

Abstract
Receptors for advanced glycation end-products (RAGE) mediate the inflammatory reaction that follows aneurysmal subarachnoid haemorrhage. Soluble RAGE (sRAGE) may function as a decoy receptor. The significance of this endogenous anti-inflammatory mechanism in subarachnoid haemorrhage (SAH) remains unknown. The present study aims to analyse sRAGE levels in the cerebrospinal fluid (CSF) of SAH patients. sRAGE levels were assayed by ELISA kit in 47 CSF samples collected on post-SAH days 0–3, 5–7, and 10–14 from 27 SAH patients with acute hydrocephalus. CSF levels of sRAGE were compared with a control group and correlated with other monitored parameters. In the control group, the CSF contained only a trace amount of sRAGE. By contrast, the CSF of 20 SAH patients collected on post-SAH days 0–3 was found to contain statistically significant higher levels of sRAGE (mean concentration 3.91 pg/mL, p < 0.001). The most pronounced difference in CSF sRAGE levels between good and poor outcome patients was found on days 0–3 post-SAH but did not reach the significance threshold (p = 0.234). CSF sRAGE levels did not change significantly during hospitalisation (p = 0.868) and correlated poorly with treatment outcome, systemic inflammatory markers, and other monitored parameters. Our study revealed an early and constant increase of sRAGE level in the CSF of SAH patients.
1. Introduction
The management of aneurysmal subarachnoid haemorrhage (SAH) has seen no significant advance since the introduction of nimodipine and the use of Guglielmi detachable coils [1, 2]. Favourable outcomes occur in only about one-third of patients admitted in a poor neurological state, and no new drugs have been approved for use in SAH in the past two decades; hence there is an urgent need to look for new therapies [3–8]. Early brain injury (EBI) is considered a promising target for future research [9, 10]. This represents the pathophysiological events occurring during the first 72 h following SAH and strongly determines the mortality and morbidity [11]. Experimental models support a number of mechanisms for EBI including inflammation [9]. Further clinical studies need to determine which of these mechanisms predominate. The present writers regard inflammation as a promising target for investigation. On a cellular level, inflammation is triggered by a ligand-receptor interaction. Among the most abundant multiligand are receptors for advanced glycation end-products (RAGE). RAGE has been shown to be present in neurons, glia, and microglia in the human hippocampus and cortex [12]. The concentration of many of its ligands (e.g., high mobility group box 1 protein, S100B protein) in plasma or cerebrospinal fluid (CSF) correlates with the clinical outcomes in patients with SAH [13–15]. Binding of these ligands to RAGE leads to the recruitment of multiple intracellular signalling molecules and eventually activates pathways responsible for acute and chronic inflammation [16]. There is a growing body of evidence that RAGE and its ligands are involved in the pathogenesis of other disorders including some cardiovascular conditions, neurodegenerative processes, and autoimmune diseases [17]. The soluble isoform of RAGE (sRAGE) corresponds to the extracellular domain of RAGE lacking cytosolic and transmembrane domains. As a decoy receptor, sRAGE is able to bind the same ligands as a membrane-bound form but unable to trigger the intracellular responses. The anti-inflammatory potential was confirmed in a mouse experimental stroke model, where intravenous administration of recombinant sRAGE significantly reduced infarct size and improved functional outcome [18]. The soluble form of RAGE has also been widely recognised as a biomarker. CSF levels of sRAGE were observed to be reduced in Guillain-Barré syndrome and multiple sclerosis [19, 20]. In view of the presence of sRAGE in CSF, its significant role in ischaemic stroke, and the role of its ligands in SAH, we aim to analyse sRAGE levels in CSF of patients with SAH requiring acute treatment of hydrocephalus.
