

Abstract
Epidermal growth factor receptor (EGFR) is an important target for cancer therapy. In this study, EGFR inhibitors were investigated to build a two-dimensional quantitative structure-activity relationship (2D-QSAR) model and a three-dimensional quantitative structure-activity relationship (3D-QSAR) model. In the 2D-QSAR model, the support vector machine (SVM) classifier combined with the feature selection method was applied to predict whether a compound was an EGFR inhibitor. As a result, the prediction accuracy of the 2D-QSAR model was 98.99% by using tenfold cross-validation test and 97.67% by using independent set test. Then, in the 3D-QSAR model, the model with q2 = 0.565 (cross-validated correlation coefficient) and r2 = 0.888 (non-cross-validated correlation coefficient) was built to predict the activity of EGFR inhibitors. The mean absolute error (MAE) of the training set and test set was 0.308 log units and 0.526 log units, respectively. In addition, molecular docking was also employed to investigate the interaction between EGFR inhibitors and EGFR.
1. Introduction
Epidermal growth factor receptor (EGFR), a transmembrane glycoprotein, is classified to the prototype of receptor tyrosine kinases (TKs) family that includes EGFR, ErbB-2, ErbB-3, and ErbB-4. EGFR is activated by its cognate ligands via forming a homodimer or heterodimer with other members of the EGFR family, such as epidermal growth factor (EGF) and transforming growth factor alpha (TGF-α) [1]. Several signal transduction cascades are initiated when EGFR is activated and then lead to DNA synthesis and cell proliferation [2, 3]. While EGFR is amplified or mutated, DNA synthesis and cell proliferation will be abnormal and lead to cancer. Currently, the amplification or mutation of EGFR has been found in human solid tumors, such as glioma, lung cancer, ovarian cancer, and breast cancer. Hence, EGFR is also considered to be a potential anticancer target in this disease [4–8]. Many EGFR inhibitors have been developed and approved by the FDA, such as lapatinib, which has been applied for the treatment of breast cancer [9]. Moreover, other EGFR inhibitors like temozolomide, lomustine, erlotinib, and gefitinib, are approved by the FDA for the treatment of glioma [10, 11]. However, the existing EGFR inhibitors are beyond people's expectation due to selectivity, toxicity, and side effect. Hence, it is necessary to design and synthesize new potential EGFR inhibitors.
Quantitative structure-activity relationship (QSAR) was a valuable tool for many different applications, including drug discovery, predictive toxicology, and risk assessment [12–14]. The applicability domain of QSAR models, defined by the Organization for Economic Co-operation and Development (OECD) according to Principle 3, includes the physicochemical, the structural, and the biological domain [15–17]. Initially, two-dimensional quantitative structure-activity relationship (2D-QSAR) was widely explored and used in medicinal chemistry study. However, some limitations spurred the appearance of three-dimensional quantitative structure-activity relationship (3D-QSAR). In the 3D-QSAR study, the correlation between 3D steric and electrostatic fields and biologically activity draws attention. For the molecular field study, CoMFA was widely used preliminarily. However, the time-consuming limit stimulates the advent of TopCoMFA. TopCoMFA overcomes the weakness and uses an objective method to fragment and align the molecules. In addition, the fragmentation process is automated except for some specific bonds that should be cleaved manually. Of course, TopCoMFA and CoMFA also have similarity that they both share QSAR PLS analysis. The details about TopCoMFA and CoMFA are in [18].
Drug development is a long process, and it requires a vast amount of material and financial resources. QSAR and molecular docking technology have been extensively employed in drug virtual screening and potential molecular targets prediction, which may shorten the cycle of the drug development [19–22]. In this work, 2D-QSAR model was employed to determine EGFR inhibitor, and the 3D-QSAR model was used to predict the activity. Finally, molecular docking was applied to investigate the binding sites.
2. Materials and Methods
2.1. CfsSubsetEval Method and Greedy Stepwise Algorithm
A data set containing n vectors has 2n possible combinations of features for the subset. A useful subset which can correctly predict other compounds is one of 2n combinations. The best way to find an optimal subset is to try all the possible feature combinations. However, this strategy is difficult to carry out due to the huge computation. In this study, the CfsSubsetEval (CFS) search method combined with Greedy Stepwise (GS) algorithm was employed to search the optimal feature subset. The main idea of the GS algorithms is to make the best choice when selecting good features. The CFS method was used to evaluate the attribute. Thus, the CFS method, combined with the GS algorithm, was employed to select the optimal subset from these 2n combinations. Additional details about the CFS method and the GS algorithm could be found in [23–25].
2.2. SVM
Support vector machine (SVM), a supervised learning algorithm, is usually used for pattern recognition classification [26]. SVM was employed for the classification and sensitivity analysis in our study due to its high performance in many studies [25, 27, 28].
