | 33.33 | Hypothetical protein |
| PA3142 | N/A | N/A | Integrase |
| PA3143 | N/A | N/A | Transposase |
| PA3144 | N/A | N/A | Transposase with Helix-turn-helix Hin domain |
| PA3145 | ACG06_09210 | 63.85 | Glycosyltransferase WbpL |
| PA3146 | ACG06_09205 | 37.78 | Probable NAD-dependent epimerase/dehydratase WbpK |
| PA3147 | ACG06_09195 | 24.10 | Probable glycosyl transferase WbpJ |
| PA3148 | N/A | N/A | UDP-N-acetylglucosamine 2-epimerase WbpI |
| PA3149 | N/A | N/A | Probable glycosyltransferase WbpH |
| PA3150 | N/A | N/A | LPS biosynthesis protein WbpG |
| PA3153 | N/A | N/A | O-antigen translocase |
| PA3154 | N/A | N/A | B-band O-antigen polymerase |
| PA3156 | N/A | N/A | UDP-2-acetamido-3-amino-2,3-dideoxy-d-glucuronic acid N-acetyltransferase, WbpD |
| PA3157 | N/A | N/A | Probable acetyltransferase, WbpC |
| PA3158 | N/A | N/A | UDP-2-acetamido-2-deoxy-d-glucuronic acid 3-dehydrogenase, WbpB |
| PA3159 | N/A | N/A | UDP-N-acetyl-d-glucosamine 6-Dehydrogenase,WbpA |
| PA3160 | ACG06_09160 | 54.95 | O-antigen chain length regulator, Wzz |
| PA3487 | N/A | N/A | Tle5,Secreted Factors (toxins, enzymes, alginate) |
| PA3498 | ACG06_28265 | 44.79 | Probable oxidoreductase |
| PA4150 | N/A | N/A | Probable dehydrogenase E1 component |
| PA4175 | N/A | N/A | Protease IV |
| PA4197 | N/A | N/A | BfiS, Two-component System |
| PA4525 | ACG06_25540 | 41.67 | Type 4 fimbrial precursor PilA |
| PA4527 | ACG06_25550 | N/A | Frameshift type 4 fimbrial biogenesis protein PilC (putative pseudogene) |
Interestingly, SNP distribution analysis in the genomes of PAO1 and ATCC 27853 revealed that a large number of non-synonymous variant sites present in the two strains are concentrated in the regions and genes that encode surface associated proteins, such as those that encode flagellar components, pyoverdine receptor, transporters, and type 4 pili (Additional file 1: Table S4 and Figure S2). These genomic differences combined suggest potentially different surface characteristics of ATCC 27853 when compared to PAO1. We therefore cultured the two strains on LB agar surface supplemented with Congo red and examined their capabilities to form colony biofilms [29]. A distinctive wrinkled colony morphology was observed in ATCC 27853 but not in PAO1 (Fig 6), suggesting a different surface pattern of ATCC 27853 compared with PAO1 and a strong capability of the strain to form biofilms. The stronger color of the ATCC 27853 biofilm compared to the biofilm of PAO1 on Congo red containing plate indicated a high level of exopolysaccharide matrix production in ATCC 27853, consistent with a stronger capability of the strain to form biofilm.
Fig. 6.
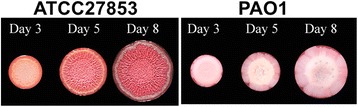
Colony morphology of P. aeruginosa ATCC 27853 and P. aeruginosa PAO1 cultured at 25 °C on LB agar plates supplemented with Congo Red
Transcriptomes of P. aeruginosa ATCC 27853 and P. aeruginosa PAO1
The distinctive pattern of colony biofilms of ATCC 27853 and PAO1 shown above prompted us to investigate the functional genome of ATCC 27853 and compare it with that of PAO1 at that growth stage. We performed RNA-seq to obtain the complete transcriptomes of both strains cultured on LB agar surface at 25 °C, condition that is identical to that of colony biofilm formation described above. Cell cultures following 48 h incubation were harvested and RNA was extracted and sequenced as described in Materials and Methods. Statistical analysis including total reads number and bases sequenced, genome coverage, CDS coverage and mapping ratio for each sample from RNA-seq analyses are presented in supplementary data (Additional file 1: Table S5). To conduct a genome wide comparative gene expression analysis, orthologous genes between ATCC 27853 and PAO1 were first identified using reciprocal blastn and the ratio of their respective expression in the two strains was calculated by DESeq (Additional file 1: Table S3) [30].
One hundred thirty seven genes with higher expression levels (log2 fold changes over 2) in ATCC 27853 than in P. aeruginosa PAO1 (Fig. 7, Additional file 1: Table S3) were identified. These include several classes of genes involved in biofilm formation, such as the type IV pili biogenesis gene cluster (pilQPONM: PA5040-PA5044) which is involved in the initiation of biofilms. Genes encoding twitching motility proteins, pilGHIJK-chpABCDE (PA0408-PA0417) were expressed at a higher level in ATCC 27853 than in PAO1 (Fig. 7, Additional file 1: Table S3). pilABCDE (PA4525-PA4528), pilTU (PA0395-PA0396) and pilSR-yfiT-fimTU-pilVWXY1Y2E (PA4546-PA4556) were also identified to display a slightly higher expression level in ATCC 27853 than in PAO1 (Additional file 1: Table S3). Expression of a proton motive force gene (pfm) (PA2950) that encodes a protein involved in energy metabolism critical for the rotation of flagellum in P. aeruginosa [31] was also higher in ATCC 27853 than in PAO1 (Fig. 7, Additional file 1: Table S3). Additionally, several other genes which are not directly involved in biofilm formation but have been reported to mediate the process were also found to be expressed at a higher level in ATCC 27853 than in PAO1, such as Chaperone-usher pathway (cup) A (PA2128-PA2132, cupA1-A4) encoding genes which were found to be required for adhesion to inert surfaces [32, 33], the cbb3-type cytochrome c oxidase cco2 gene cluster (ccoN2O2Q2P2, PA1555-PA1557) which has been shown to promote biofilm formation under hypoxia through NO induction and its effect on cell elongation [34], as well as pyeR (PA4354) that encodes a non-classical ArsR family member of transcriptional regulators modulating biofilm formation in P. aeruginosa [35] (Fig. 7, Additional file 1: Table S3). All these genetic and transcriptional data support the distinct colony morphogenesis observed in ATCC 27853.