2. Materials and Methods
2.1. Study Population
This single-centre, observational, prospective study was conducted in accordance with the Declaration of Helsinki and its protocol was approved by the local bioethics committee. Between January 2015 and September 2016, twenty-seven patients met the enrollment criteria which are as follows: (1) SAH confirmed by computed tomography (CT), (2) early (<24 h) endovascular treatment, (3) acute hydrocephalus diagnosed on CT and managed with external ventricular drainage (EVD) < 48 h, and (4) informed consent (by patient or family). Patients below the age of 18 were excluded due to physiological differences in CSF content as well as distinct aSAH presentation, aneurysm morphology, and outcome [21, 22]. Also excluded were patients with central nervous system (CNS) disease and those with active systemic diseases (diabetes mellitus, rheumatoid arthritis, malignancy, cirrhosis, and renal failure). Meticulous care was taken to rule out patients with signs of EVD infection. CSF cell count was checked at least twice per patient and CSF culture was ordered at least once on post-SAH days 10–14. SAH management in our unit involves the continuous intravenous infusion of nimodipine for at least ten days, whilst avoiding hypotension by means of vasopressors. CT scan of the head was carried out at least twice in every patient: on post-SAH days 2-3 to assess procedure related injury and before discharge to assess delayed cerebral ischaemia. The control samples of CSF were obtained during anaesthesia from 20 patients with a negative history of CNS disease.
2.2. End Points
Subjects were followed until death or the completion of 3 months following SAH. The primary outcome was the functional state after 3 months, and the secondary outcome was in-hospital mortality. The functional outcome was defined using the Glasgow Outcome Scale (GOS); these were dichotomized as good (GOS 4-5) or poor (GOS 1–3) outcomes.
2.3. Sample Collection and Assays
CSF samples were collected from the EVD at three time points, on post-SAH days 0–3, 5–7, and 10–14. The final sRAGE assays comprised twenty samples from days 0–3, sixteen from days 5–7, and eleven from days 10–14. A complete set of three samples was obtained from only five patients on account of suspected EVD infection, EVD obstruction, or early EVD removal. Each sample was centrifuged and stored at −80°C until assayed. The sRAGE assays were carried out using ELISA commercial kit RAB0007-1KT (Sigma Aldrich, St. Louis, USA). The minimum detectable level of sRAGE was 2.06 pg/mL, and linearity was conserved between 2.06 pg/mL and 1500 pg/mL. Haemoglobin (Hgb) level, C-reactive protein (CRP) level, and white blood cell (WBC) count and fibrinogen level were assessed daily by automatic analysers XT 2000i (Sysmex, Japan), Cobas 6000 (Roche Diagnostic, USA), and ACL TOP 500 (Instrumentation Laboratory, Italy).
2.4. Statistical Analysis
In the tables, values for numerical data have been expressed as mean and standard deviations; for ordinal numerical data, they are expressed as median and interquartile range and for categorical data as counts and percentages. In the figures, all data are presented as mean and standard deviations. The normality of data distribution was assessed using the Shapiro-Wilk test. The correlations were assessed by Spearman's test, and correlation coefficient (cc) > 0.6 (cc < −0.6) was considered significant. A value of p < 0.05 was considered statistically significant when comparing. Data were analysed using Statistica 10 (Statsoft, Inc., Tulsa, OK, USA).
3. Results
The control group consisted of twenty-five patients free of CNS disease. Detectable levels of sRAGE were found in only two members of this group (CSF sRAGE of 2.1 and 1.87 pg/mL). The details of the study group are presented in Table 1. CSF collected on days 0–3 following aneurysmal rupture in twenty of these patients contained statistically significant higher levels of sRAGE (p < 0.001) (Figure 1). No sRAGE was found in four of these twenty. Mean concentration varied significantly (0–15.22 pg/mL) but failed to differentiate good and poor outcome. The most pronounced difference between good and poor outcome was found at this stage but did not achieve statistical significance (p = 0.234) (Figure 2). The p values for days 5–7 and 10–14 were 0.291 and 0.490, respectively. Furthermore, CSF sRAGE levels did not change significantly during hospitalisation (p = 0.868) (Figure 3). Spearman's test revealed that the strongest correlation with outcome (measured by GOS at 3 months) were the admission grades Hunt and Hess (HH) (cc = −0.656), Glasgow Coma Scale (GCS) (cc = 0.688), and World Federation of Neurosurgical Societies (WFNS) (cc = −0.741), together with the fibrinogen level on days 10–14 post-SAH (cc = −0.626). sRAGE levels, haemoglobin, and blood inflammatory markers (CRP, WBC) showed poor correlation with treatment outcome (Table 2). The sRAGE levels of patients scoring 5 on the WFNS scale at days 0–3 showed a stronger correlation (cc = 0.485) with treatment outcome than those scoring 5 on the HH scale (cc = 0.176).