2.3. Topomer CoMFA
Topomer CoMFA, possessing both the topomer technique and CoMFA technology, can overcome the alignment problem of CoMFA [18, 29]. Partial least squares (PLS) regression is employed to build the topomer CoMFA model, and the leave-one-out (LOO) cross-validation is used to evaluate the model. Additional details about the topomer CoMFA can be found in [29–31].
2.4. Data Preparation
100 inhibitors derived from the literature and 185 noninhibitors downloaded from the DUD database (http://dud.docking.org) were collected [32–41]. For 2D-QSAR study, the data set containing inhibitors and noninhibitors was randomly divided into three training sets which accounted for 75%, 70%, and 50% of the whole data set, respectively (see Supplementary Material 1, available online at https://doi.org/10.1155/2017/4649191). For 3D-QSAR study, the 100 inhibitors were randomly divided into a training set (77 molecules) and an independent test set (23 molecules).
2.5. Molecular Descriptor Calculation
Molecular descriptor can reflect physicochemical and geometric properties of the compounds. In this study, forty-five molecular descriptors calculated by the ChemOffice were applied to represent compounds [42]. First, three-dimensional structures of the molecules were optimized by MM+ force field with the Polak-Ribiere algorithm until the root-mean-square gradient became less than 0.1 Kcal/mol. Then, quantum chemical parameters were obtained for the most stable conformation of each molecule by using PM3 semiempirical molecular orbital method at the restricted Hartree-Fock level with no configuration interaction.
2.6. Validation Methods for Prediction Results
In this study, tenfold cross-validation test and independent set test were applied to evaluate the prediction ability of the 2D-QSAR model. For the tenfold cross-validation test, the data set was divided into ten subsets. Nine subsets were used as the training set and the left subset was predicted. In turn, each subset was omitted in order to be predicted, and the correct rate was obtained from each trial. The average of the correct rate from ten trials was used to estimate the accuracy of the algorithm [43–45].
2.7. Prediction Measurement
Sensitivity (SN), specificity (SP), overall accuracy (ACC), and Matthew's correlation coefficient (MCC) were employed to evaluate the 2D prediction model. The SN, SP, ACC, and MCC can be represented as
| (1) |
TP, TN, FP, and FN are true positives, true negatives, false positives, and false negatives, respectively.
In the topomer CoMFA model, q2, r2, and MAE were applied to evaluate the model [46]. The cut-off value of q2 is 0.5. The MAE of the test set was less than 0.1 × training set range and MAE + 3 × σ according to the MAE based criteria. The optimized model was determined by the highest q2, and the validity of the model depends on r2 value [47].
2.8. Steric and Electrostatic Field Analysis
Topomer CoMFA analysis is an effective approach which has been applied in drug design for HIV, central nervous system diseases, and other tumors [48–50]. In the topomer CoMFA model, there are two different ways to calculate the molecular field. One way is to reduce the field contributions of fragmenting atoms; the other way is to calculate the steric and electrostatic fields on a regularly spaced grid. For detailed information, see [51]. Topomer CoMFA analysis is used to calculate the steric field and electrostatic fields of R1 and R2 groups. Steric and electrostatic field analysis may help design novel EGFR drugs.
2.9. Molecular Docking
SYBYL X-2.0 was used for molecular docking based on its Surflex-Dock module [52]. The crystal structure of EGFR with the resolution of 2.6 Å was downloaded from the Protein Data Bank (PDB ID: 1M17) [53]. Protein was prepared with protein structure preparation module of the SYBYL X-2.0. All the water molecules and ligands were deleted, and hydrogen atoms were added to the crystal structure. In addition, positive and negative charges were added to N-terminal and C-terminal regions of the EGFR which became NH3+ and COO−. EGFR inhibitors were minimized at physiological pH 7.0 with hydrogen atoms and charge by using Powell energy gradient method and the Gasteiger-Huckel system.
3. Results
3.1. Feature Selection and the 2D-QSAR Prediction Model
A feature subset containing nine molecular descriptors (DPLL, H, HF, HOMO, MR, Pc, TIndx, VP, and WIndx) was obtained based on CFS combined with GS algorithms. Sensitivity analysis was applied to these nine descriptors to evaluate how they affected the activity of EGFR inhibitors (see Figure 1).
Figure 1.

(a) Activity value versus DPLL. (b) Activity value versus H. (c) Activity value versus HF. (d) Activity value versus HOMO. (e) Activity value versus MR. (f) Activity value versus Pc. (g) Activity value versus TIndx. (h) Activity value versus VP. (i) Activity value versus WIndx.
Based on the optimal features subset, the SVM classifier method was used to build the 2D-QSAR prediction model. As a result, the prediction accuracy of these models whose data set accounted for 75%, 70%, and 50% of the whole data set was 98.13%, 98.99%, and 91.24%, respectively, by tenfold cross-validation test. The sensitivity, specificity, and overall accuracy of these three models were more than 90%, which indicated that changing the size of the training set had a little impact on the quality of the 2D-SAR models (see Table 1). The model built via the data set accounting for 70% of the whole data set was chosen finally due to its higher prediction accuracy and smaller size. Although the result of the tenfold cross-validation test was well, it was not good enough for evaluating the classifier as the SVM classifier might be overfitted. To validate the reliability of the classifier, an independent test set was further employed in this study. As a result, the prediction accuracy of the independent set test was 97.67%.