Fig. 7.
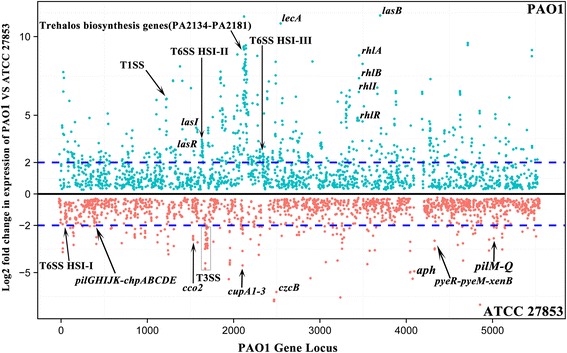
The genome wide transcriptomic profile of P. aeruginosa ATCC 27853 and PAO1. Green dots represent genes with higher relative expression level in PAO1 and red dots indicate genes with higher relative expression levels in ATCC 27853. The blue dashed lines represent log2-fold changes in expression. Selective genes and operons with distinctive expression patterns in the two strains are indicated
On the other hand, a much larger number (532 genes vs 137 as mentioned above) of genes with higher expression levels (log2 fold changes over 2) in P. aeruginosa PAO1 than in ATCC 27853 were observed (Fig. 7, Additional file 1: Table S3). Of particular prominence is a large fragment (PA2134-PA2181) of genes encoding trehalose biosynthesis. The homologous genes of this fragment in PA14 have been demonstrated to be involved in infection of plants [36]. Genes encoding several other virulence factors, such as lecA (encoding galactophilic lectin LecA) and lasB (encoding elastase LasB) were expressed at a higher level in PAO1 than in ATCC 27853 (Additional file 1: Table S3). It was also noticed that several transcriptional regulators which are quorum sensing genes mediating virulence factor production such as LasI, LasR, and RhlI and RhlR were also expressed at a higher level in PAO1 than in ATCC 27853 (Fig. 8).
Fig. 8.
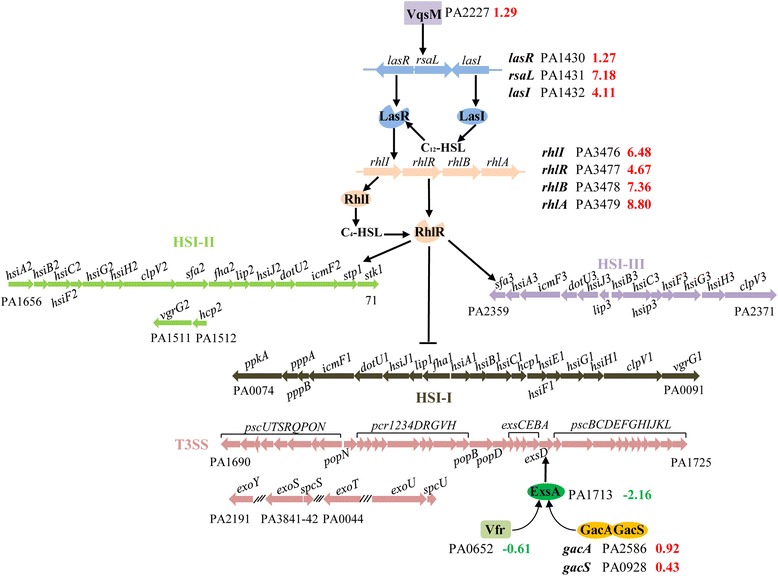
Differential expression of the genes involved in the type III and type VI secretion systems and their regulators in P. aeruginosa ATCC 27853 and PAO1. Gene locus tags in PAO1 are shown. Values following gene locus tags of regulators indicate Log2 gene expression changes in PAO1 relative to that in ATCC 27853 (red color indicates higher expression in PAO1 than in ATCC27853, green color indicates higher expression level in ATCC 27853 than in PAO1). The full list of genes displaying differential expression in the two strains and their values are provided in supplementary Additional file 1: Table S3
An interesting observation is the expression patterns of the genes encoding various secretion systems in P. aeruginosa species. The components of type III secretion systems (T3SSs), such as genes in psc, pcr and exs gene clusters, display remarkably higher expression levels in ATCC 27853 than in PAO1 (Figs. 7 and 8), whereas those of the type I secretion system, namely T1SS, display a relatively higher expression level in PAO1 than in ATCC 27853. In the case of the type VI secretion system (T6SS) which includes three hemolysin co-regulated protein (Hcp) secretion islands HSI-I, II, III, while HSI-I was found to display a higher relative expression level in ATCC 27853 than in PAO1, that of HSI-II and III is opposite, i.e., they are expressed at higher level in PAO1 than in ATCC 27853 (Figs. 7 and 8).
Discussion
Morphogenesis in PAO1 and ATCC 27853
Surface characteristics play an important role in the morphogenesis of bacteria. P. aeruginosa is a well established model strain to study biofilms [37]. Outer membrane LPS and extracellular appendages, such as flagella, type IV pili and Cup fimbriae, are involved in the initial attachment of bacteria to a surface [38]. The present comparative genomic and transcriptomic study on P. aeruginosa ATCC 27853 and PAO1 revealed distinct genetic and expression pattern of surface associated proteins in ATCC 27853. Lacking of the B-band O-antigen (A+B−) has been reported to lead to an increased hydrophobicity of the cell surface and an enhanced adherence to polystyrene materials [39]. Increased expression of type IV pili biosynthesis genes and flagella motility genes also enhances bacterial adherence to various surfaces during the initiation of a biofilm. Our transcriptome analysis supports the expression patterns of these genes in ATCC 27853 which is consistent with the observed enhanced colony biofilm formation of the strain.