Table 1.
Study group patients' characteristic.
| Male | 15 (56%) | ||
| Age (years) | 58.07 ± 15.8 | ||
|
| |||
| Aneurysm location | |||
|
| |||
| Middle cerebral artery | 7 (26%) | ||
| Anterior communicating artery | 7 (26%) | ||
| Anterior cerebral artery | 4 (15%) | ||
| Basilar artery | 4 (15%) | ||
| Internal carotid artery | 3 (11%) | ||
| Posterior cerebral artery | 2 (7%) | ||
|
| |||
| Aneurysmal size (mm) | 5.08 ± 1.8 | ||
| Cerebral infarction due to DCI on CT | 20 (74%) | ||
| Intracerebral haemorrhage on CT | 14 (52%) | ||
| Intraventricular blood on CT | 26 (96%) | ||
| Fisher CT score | 4 (4-4) | ||
| Modified Fisher CT score | 4 (2–4) | ||
| WFNS score on admission | 5 (3–5) | ||
| HH score on admission | 4 (4-5) | ||
| GCS on admission | 5 (4–10) | ||
|
| |||
| Post-SAH days 0–3 | Post-SAH days 5–7 | Post-SAH days 10–14 | |
|
| |||
| CRP level (mg/L) | 106.90 ± 88.9 | 129.68 ± 96.8 | 75.09 ± 84.3 |
| WBC count (106/mm3) | 13.81 ± 5,4 | 12.01 ± 5.0 | 14.35 ± 6.3 |
| Hgb level (mg/dL) | 12.52 ± 1.7 | 12.10 ± 1.6 | 10.84 ± 1.2 |
| Fibrinogen (mg/dL) | 415.61 ± 163.0 | 617.63 ± 241.1 | 600.33 ± 247.7 |
| sRAGE (pg/mL) | 3.91 ± 4.0 | 4.24 ± 3.9 | 4.05 ± 3.8 |
|
| |||
| Treatment outcome (according to GOS at 3 months) | |||
|
| |||
| Low disability (score of 5) | 5 (19%) | ||
| Moderate disability (score of 4) | 2 (7%) | ||
| Severe disability (score of 3) | 4 (15%) | ||
| Persistent vegetative state (score of 2) | 5 (19%) | ||
| Death (score of 1) | 11 (41%) | ||
Figure 1.

sRAGE level in study and control group. Mann–Whitney test revealed significantly higher (p < 0.001) levels of sRAGE in CSF of SAH patients.
Figure 2.

sRAGE level in patients with poor and good treatment outcome on days 0–3 post-SAH. Mann–Whitney test revealed no significant difference (p = 0.234) in sRAGE level between patients with good and poor treatment outcome.
Figure 3.
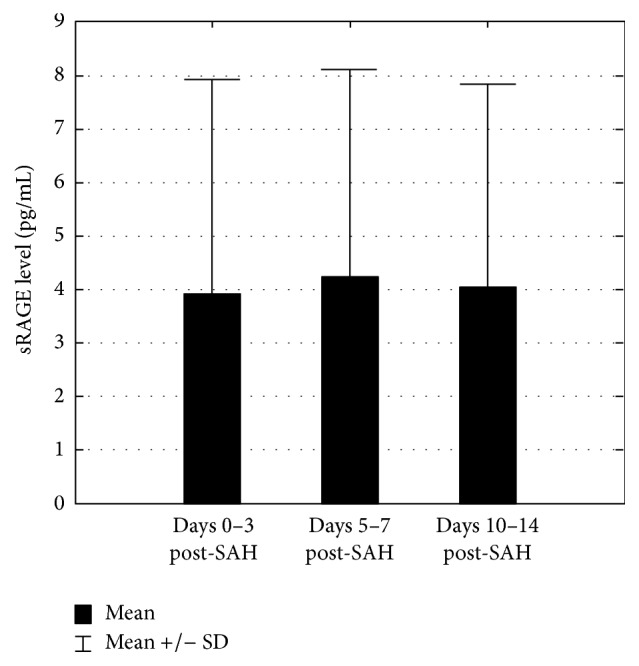
sRAGE level changes in time in study group. Skillings-Mack test indicates no significant change of CSF sRAGE level during hospitalisation (p = 0.868).