Three types of exopolysaccharides, alginate, Psl and Pel, play an important role in the biofilm maturation and development stage. Alginate has been proposed not to be a critical component of the extracellular polysaccharide matrix in nonmucoid P. aeruginosa strains [40]. The low expression levels of alginate biosynthesis genes in PAO1 and ATCC 27853 are consistent with the nonmucoid colony morphologies of the two strains. Previous studies demonstrated that Pel and Psl have distinct physical properties and functional roles during biofilm maturation and development [41]. The pel locus (referring to pellicle, a biofilm formed at the air-medium interface), containing the genes pelA-G, is responsible for synthesis of the glucose-rich component of the matrix, whereas the psl locus (polysaccharide synthesis locus), containing the genes pslA-O, is responsible for the mannose- and galactose-rich component which forms a fiber-like matrix to enmesh bacterial communities [42]. Pel is required for close association of the two species in mixed-species microcolonies. In contrast, Psl is important for P. aeruginosa to form single-species biofilms. In the present study, expression of Pel biosynthesis genes were detected at a low level in both strains, however, a higher expression level of psl genes in ATCC 27853 compared to PAO1 was observed indicating a role of Psl in the development of ATCC 27853 colony biofilm. This result is also in agreement with a lower expression level of amrZ (PA3385) in ATCC 27853 than in PAO1, as the AmrZ transcriptional repressor controls switching between an alginate-producing mucoid state and a Pel-producing biofilm state through repression of psl genes [43, 44]. Another important signaling molecule which level in the cell correlates with the capability of the bacterium to form biofilms is the second messenger c-di-GMP. However, expression of several genes encoding diguanylate cyclase and phosphodiesterases which are involved in c-di-GMP production [45] was shown to be similar in PAO1 and ATCC 27853 in our comparative transcriptome analysis, suggesting that c-di-GMP did not play an important role in the distinctive colony biofilm formation observed in ATCC 27853 in comparison with that of PAO1.
Contribution of the phenazine compounds to the biofilm development of P. aeruginosa has also been reported [24, 46–48]. Recently, it was found that PYO can promote biofilm development of the bacterium by binding to extracellular DNA and enhancing the formation of extracellular matrix of biofilms [48]. Higher level of PYO production in ATCC 27853 than in PAO1 was observed in the present study. Thus, PYO may also contribute to the enhanced biofilm formation in ATCC 27853. The last step of PYO biosynthesis is the conversion of the zwitterionic intermediate 5-Me-PCA to the less charged PYO via hydroxylative decarboxylation. Interestingly, 5-Me-PCA, which is exported out of cells by the MexGHI-OmpD RND type efflux pump, was also shown to mediate the biofilm formation of P. aeruginosa in PA 14 [24]. It was proposed that export of 5-Me-PCA serves as a detoxification means in P. aeruginosa, likewise the conversion of this molecule to PYO which decreases the charge of the molecule and allows the transport of the product (PYO) across the membrane without the assistance of an efflux pump [24]. Indeed, PYO was shown not to be the substrate of the MexGHI-OmpD pump. The mexGHI-ompD system is present in both PAO1 and PA14, but is lacking in ATCC 27853. Yet, a higher level of PYO is observed in ATCC 27853 than in PAO1. This suggests that ATCC 27853 may contain other detoxification means allowing production of PYO in high level but minimizing the potential cytotoxicity of the intermediate 5-Me-PCA. Indeed, our genomic analysis revealed considerable differences of the two strains in terms of the numbers (122) of COGs. There are 71 unique COGs present in ATCC 27853 but are absent in PA14 and 51 COGs present in PA14 are lacking in ATCC 27853 (Fig. 2). These interesting observations warrant a comparative, molecular analysis of the PYO biosynthesis in ATCC 27853, PAO1, and PA14.
Phylogenetic relations and accessory genes of P. aeruginosa ATCC 27853
In the phylogenetic tree constructed (Fig. 1), ATCC 27853 was shown to be extraordinarily closely related to three strains, P. aeruginosa T38079, P. aeruginosa F9670 and P. aeruginosa S86968. This phenomenon is interesting. Sequences of the three strains, T38079, F9670 and S86968, became available only very recently in the NCBI GenBank, and we included them in our phylogenetic analysis. However, this observation does not necessarily mean that these four strains are almost identical. This is because the SNPs utilized to construct the phylogenetic tree were extracted from the core genomic regions of all 59 strains which complete genome sequences are available. The SNPs do not cover the accessory genomes which are unique to each of the strains. Thus, the resulting relatedness of the strains in the phylogenetic tree does not reflect their associations at the complete genome level. Nevertheless, in the dataset we extracted, only 146 SNPs among these four strains were identified. Furthermore, the three strains and ATCC 27853 are assigned to the same multi-locus sequence type (MLST, https://pubmlst.org/paeruginosa/) and the same phylogenetic group based on NCBI GenBank, indicating very similar genomic contents of these four strains.
Core genome and accessory genes are two main components of the genomes of different P. aeruginosa strains [2]. Accessory genes are associated with genomic islands and islets that are attributed to diversification of strains within the species. This is termed as diversifying selection. Certain selective pressure might be responsible for the acquiring of these accessory genes and the resulting genome diversity among the different strains within the same species.
With the complete genome of P. aeruginosa ATCC 27853 on hand, its accessory genes were extensively examined in the current study. Within these accessory genes, the most prominent observation was the presence of seven prophages. Prophages contained in the genome of bacteria have been shown to play important roles in the physiology of the host bacterial species [49]. For example, two tandem defective phage (pyocin) islands on the P. aeruginosa PAO1 genome are the determinants of fluoroquinolone susceptibility of the strain [4]. Another study on P. aeruginosa LESB58 (Liverpool Epidemic Strain) demonstrated that the four prophages present in its genome could enhance competitiveness of the strain in a chronic rat lung infection model [5]. The abundance of prophages in the genome of ATCC 27853 implies the complexity and strong fitness potential of the strain. However, expression of these prophages was found to be low or non-detectable in the present study based on the transcriptome data (Fig. 7). This probably was due to the rich growth medium used in this study. Elucidating the functions of the genes within these prophages especially those encoding several transcriptional factors may help to disclose the potential roles of the prophages in the fitness of ATCC 27853 to the non-laboratory, harsh environments in nature and in animal hosts.
Secretion systems
Secretion systems are important for the adaptation and pathogenesis of P. aeruginosa through dedicated secretion of specific exoproteins [50]. It has been shown that type III secretion systems (T3SSs) are correlated with acute infections in P. aeruginosa, while type VI secretion systems are often associated with chronic infections and biofilm formation of the species [47]. In the present study, genes encoding T3SS were found to be expressed at a remarkably higher level in ATCC 27853 than in PAO1 (Figs. 7 and 8). The genes encoding transcriptional activators of T3SS, e.g. exsA were also expressed at higher level in ATCC 27853 than in PAO1. Interestingly, a differential expression pattern of the three Hcp islands of T6SS was observed in these two strains, while HSI-II and HSI-III was expressed at a higher level in PAO1, HSI-I expression was higher in ATCC 27853 (Fig. 8). The three Hcp islands of P. aeruginosa have been assigned to different phylogenetic groups based on phylogenetic analysis, indicating a distinct evolutionary history of the three components [51]. This also suggests different roles of these three HSI islands during pathogenesis of P. aeruginosa. In addition, previous studies have demonstrated that the expression of these three Hcp islands of T6SS is mediated by different regulators [47]. LasR and RhlR positively regulate the expression of HSI-II and HSI-III gene clusters and LasR negatively regulates the HSI-I gene cluster in P. aeruginosa [47]. This is consistent with the higher expression level of LasR and RhlR in PAO1 compared with that in ATCC 27853 (Fig. 8). These observations indicate the complex expression patterns and functional roles of these secretion systems in the physiology and pathogenicity of different P. aeruginosa strains.
Conclusions
In summary, several genomic features of P. aeruginosa ATCC 27853 were identified based on the complete genome sequence generated using Pacific Biosciences SMRT (PacBio) technology. Comparing with the genomes of the other two frequently used model strains P. aeruginosa PAO1 and PA14, three unique genomic islands were present in P. aeruginosa ATCC 27853 which contain genes possibly related to the metabolisms of aromatic compounds. Seven prophages are predicted including the prophage 2 which is located adjacent to the phz1 phenazine biosynthesis gene clusters. Survey of virulence related genes revealed the lack of a gene cluster encoding the B-band O-antigen of LPS in P. aeruginosa ATCC 27853 which is important in evading of host immune responses and biofilm formation. Transcriptome analysis revealed differential gene expression of several groups of surface associated proteins and those involved in cellular redox metabolism, and the type I, III and VI secretion systems, confirming the different surface characteristics of ATCC 27853 from that of PAO1 and suggesting unique physiological and pathogenic potentials of ATCC 27853. These information provides genetic basis for the comprehensive understanding of the physiology, pathogenicity, and virulence of the strain.
Methods
Culture of bacterial cells and genomic DNA extraction
P. aeruginosa ATCC 27853 used in the present study was a gift obtained from Chinese University of Hong Kong (CUHK). It was cultivated in Luria-Bertani (LB) broth overnight with shaking (150 rpm) at 37 °C. Bacterial cells were harvested from 1 ml liquid culture via centrifugation at 10,000 rpm for 10 min. Genomic DNA of P. aeruginosa ATCC 27853 was extracted using QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). The concentration and quality of genomic DNA was determined by NanoDrop and gel electrophoresis.
Colony morphology assay
Congo red plates were prepared following the protocol described by Dietrich et al. with slight modifications [29]. Briefly, 1% tryptone and 1% agar were mixed with 40 μg/ml Congo red and 20 μg/ml Coomassie blue and poured on the square petri dish. 10 μl of overnight culture of P. aeruginosa inoculated from single colonies was spotted onto the square agar plates followed by incubation at 25 °C up to 9 days. Colony morphologies were recorded daily.
Extraction and quantification of pyocyanin
Pyocyanin from liquid cultures harvested at different time point were extracted and measured following the protocol used by Recinos et al. and Apidianakis et al. with slight modification [26, 52]. Supernatant was collected after centrifugation at 13,000 rpm for 5 min and mixed with 0.6 volume of chloroform following vortex for 10 s twice. After centrifugation at 13,000 rpm for 5 min, blue layer at the bottom was transferred to a new tube and mixed with 0.5 volume of 0.2 M HCl with vortex for 10 s twice. 0.1 ml of the pink layer was transferred to a 96-well plate after 13,000 rpm for 5 min. Absorbance was determined at 510 nm.
RNA preparation
Total RNA was extracted from triplicates of both ATCC 27853 and PAO1. 10 μl of overnight culture of P. aeruginosa PAO1 and ATCC 27853 in LB Broth inoculated from single colonies was spotted on the LB agar surface and incubated at 25 °C for 2 days. Cell patches were scraped from the plates and resuspended in 1 ml LB medium. 0.125 ml ice-cold phenol/ethanol stop solution (5:95, v/v, Ambion™ water saturated phenol at pH 6.6) was mixed with bacterial culture and placed on ice for 10 min to stop mRNA degradation. The mixture was subsequently centrifuged at 4800 rpm for 10 min at 4 °C. The supernatant was removed and cell pellet was stored at −80 °C for RNA extraction. RNA extraction was following the manufacture’s instructions using RNeasy Mini kit (Qiagen, Hilden, Germany). The quality of the extracted RNA has passed the Agilent Bioanalyzer analysis in Genome Research Centre of The University of Hong Kong (all RNA Integrity Number, RIN, are over 7). Stranded libraries for all RNA samples were constructed with Kapa Biosystems RNA library preparation chemistry in Georgia Genomics Facility at University of Georgia.