Table 2.
Spearman's correlation between treatment outcome and monitored parameters.
| Parameter | Correlation coefficient | p value | |
|---|---|---|---|
| Days 0–3 post-SAH | CRP | −0.273 | 0.257 |
| WBC | −0.433 | 0.063 | |
| Hgb | 0.118 | 0.628 | |
| Fibrinogen | −0.286 | 0.248 | |
| sRAGE | −0.177 | 0.454 | |
|
| |||
| Days 5–7 post-SAH | CRP | −0.339 | 0.198 |
| WBC | −0.333 | 0.207 | |
| Hgb | 0.001 | 0.995 | |
| Fibrinogen | −0.484 | 0.057 | |
| sRAGE | −0.302 | 0.254 | |
|
| |||
| Days 10–14 post-SAH | CRP | −0.480 | 0.134 |
| WBC | −0.086 | 0.800 | |
| Hgb | 0.493 | 0.122 | |
| Fibrinogen | −0.626 | 0.070 | |
| sRAGE | 0.139 | 0.682 | |
|
| |||
| WFNS on admission | −0.741 | <0.001 | |
| HH on admission | −0.656 | <0.001 | |
| GCS on admission | 0.688 | <0.001 | |
4. Discussion
Our study demonstrated elevation of CSF sRAGE in patients with poor grade SAH requiring EVD insertion. Clinical studies of sRAGE in patients with neurological disorders have thus far revealed the following: (1) serum sRAGE elevation in ischaemic stroke patients [18], (2) serum sRAGE correlation with severity of the axonal subtype of Guillain-Barré syndrome [19], and (3) CSF sRAGE decrease in patients with multiple sclerosis and Guillain-Barré syndrome [19, 20]. We are not aware of any previous reports demonstrating elevation of sRAGE in CSF following pathological processes, particularly in patients with SAH.
RAGE is a transmembrane protein that belongs to the immunoglobulin superfamily [18, 23–25]. The human RAGE gene is located on chromosome 6 and its expression leads to production of a 55-kDa type I membrane glycoprotein [24]. Soluble isoforms of RAGE are formed either by (1) removal of the transmembrane region from the pre-RNA during alternative splicing (leading to the production of endogenous sRAGE) or (2) proteolytic cleavage of the full-length membrane form of RAGE protein (mRAGE) by a membrane metalloproteinase called ADAM 10 or an extracellular matrix metalloproteinase 9 (MMP-9) [25–27]. ADAM 10 is a representative of sheddases, membrane-bound enzymes that cleave extracellular portions of transmembrane proteins, releasing the soluble ectodomains from the cell surface. In healthy population, mean blood plasma sRAGE concentration ranges from 800 to 1500 pg/mL [28, 29]. In our study, CSF sRAGE levels in control patients without a history of neurological disorder were undetectable. This finding is in accordance with the recent findings of Zhang et al. [19]. A rat experimental SAH model revealed significant increases in RAGE protein and mRNA levels in neurons and microglia [30]. Furthermore, an increase of MMP-9 levels in both CSF and serum was observed during SAH [31]. Based on these findings, we suspect that three mechanisms are leading to an increase of sRAGE levels in the CSF in our patients. Firstly, SAH-induced expression of RAGE leads to overexpression of all its isoforms, including endogenous sRAGE. This explanation follows Tang et al. hypothesis that high levels of plasma sRAGE at 48 h after stroke may reflect the rapid activation of mRAGE expression induced by the cerebral ischaemia [18]. A second possible mechanism is an excessive cleavage of membrane-bound RAGE. Increase of RAGE expression on cell membranes and rise of MMP-9 level (both observed during SAH) support this hypothesis [30, 31]. A third expected mechanism is introduction of free plasma sRAGE during aneurysm rupture and blood extravasation to subarachnoid space. Estimated total SAH volume equals 35 mL and as a blood contains 200 to 400 times higher sRAGE levels than those measured in CSF, we would expect more significant elevation of sRAGE levels originating from the SAH patients analysed in our study [32].