Sequencing and de novo assembly
The whole genome sequencing of P. aeruginosa ATCC 27853 was performed using the PacBio RS II single-molecule, real-time sequencing system (SMRT) platform using 20 kb insert library and P6-C4 chemistry (Pacific Biosciences, Menlo Park, CA) by Macrogen(Korean). Raw SMRT reads were error corrected, de novo assembled the polished using the SMRT Analysis workflow [53] from Pacific Biosciences. The genome was checked for circularization by self-aligning the contig and inspecting the dotplot for sticky edges (dotplot was created in Geopard [54]). Circularization was carried out by trimming one end of the contig then collapsing using Minimus2 [55]. The genome of P. aeruginosa ATCC 27853 and transcriptomes of the two strains, PAO1 and P. aeruginosa ATCC 27853 were sequenced on the Illumina NextSeq platform (Illumina, San Diego, California, USA) using a run of 300 Cycles PE150 High Output Flow Cell in the Georgia Genomics Facility at the University of Georgia. DNA-seq raw reads from P. aeruginosa ATCC 27853 were aligned to the single PacBio contig and the Variant Call Format (VCF) file was generated with SAMtools [56].
Genome annotations
Automated gene calling and annotation was carried out using the National Center for Biotechnology Information (NCBI)‘s Prokaryotic Genome Annotation Pipeline 2.0 (PGAP) [57]. We assessed and validated the annotation by comparing to that from the Rapid Annotations using Subsystems Technology (RAST) Server [58] as well as that from Prokka [59]. tRNA genes were predicted using tRNAscan-SE 1.3.1 [60] and rRNA genes using RNAmmer 1.2 [61]. Metabolic pathways were predicted in silico using KAAS [62]. Protein sequences of P. aeruginosa ATCC 27853 were BLAST-ed against the Clusters of Orthologous Groups (COG) database with an e-value score of 1e-5 [63].
Prediction of prophage and genomic islands
Prophages in the genome of P. aeruginosa ATCC 27853 were predicted using the online softwares Prophinder with parameters (Scanning window size: 20,50,100,200,300; Minimum nb of CDS in prophages: 20; Minimum nb of ACLAME hits: 20; Blast Eval threshold: 1e-5; Minimum DR size: 10) [18] and PHAST [19]. IslandViewer was used with two methods including SIGI-HMM and IslandPath-DIMOB [15] to predict genomic islands (GIs). Hypothetic genes in prophages or GIs annotated by methods mentioned above were also blasted against the Pfam database constructed based on protein modules to improve annotations [64]. In addition, all available complete genomes of P. aeruginosa in Genbank were surveyed with PHAST to predict prophages [19].
Virulence gene prediction
In P. aeruginosa PAO1, 273 virulence genes were identified based on a conserved list of 369 virulence genes in Pseudomonas species obtained from the Virulence Factor Database (VFDB) [27], Victors Virulence Factors (PHIDIAS) (http://www.phidias.us/victors/index.php), and curation by the Pseudomonas Genome Database (PseudoCAP) [65] with a primary focus on P. aeruginosa PAO1 and P. aeruginosa PA14. These 273 virulence proteins were blasted against all proteins in ATCC 27853 through BLASTp with 1e-5 e-value. Those without positive result of the blast search were recognized as absent in ATCC 27853. All the protein sequences of ATCC 27853 were also blasted against this conserved list of virulence genes with 1e-5 e-value.
Comparative analysis of genomes
Four draft genomes of P. aeruginosa ATCC 27853 were recruited from Genbank (Table 1) [9–12] and compared with the complete genome obtained in the current study. 58 complete genomes of P. aeruginosa were also retrieved from Genbank and were compared with ATCC 27853 using progressiveMauve with default settings [66]. Proteins present exclusively in an individual strain and those shared between two or three strains based on Mauve and COG blast analysis were counted and represented in Venn diagrams generated by VennDiagram in R-platform [67]. For single nucleotide polymorphisms (SNPs) calling between PAO1 and ATCC 27853, VCF was first generated using Parsnp from Harvest tools [13]. VCF was annotated using SnpEff using PAO1 as reference genome [68].
Phylogenetic analyses
The phylogenetic analysis was performed to validate the phylogenetic position of P. aeruginosa ATCC 27853. Parsnp from Harvest tools [13] was employed with default settings to collect single nucleotide polymorphisms (SNPs) from all currently available complete genomes of P. aeruginosa and 269,561 SNPs were submitted for phylogenetic analysis with a maximum likelihood (ML) criterion in MEGA [14]. Parameters for this analysis included: Tamura-Nei substitution model, Gamma Distributed Rates among sites, Nearst-Neighbor-Interchange (NNI) ML Heuristic method for tree inference options, using automatically generated initial tree with NJ method, and 100 times bootstrap test.
RNA-seq quality processing
We performed quality control (QC) on the raw Illumina RNA-Seq data using BBduk2 (BBMap short read aligner, http://sourceforge.net/projects/bbmap). Reads were culled based on a minimum average quality of 20 over a window of 7 bp. Low quality read edges were trimmed and reads containing more than two ambiguous bases were removed. Finally, read pairs were trimmed evenly and a minimum length of 60 bp was enforced.
RNA-seq read mapping
QC reads were mapped to their respective reference genomes in two stages. First, QC reads were aligned using BWA-MEM with default parameters [69]. The second round of read mapping was conducted using Stampy with the output from BWA-MEM (with Stampy’s --bamkeepgoodreads -M options) [70]. SAMtools and BamTools were used for format conversions, statistics, and quality assessment and control [53, 71]. IGV tools were also used to visually inspect mapping quality to ensure its accuracy [72].
Fragment counts and statistics
Fragment (our RNA-Seq data are stranded) counting per genomic features (genes) was performed using featureCounts [73]. Reads that mapped with MAPQ scores below 10 were removed. Enforcing a MAPQ score below 10 also excludes multi-mapped reads albeit the percentage of this category is low (data not shown). Multi-mapping was determined using default parameters. Read pairs were checked for proper pairing as well as the proper insert size. Counting was performed for each gene based on its locus_tag. Read counts were used as input for DESeq analysis [30]. Genes with mean normalized expression <50 reads in all samples were considered as transcriptional noise and filtered out from the analysis. In DESeq, fold changes (log2(fold-change) ≥ 2or ≤2) for each expression gene and p-value < 0.05 [cut-off at 5% false discovery rate (FDR)] was employed as threshold for the statistics analysis.
Acknowledgements
This work was supported by the Hong Kong University Grants Council General Research Fund (HKU17142316) and Seed Funding for Basic Research Scheme of The University of Hong Kong (201411159065) to Dr. Aixin Yan and the postdoctoral fellowship from The University of Hong Kong to Dr. Huiluo Cao. We thank the Centre for Genomic Sciences at the University of Hong Kong for the sequencing and bioinformatics analysis assistance of the project. We thank Prof. Christopher Rensing (Fujian Agriculture & Forestry University) for proof-reading our manuscript.
Availability of data and materials
The datasets supporting the conclusions of this article are available in the Genbank repository under the accession number CP011857 for the complete genome of P. aeruginosa ATCC 27853 and the project accession number PRJNA377172 for the transcriptome data of P. aeruginosa PAO1 and ATCC 27853.
Authors’ contributions
HC and AY designed the experiment. HC, YL, SB, and ZX did the experiment. HC and AY analyzed the data and wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interest.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- COG
Clusters of orthologous group
- GI
Genomic islands
- Hcp
Hemolysin co-regulated protein
- HGAP
Hierarchical Genome Assembly Process
- HSI
Hcp secretion island
- NCBI
The National Center for Biotechnology Information
- ORFs
Open reading frames
- RAST
Rapid Annotations using Subsystems Technology
- SMRT
Single Molecule Real-Time
- T3SS
Type III secretion systems
- T6SS
Type VI secretion system
Additional file
A list of complete genomes of Pseudomonas aeruginosa employed in the present study. Table S2. Annotations of ORFs in Prophage 2 predicted in P. aeruginosa ATCC 27853. Table S3. Table S3 Differentially expressed genes in P. aeruginosa ATCC 2853 and PAO1 revealed by DESeq of the RNA-seq data (see supplemented excel file). Table S4 Top 50 ranked genes with numbers of non-synonymous variants between Pseudomonas aeruginosa ATCC 2853 and PAO1 with function description. Table S5 RNA-seq statistics and coverage after quality filtering for PAO1 and ATCC 27853. Fig. S1. Location of prophage B3 in four P. aeruginosa genomes: ATCC 27853, P. aeruginosa NCGM2.S1, P. aeruginosa VRFPA04 and P. aeruginosa Carb01_63. Fig. S2 Distribution of nucleotide change numbers in the genomes of P. aeruginosa ATCC 27853 and PAO1. (ZIP 718 kb)
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3842-z) contains supplementary material, which is available to authorized users.
Contributor Information
Huiluo Cao, Phone: (852) 22990864, Email: hcao@hku.hk.
Yong Lai, Email: ylai@connect.hku.hk.
Salim Bougouffa, Email: salim.bougouffa@gmail.com.
Zeling Xu, Email: zelingxu@connect.hku.hk.
Aixin Yan, Phone: (852) 22990864, Email: ayan8@hku.hk.
References
- 1.Turner KH, Wessel AK, Palmer GC, Murray JL, Whiteley M. Essential genome of Pseudomonas aeruginosa in cystic fibrosis sputum. Proc Natl Acad Sci U S A. 2015;112:4110–4115. doi: 10.1073/pnas.1419677112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Valot B, Guyeux C, Rolland JY, Mazouzi K, Bertrand X, Hocquet D. What it takes to be a Pseudomonas aeruginosa? The core genome of the opportunistic pathogen updated. PLoS One. 2015;10:e0126468. doi: 10.1371/journal.pone.0126468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ozer EA, Allen JP, Hauser AR. Characterization of the core and accessory genomes of Pseudomonas aeruginosa using bioinformatic tools spine and AGEnt. BMC Genomics. 2014;15:737. doi: 10.1186/1471-2164-15-737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brazas MD, Hancock RE. Ciprofloxacin induction of a susceptibility determinant in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2005;49:3222–3227. doi: 10.1128/AAC.49.8.3222-3227.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Winstanley C, Langille MG, Fothergill JL, Kukavica-Ibrulj I, Paradis-Bleau C, Sanschagrin F, et al. Newly introduced genomic prophage islands are critical determinants of in vivo competitiveness in the Liverpool epidemic strain of Pseudomonas aeruginosa. Genome Res. 2009;19:12–23. doi: 10.1101/gr.086082.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Medeiros AA, O’Brien TF, Wacker WE, Yulug NF. Effect of salt concentration on the apparent in-vitro susceptibility of Pseudomonas and other gram-negative bacilli to gentamicin. J Infect Deseases. 1971;124(Suppl):S59–S64. doi: 10.1093/infdis/124.Supplement_1.S59. [DOI] [PubMed] [Google Scholar]
- 7.Pollock HM, Minshew BH, Kenny MA, Schoenknecht FD. Effect of different lots of Mueller-Hinton agar on the interpretation of the gentamicin susceptibility of Pseudomonas Aeruginosa. Antimicrob Agents Chemother. 1978;14:360–367. doi: 10.1128/AAC.14.3.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cavalieri SJ, Microbiology ASf. Manual of Antimicrobial Susceptibility Testing (American Society for Microbiology). 2009.