As our understanding of the SAH complications has improved, identifying mediators of its critical pathways and designing new targeted therapies becomes of primary importance in neuroproteomics research [33]. The inflammatory reaction, which contributes to SAH-induced brain injury is characterised by complex, multilevel interactions between its separate components. The activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) observed in SAH leads to excessive inflammation and subsequent brain injury [34, 35]. NF-kB activation is mediated by numerous upstream pathways, including those starting at RAGE and Toll-like receptors 2 and 4 (TLR2/TLR4). Results of our previous studies on soluble TLR2 and 4 suggested that these played only a minor role in this inhibitory mechanism [36]. Tang et al. in their study of sRAGE in human stroke patients found sRAGE to be an independent predictor of functional outcome. In experimental settings, administration of recombinant sRAGE significantly improved the outcome after ischaemic stroke in mice. The suspected protective mechanism depends on high mobility group box 1 binding [18]. Quade-Lyssy et al. have reported that atorvastatin increased the levels of serum sRAGE [37], whilst Cheng et al. [38] and Potey et al. [39] have shown some evidence that atorvastatin ameliorates vasospasm and EBI after SAH. However, the STASH failed to detect any benefit to the long- or short-term outcome using simvastatin in aneurysmal SAH [40]. In the most recent report by Wang et al., administration of recombinant sRAGE significantly reduced the number of positive TUNEL staining cells in SAH rat and improved cell viability in post-SAH CSF-treated cultured neurons [41]. In our study, sRAGE levels failed to differentiate between good and poor outcome patients. These preliminary findings are similar to the results obtained with soluble TLR2/TLR4 and might suggest that sRAGE has limited significance as a prognostic biomarker. Yet, further investigations (addressing limitations of the current study) are essential to assess role of sRAGE in the endogenous anti-inflammatory mechanism. Our study showed no correlation between sRAGE and systemic inflammatory mediators. Although CRP is able to augment mRNA expression of RAGE genes, the levels to which this can be achieved are not known [42].
Conclusions are limited by the small number of patients involved in the study and lack of consecutive sampling in most cases. The enrolled patients do not represent the full spectrum of SAH as hydrocephalus was an inclusion criterion and could have contributed to brain injury before EVD insertion. Despite careful monitoring, EVD infection remains a potential bias. In addition, only a few elements of the inflammatory pathway were investigated, and further investigation will be required to elucidate the larger picture of post-SAH inflammation.
5. Conclusions
CSF levels of sRAGE increase early in patients with SAH who require acute treatment of hydrocephalus and remain elevated but do not correlate with treatment outcome. The significance of sRAGE as an endogenous anti-inflammatory mechanism requires further investigation.
Acknowledgments
The authors would like to thank Mr. Paul H. Walter, Consultant Neurosurgeon, for the thorough review; Ms. Barbara Więckowska (Department of Computer Science and Statistics, Poznan Medical University) for her valuable statistical assistance. They are also grateful to the nursing staff of ICU of Heliodor Święcicki Clinical Hospital of the Poznan University of Medical Sciences. This study was supported in part by a Diamond Grant no. DI2014 005044 from the Polish Ministry of Science and Higher Education (years 2015 and 2016).
Conflicts of Interest
The authors declare that there are no conflicts of interest regarding the publication of this paper. Received Funding no. DI2014 005044 did not lead to any conflicts of interest regarding the publication of this manuscript.
References
- 1.Pickard J. D., Murray G. D., Illingworth R., et al. Effect of oral nimodipine on cerebral infarction and outcome after subarachnoid haemorrhage: British aneurysm nimodipine trial. The British Medical Journal. 1989;298:636–642. doi: 10.1136/bmj.298.6674.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guglielmi G., Vinuela F., Dion J., Duckwiler G. Electrothrombosis of saccular aneurysms via endovascular approach. Part 2: Preliminary clinical experience. Journal of Neurosurgery. 1991;75(1):8–14. doi: 10.3171/jns.1991.75.1.0008. [DOI] [PubMed] [Google Scholar]
- 3.de Oliveira Manoel A. L., Mansur A., Silva G. S., Germans M. R., Jaja B. N. R., Kouzmina E., et al. Functional Outcome After Poor-Grade Subarachnoid Hemorrhage: A Single-Center Study and Systematic Literature Review. Neurocritical Care, Springer US. 2016;25:338–350. doi: 10.1007/s12028-016-0305-3. [DOI] [PubMed] [Google Scholar]
- 4.King M. D., Laird M. D., Ramesh S. S., et al. Elucidating novel mechanisms of brain injury following subarachnoid hemorrhage: an emerging role for neuroproteomics. Neurosurgical Focus. 2010;28(1):p. E10. doi: 10.3171/2009.10.focus09223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sabri M., Lass E., MacDonald R. L. Early brain injury: A common mechanism in subarachnoid hemorrhage and global cerebral ischemia. Stroke Research and Treatment. 2013 doi: 10.1155/2013/394036.394036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fujii M., Yan J., Rolland W. B., Soejima Y., Caner B., Zhang J. H. Early brain Injury, an evolving frontier in subarachnoid hemorrhage research. Translational Stroke Research. 2013;4(4):432–446. doi: 10.1007/s12975-013-0257-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cahill J., Zhang J. H. Subarachnoid hemorrhage: Is it time for a new direction? Stroke. 2009;40(3):S86–S87. doi: 10.1161/STROKEAHA.108.533315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang J. H., Pluta R. M., Hansen-Schwartz J., et al. Cerebral vasospasm following subarachnoid hemorrhage: time for a new world of thought. Neurological Research. 2009;31(2):151–158. doi: 10.1179/174313209X393564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sehba F. A., Pluta R. M., Zhang J. H. Metamorphosis of subarachnoid hemorrhage research: From delayed vasospasm to early brain injury. Molecular Neurobiology. 2011;43(1):27–40. doi: 10.1007/s12035-010-8155-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brathwaite S., Macdonald R. L. Current management of delayed cerebral ischemia: update from results of recent clinical trials. Translational Stroke Research. 2014;5(2):207–226. doi: 10.1007/s12975-013-0316-8. [DOI] [PubMed] [Google Scholar]
- 11.Kusaka G., Ishikawa M., Nanda A., Granger D. N., Zhang J. H. Signaling pathways for early brain injury after subarachnoid hemorrhage. Journal of Cerebral Blood Flow & Metabolism. 2004;24:916–925. doi: 10.1097/01.WCB.0000125886.48838.7E. [DOI] [PubMed] [Google Scholar]
- 12.Choi B. R., Cho W. H., Kim J., et al. Increased expression of the receptor for advanced glycation end products in neurons and astrocytes in a triple transgenic mouse model of Alzheimer's disease. Experimental and Molecular Medicine. 2014;46, article e75 doi: 10.1038/emm.2013.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lai P. M. R., Du R. Association between S100B levels and long-term outcome after aneurysmal subarachnoid hemorrhage: Systematic review and pooled analysis. PLoS ONE. 2016;11(3) doi: 10.1371/journal.pone.0151853.e0151853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakahara T., Tsuruta R., Kaneko T., et al. High-mobility group box 1 protein in CSF of patients with subarachnoid hemorrhage. Neurocritical Care. 2009;11(3):362–368. doi: 10.1007/s12028-009-9276-y. [DOI] [PubMed] [Google Scholar]