- 9.Fang X, Fang Z, Zhao J, Zou Y, Li T, Wang J, et al. Draft genome sequence of Pseudomonas aeruginosa strain ATCC 27853. J Bacteriol. 2012;194:3755. doi: 10.1128/JB.00690-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu C, Hu J, Fang X, Zhang D, Chang D, Wang J, et al. Genome sequence of Pseudomonas aeruginosa strain LCT-PA41, with changed metabolism after space flight. Genome Announc. 2014;2(1):e01124–e01113. doi: 10.1128/genomeA.01124-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Minogue TD, Daligault HE, Davenport KW, Broomall SM, Bruce DC, Chain PS, et al. Draft genome assembly of Pseudomonas aeruginosa quality control reference strain Boston 41501. Genome Announc. 2014;2(5):e00960–e00914. doi: 10.1128/genomeA.00960-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu G, Hu J, Fang X, Fang X, Zhang X, Wang J, et al. Genome sequence of Pseudomonas aeruginosa strain LCT-PA220, which was selected after space flight by using Biolog's powerful carbon source utilization technology. Genome Announc. 2014;2:e00169–e00114. doi: 10.1128/genomeA.00169-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Treangen TJ, Ondov BD, Koren S, Phillippy AM. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 2014;15:524. doi: 10.1186/s13059-014-0524-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dhillon BK, Laird MR, Shay JA, Winsor GL, Lo R, Nizam F, et al. IslandViewer 3: more flexible, interactive genomic island discovery, visualization and analysis. Nucleic Acids Res. 2015;43(W1):W104–W108. doi: 10.1093/nar/gkv401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Waack S, Keller O, Asper R, Brodag T, Damm C, Fricke WF, et al. Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinformatics. 2006;7:142. doi: 10.1186/1471-2105-7-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsiao WW, Ung K, Aeschliman D, Bryan J, Finlay BB, Brinkman FS. Evidence of a large novel gene pool associated with prokaryotic genomic islands. PLoS Genet. 2005;1(5) doi: 10.1371/journal.pgen.0010062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lima-Mendez G, Van Helden J, Toussaint A, Leplae R. Prophinder: a computational tool for prophage prediction in prokaryotic genomes. Bioinformatics. 2008;24:863–865. doi: 10.1093/bioinformatics/btn043. [DOI] [PubMed] [Google Scholar]
- 19.Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic Acids Res. 2011;39:W347–W352. doi: 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arndt D, Grant J, Marcu A, Sajed T, Pon A, Liang Y, et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016;44(W1):W16–W21. doi: 10.1093/nar/gkw387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Braid MD, Silhavy JL, Kitts CL, Cano RJ, Howe MM. Complete genomic sequence of bacteriophage B3, a mu-like phage of Pseudomonas aeruginosa. J Bacteriol. 2004;186:6560–6574. doi: 10.1128/JB.186.19.6560-6574.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fuqua C, Winans SC, Greenberg EP. Census and consensus in bacterial ecosystems: the LuxR-LuxI family of quorum-sensing transcriptional regulators. Annu Rev Microbiol. 1996;50:727–751. doi: 10.1146/annurev.micro.50.1.727. [DOI] [PubMed] [Google Scholar]
- 23.Mavrodi DV, Parejko JA, Mavrodi OV, Kwak YS, Weller DM, Blankenfeldt W, et al. Recent insights into the diversity, frequency and ecological roles of phenazines in fluorescent Pseudomonas spp. Environ Microbiol. 2013;15:675–686. doi: 10.1111/j.1462-2920.2012.02846.x. [DOI] [PubMed] [Google Scholar]
- 24.Sakhtah H, Zhang Y, Morales DK, Fields BL, Price-Whelan A, Hogan DA, et al. The Pseudomonas aeruginosa efflux pump MexGHI-OpmD transports a natural phenazine that controls gene expression and biofilm development. Proc Natl Acad Sci U S A. 2016;113(25):E3538–E3547. doi: 10.1073/pnas.1600424113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cui Q, Lv H, Qi Z, Jiang B, Xiao B, Liu L, et al. Cross-regulation between the phz1 and phz2 Operons maintain a balanced level of Phenazine biosynthesis in Pseudomonas aeruginosa PAO1. PLoS One. 2016;11(1):e0144447. doi: 10.1371/journal.pone.0144447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Recinos DA, Sekedat MD, Hernandez A, Cohen TS, Sakhtah H, Prince AS, et al. Redundant phenazine operons in Pseudomonas aeruginosa exhibit environment-dependent expression and differential roles in pathogenicity. Proc Natl Acad Sci U S A. 2012;109(47):19420–19425. doi: 10.1073/pnas.1213901109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen L, Yang J, Yu J, Yao Z, Sun L, Shen Y, et al. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 2005;33:D325–D328. doi: 10.1093/nar/gki008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Larkin A, Imperiali B. Biosynthesis of UDP-GlcNAc(3NAc)a by WbpB, WbpE, and WbpD: enzymes in the Wbp pathway responsible for O-antigen assembly in Pseudomonas aeruginosa PAO1. Biochemistry. 2009;48:5446–5455. doi: 10.1021/bi900186u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dietrich LE, Okegbe C, Price-Whelan A, Sakhtah H, Hunter RC, Newman DK. Bacterial community morphogenesis is intimately linked to the intracellular redox state. J Bacteriol. 2013;195(7):1371–1380. doi: 10.1128/JB.02273-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bai F, Li Y, Xu H, Xia H, Yin T, Yao H, et al. Identification and functional characterization of pfm, a novel gene involved in swimming motility of Pseudomonas aeruginosa. Gene. 2007;401(1–2):19. doi: 10.1016/j.gene.2007.06.019. [DOI] [PubMed] [Google Scholar]
- 32.Vallet I, Olson JW, Lory S, Lazdunski A, Filloux A. The chaperone/usher pathways of Pseudomonas aeruginosa: identification of fimbrial gene clusters (cup) and their involvement in biofilm formation. Proc Natl Acad Sci U S A. 2001;98:6911–6916. doi: 10.1073/pnas.111551898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vallet I, Diggle SP, Stacey RE, Cámara M, Ventre I, Lory S, et al. Biofilm formation in Pseudomonas aeruginosa: fimbrial cup gene clusters are controlled by the transcriptional regulator MvaT. J Bacteriol. 2004;186(9):2880–2890. doi: 10.1128/JB.186.9.2880-2890.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hamada M, Toyofuku M, Miyano T, Nomura N. cbb3-type cytochrome c oxidases, aerobic respiratory enzymes, impact the anaerobic life of Pseudomonas aeruginosa PAO1. J Bacteriol. 2014;196(22):3881–3889. doi: 10.1128/JB.01978-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mac Aogáin M, Mooij MJ, McCarthy RR, Plower E, Wang YP, Tian ZX, et al. The non-classical ArsR-family repressor PyeR (PA4354) modulates biofilm formation in Pseudomonas aeruginosa. Microbiology. 2012;158(Pt 10):2598–2609. doi: 10.1099/mic.0.058636-0. [DOI] [PubMed] [Google Scholar]
- 36.Djonović S, Urbach JM, Drenkard E, Bush J, Feinbaum R, Ausubel JL, et al. Trehalose biosynthesis promotes Pseudomonas aeruginosa Pathogenicity in plants. PLoS Pathog. 2013;9(3):e1003217. doi: 10.1371/journal.ppat.1003217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Siryaporn A, Kuchma SL, O'Toole GA, Gitai Z. Surface attachment induces Pseudomonas aeruginosa virulence. Proc Natl Acad Sci U S A. 2014;111(47):16860–16865. doi: 10.1073/pnas.1415712111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mikkelsen H, Sivaneson M, Filloux A. Key two-component regulatory systems that control biofilm formation in Pseudomonas aeruginosa. Environ Microbiol. 2011;13(7):1666–1681. doi: 10.1111/j.1462-2920.2011.02495.x. [DOI] [PubMed] [Google Scholar]
- 39.Makin SA, Beveridge TJ. The influence of A-band and B-band lipopolysaccharide on the surface characteristics and adhesion of Pseudomonas aeruginosa to surfaces. Microbiology. 1996;142(Pt 2):299–307. doi: 10.1099/13500872-142-2-299. [DOI] [PubMed] [Google Scholar]
- 40.Wozniak DJ, Wyckoff TJ, Starkey M, Keyser R, Azadi P, O'Toole GA, et al. Alginate is not a significant component of the extracellular polysaccharide matrix of PA14 and PAO1 Pseudomonas aeruginosa biofilms. Proc Natl Acad Sci U S A. 2003;100(13):7907–7912. doi: 10.1073/pnas.1231792100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chew SC, Kundukad B, Seviour T, van der Maarel JRC, Yang L, Rice SA, et al. Dynamic remodeling of microbial biofilms by functionally distinct exopolysaccharides. MBio. 2014;5(4):e01536–e01514. doi: 10.1128/mBio.01536-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karatan E, Watnick P. Signals, regulatory networks, and materials that build and break bacterial biofilms. Microbiol Mol Biol Rev. 2009;73:310–347. doi: 10.1128/MMBR.00041-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jones CJ, Newsom D, Kelly B, Irie Y, Jennings LK, Xu B, et al. ChIP-Seq and RNA-Seq reveal an AmrZ-mediated mechanism for cyclic di-GMP synthesis and biofilm development by Pseudomonas aeruginosa. PLoS Pathog. 2014;10:e1003984. doi: 10.1371/journal.ppat.1003984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jones CJ, Ryder CR, Mann EE, Wozniak DJ. AmrZ modulates Pseudomonas aeruginosa biofilm architecture by directly repressing transcription of the psl operon. J Bacteriol. 2013;195(8):1637–1644. doi: 10.1128/JB.02190-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cotter PA, Stibitz S. C-di-GMP-mediated regulation of virulence and biofilm formation. Curr Opin Microbiol. 2007;10(1):17–23. doi: 10.1016/j.mib.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 46.Okegbe C, Price-Whelan A, Dietrich LE. Redox-driven regulation of microbial community morphogenesis. Curr Opin Microbiol. 2014;18:39–45. doi: 10.1016/j.mib.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Silverman JM, Brunet YR, Cascales E, Mougous JD. Structure and regulation of the type VI secretion system. Annu Rev Microbiol. 2012;66:453–472. doi: 10.1146/annurev-micro-121809-151619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Das T, Kutty SK, Tavallaie R, Ibugo AI, Panchompoo J, Sehar S, et al. Phenazine virulence factor binding to extracellular DNA is important for Pseudomonas aeruginosa biofilm formation. Sci Rep. 2015;5:8398. doi: 10.1038/srep08398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nanda AM, Thormann K, Frunzke J. Impact of spontaneous prophage induction on the fitness of bacterial populations and host-microbe interactions. J Bacteriol. 2015;197(3):410–419. doi: 10.1128/JB.02230-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Filloux A. Protein secretion Systems in Pseudomonas aeruginosa: an essay on diversity, evolution, and function. Front Microbiol. 2011;2:155. doi: 10.3389/fmicb.2011.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bingle LE, Bailey CM, Pallen MJ. Type VI secretion: a beginner's guide. Curr Opin Microbiol. 2008;11:3–8. doi: 10.1016/j.mib.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 52.Apidianakis Y, Pitsouli C, Perrimon N, Rahme L. Synergy between bacterial infection and genetic predisposition in intestinal dysplasia. Proc Natl Acad Sci U S A. 2009;106(49):20883–20888. doi: 10.1073/pnas.0911797106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods. 2013;10:563–569. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 54.Krumsiek J, Arnold R, Rattei T. Gepard: a rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics. 2007;23(8):1026–1028. doi: 10.1093/bioinformatics/btm039. [DOI] [PubMed] [Google Scholar]
- 55.Sommer DD, Delcher AL, Salzberg SL, Pop M. Minimus: a fast, lightweight genome assembler. BMC Bioinformatics. 2007;8:64. doi: 10.1186/1471-2105-8-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Angiuoli SV, Gussman A, Klimke W, Cochrane G, Field D, Garrity G, et al. Toward an online repository of standard operating procedures (SOPs) for (meta)genomic annotation. OMICS. 2008;12:137–141. doi: 10.1089/omi.2008.0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, et al. The SEED and the rapid annotation of microbial genomes using Subsystems technology (RAST) Nucleic Acids Res. 2014;42:D206–D214. doi: 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 60.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:W182–W185. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, et al. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Winsor GL, Lam DK, Fleming L, Lo R, Whiteside MD, Yu NY, et al. Pseudomonas genome database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res. 2011;39:D596–D600. doi: 10.1093/nar/gkq869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Darling AE, Mau B, Perna NT. ProgressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen H, Boutros PC. VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics. 2011;12:35. doi: 10.1186/1471-2105-12-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6(2):80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv Prepr. 2013;0(0):1–2.
- 70.Lunter G, Goodson M. Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 2011;21(6):936–939. doi: 10.1101/gr.111120.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Barnett DW, Garrison EK, Quinlan AR, Strömberg MP, Marth GT. BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics. 2011;27(12):1691–1692. doi: 10.1093/bioinformatics/btr174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14(2):178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2013;30(7):923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets supporting the conclusions of this article are available in the Genbank repository under the accession number CP011857 for the complete genome of P. aeruginosa ATCC 27853 and the project accession number PRJNA377172 for the transcriptome data of P. aeruginosa PAO1 and ATCC 27853.