- 15.Sokół B., Woźniak A., Jankowski R., et al. HMGB1 Level in Cerebrospinal Fluid as a Marker of Treatment Outcome in Patients with Acute Hydrocephalus Following Aneurysmal Subarachnoid Hemorrhage. Journal of Stroke and Cerebrovascular Diseases. 2015;24(8):1897–1904. doi: 10.1016/j.jstrokecerebrovasdis.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 16.Sparvero L. J., Asafu-Adjei D., Kang R., et al. RAGE (Receptor for advanced glycation endproducts), RAGE ligands, and their role in cancer and inflammation. Journal of Translational Medicine. 2009;7, article no. 17 doi: 10.1186/1479-5876-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramasamy R., Yan S. F., Herold K., Clynes R., Schmidt A. M. Receptor for advanced glycation end products. Fundamental roles in the inflammatory response: Winding the way to the pathogenesis of endothelial dysfunction and atherosclerosis. Annals of the New York Academy of Sciences. 2008;1126:7–13. doi: 10.1196/annals.1433.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang S., Wang Y., Li Y., Lin H., Manzanero S., Hsieh Y., et al. Functional Role of Soluble Receptor for Advanced Glycation End Products in Stroke. Arteriosclerosis, Thrombosis, and Vascular Biology. 2013:585–595. doi: 10.1161/ATVBAHA.112.300523. [DOI] [PubMed] [Google Scholar]
- 19.Zhang D. Q., Wang R., Li T., et al. Reduced soluble RAGE is associated with disease severity of axonal Guillain-Barré syndrome. Scientific Reports Nature Publishing Group. 2016;(6) doi: 10.1038/srep21890.21890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glasnović A., Cvija H., Stojić M., et al. Decreased level of sRAGE in the cerebrospinal fluid of multiple sclerosis patients at clinical onset. NeuroImmunoModulation. 2014;21(5):226–233. doi: 10.1159/000357002. [DOI] [PubMed] [Google Scholar]
- 21.Shah S. S., Ebberson J., Kestenbaum L. A., Hodinka R. L., Zorc J. J. Age-specific reference values for cerebrospinal fluid protein concentration in neonates and young infants. Journal of Hospital Medicine. 2011;6(1):22–27. doi: 10.1002/jhm.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koroknay-Pál P., Lehto H., Niemelä M., Kivisaari R., Hernesniemi J. Long-term outcome of 114 children with cerebral aneurysms. Journal of Neurosurgery: Pediatrics. 2012;9(6):636–645. doi: 10.3171/2012.2.PEDS11491. [DOI] [PubMed] [Google Scholar]
- 23.Lee E. J., Park J. H. Receptor for advanced glycation endproducts (RAGE), its ligands, and soluble rage: potential biomarkers for diagnosis and therapeutic targets for human renal diseases. Genomics & Informatics. 2013;11:224–229. doi: 10.5808/GI.2013.11.4.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neeper M., Schmidt M A., Brett J., Yan S. D., Wang F., Pan Y. C., et al. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. Journal of Biological Chemistry. 1992;267:14998–5004. [PubMed] [Google Scholar]
- 25.Tang S. C., Wang Y. C., Li Y. I., Lin H. C., Manzanero S., Hsieh Y. H., et al. Functional role of soluble receptor for advanced glycation end products in stroke. Arteriosclerosis, Thrombosis, and Vascular Biology. 2013;33:585–594. doi: 10.1161/ATVBAHA.112.300523. [DOI] [PubMed] [Google Scholar]
- 26.Chuah Y. K., Basir R., Talib H., Tie T. H., Nordin N. Receptor for advanced glycation end products and its involvement in inflammatory diseases. International Journal of Inflammation. 2013;2013:15. doi: 10.1155/2013/403460.403460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang L., Bukulin M., Kojro E., et al. Receptor for advanced glycation end products is subjected to protein ectodomain shedding by metalloproteinases. Journal of Biological Chemistry. 2008;283(51):35507–35516. doi: 10.1074/jbc.m806948200. [DOI] [PubMed] [Google Scholar]
- 28.Gopal P., Rutten E. P. A., Dentener M. A., Wouters E. F. M., Reynaert N. L. Decreased plasma sRAGE levels in COPD: Influence of oxygen therapy. European Journal of Clinical Investigation. 2012;42(8):807–814. doi: 10.1111/j.1365-2362.2012.02646.x. [DOI] [PubMed] [Google Scholar]
- 29.Gopal P., Reynaert N. L., Scheijen J. L. J. M., et al. Association of plasma sRAGE, but not esRAGE with lung function impairment in COPD. Respiratory Research. 2014;15(1, article no. 24) doi: 10.1186/1465-9921-15-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H., Wu W., Sun Q., et al. Expression and cell distribution of receptor for advanced glycation end-products in the rat cortex following experimental subarachnoid hemorrhage. Brain Research. 2014;1543:315–323. doi: 10.1016/j.brainres.2013.11.023. [DOI] [PubMed] [Google Scholar]
- 31.Chou S. H.-Y., Lee P.-S., Konigsberg R. G., et al. Plasma-type gelsolin is decreased in human blood and cerebrospinal fluid after subarachnoid hemorrhage. Stroke. 2011;42(12):3624–3627. doi: 10.1161/STROKEAHA.111.631135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Whitmore R. G., Grant R. A., Leroux P., El-Falaki O., Stein S. C. How large is the typical subarachnoid hemorrhage? A review of current neurosurgical knowledge. World Neurosurgery. 2012;77(5-6):686–697. doi: 10.1016/j.wneu.2011.02.032. [DOI] [PubMed] [Google Scholar]
- 33.Santilli F., Vazzana N., Bucciarelli L. G., Davì G. Soluble forms of RAGE in human diseases: Clinical and therapeutical implications. Current Medicinal Chemistry. 2009;16(8):940–952. doi: 10.2174/092986709787581888. [DOI] [PubMed] [Google Scholar]
- 34.Zhou M., Shi J., Hang C., et al. Potential contribution of nuclear factor-κB to cerebral vasospasm after experimental subarachnoid hemorrhage in rabbits. Journal of Cerebral Blood Flow and Metabolism. 2007;27(9):1583–1592. doi: 10.1038/sj.jcbfm.9600456. [DOI] [PubMed] [Google Scholar]
- 35.You W.-C., Wang C.-X., Pan Y.-X., et al. Activation of Nuclear Factor-κB in the Brain after Experimental Subarachnoid Hemorrhage and Its Potential Role in Delayed Brain Injury. PLoS ONE. 2013;8(3) doi: 10.1371/journal.pone.0060290.e60290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sokół B., Wąsik N., Jankowski R., Hołysz M., Wiȩckowska B., Jagodziński P. Soluble toll-like receptors 2 and 4 in cerebrospinal fluid of patients with acute hydrocephalus following aneurysmal subarachnoid haemorrhage. PLoS ONE. 2016;11(5) doi: 10.1371/journal.pone.0156171.e0156171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quade-Lyssy P., Kanarek A. M., Baiersdörfer M., Postina R., Kojro E. Statins stimulate the production of a soluble form of the receptor for advanced glycation end products. Journal of Lipid Research. 2013;54(11):3052–3061. doi: 10.1194/jlr.M038968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheng G., Wei L., Zhi-Dan S., Shi-Guang Z., Xiang-Zhen L. Atorvastatin ameliorates cerebral vasospasm and early brain injury after subarachnoid hemorrhage and inhibits caspase-dependent apoptosis pathway. BMC Neuroscience. 2009;10, article no. 7 doi: 10.1186/1471-2202-10-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Potey C., Ouk T., Petrault O., et al. Early treatment with atorvastatin exerts parenchymal and vascular protective effects in experimental cerebral ischaemia. British Journal of Pharmacology. 2015;172(21):5188–5198. doi: 10.1111/bph.13285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kirkpatrick P. J., Turner C. L., Smith C., Hutchinson P. J., Murray G. D. Simvastatin in aneurysmal subarachnoid haemorrhage (STASH): a multicentre randomised phase 3 trial. The Lancet Neurology. 2014;13:666–675. doi: 10.1016/s1474-4422(14)70084-5. [DOI] [PubMed] [Google Scholar]
- 41.Wang K. C., Tang S. C., Lee J. E., Li Y. I., Huang Y. S., Yang W. S., et al. Cerebrospinal fluid high mobility group box 1 is associated with neuronal death in subarachnoid hemorrhage. Journal of Cerebral Blood Flow & Metabolism. 2017;37:435–443. doi: 10.1177/0271678X16629484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mahajan N., Bahl A., Dhawan V. C-reactive protein (CRP) up-regulates expression of receptor for advanced glycation end products (RAGE) and its inflammatory ligand EN-RAGE in THP-1 cells: Inhibitory effects of atorvastatin. International Journal of Cardiology. 2010;142(3):273–278. doi: 10.1016/j.ijcard.2009.01.008. [DOI] [PubMed] [Google Scholar]