Table 1.
The results of prediction accuracy for different data sets containing 9 molecular descriptors using SVM classifier. DS and EP present data set and evaluation parameters, respectively.
| EP | DS | |||
|---|---|---|---|---|
| Train set (75%) | Train set (70%) | Train set (50%) | Test set (30%) | |
| SN (%) | 97.22 | 98.55 | 91.94 | 96.77 |
| SP (%) | 98.59 | 99.23 | 90.67 | 98.18 |
| ACC (%) | 98.13 | 98.99 | 91.24 | 97.67 |
| MCC | 0.958 | 0.978 | 0.824 | 0.950 |
3.2. 3D-QSAR Prediction Model
The training set was employed to build the topomer CoMFA model by fragmenting EGFR inhibitors into R1 and R2 groups. Two topomer CoMFA models were generated by two cutting ways. The topomer CoMFA model 2 with higher q2 and r2 values was selected to analyze and predict EGFR inhibitors' activities (see Table 2).
Table 2.
Results from two topomer CoMFA model studies.
| Dataset | Topomer CoMFA model 1 | Topomer CoMFA model 2 |
|---|---|---|
| Cutting model |
|
|
| q 2 | 0.483 | 0.565 |
| r 2 | 0.773 | 0.888 |
The experimental and predicted activities of the training set and the independent test set were listed in Table 3 and Figure 2. As a result, the MAE and r2 of the training set were 0.308 and 0.888, respectively. The training set range was 7.32. To estimate the reliability of model 2, the independent set test was used to evaluate the model. The MAE and r2 of the test set were 0.526 and 0.681, respectively. The MAE of the test set was less than 0.732 (0.1 × training set range) and 1.903 (MAE(training set) + 3 × σ).
Table 3.
Experimental and predicted PIC50 for topomer CoMFA model 2.
| Compound | Exp | Pre |
|---|---|---|
| Training set | ||
| 2 | 7.64 | 6.62 |
| 4 | 6.24 | 6.2 |
| 5 | 6.04 | 6.45 |
| 7 | 6 | 6.16 |
| 8 | 8 | 8.15 |
| 10 | 7.25 | 7.05 |
| 11 | 6.11 | 6.62 |
| 13 | 7 | 6.31 |
| 15 | 6.09 | 6.06 |
| 16 | 6.26 | 6.14 |
| 17 | 7.53 | 8.02 |
| 18 | 9.5 | 9.06 |
| 20 | 8.39 | 8.28 |
| 22 | 7.92 | 8.01 |
| 23 | 8.32 | 7.59 |
| 24 | 8.15 | 8.05 |
| 25 | 7.92 | 8.22 |
| 26 | 7.95 | 7.78 |
| 27 | 9.16 | 8.64 |
| 29 | 8.42 | 8.87 |
| 30 | 8.18 | 8.3 |
| 31 | 7.82 | 8.03 |
| 32 | 7.6 | 7.26 |
| 33 | 9.76 | 9.6 |
| 34 | 9.01 | 8.05 |
| 36 | 8.11 | 7.94 |
| 37 | 7.74 | 7.43 |
| 38 | 7.35 | 7.31 |
| 40 | 8.01 | 8.59 |
| 41 | 8.36 | 8.46 |
| 42 | 7.45 | 7.71 |
| 43 | 7.88 | 7.7 |
| 45 | 6.6 | 6.36 |
| 46 | 7.39 | 7.84 |
| 47 | 8 | 7.5 |
| 48 | 7.04 | 6.87 |
| 50 | 6.88 | 6.82 |
| 51 | 6.17 | 6.08 |
| 53 | 5.74 | 6.36 |
| 54 | 5.31 | 5.72 |
| 55 | 6.07 | 7.21 |
| 56 | 6.92 | 7.4 |
| 57 | 7.39 | 6.9 |
| 58 | 7.29 | 7.14 |
| 60 | 6.9 | 7.15 |
| 61 | 8.58 | 8.47 |
| 63 | 6.16 | 5.85 |
| 64 | 6.02 | 6.36 |
| 65 | 7.28 | 6.86 |
| 66 | 6.48 | 6.54 |
| 67 | 6.58 | 7 |
| 69 | 7.08 | 7.57 |
| 70 | 8.82 | 8.38 |
| 71 | 9.11 | 8.97 |
| 72 | 9.02 | 8.97 |
| 73 | 8.42 | 8.96 |
| 75 | 8.53 | 9.2 |
| 76 | 8.63 | 8.35 |
| 77 | 6.42 | 6.97 |
| 78 | 7.76 | 7.78 |
| 79 | 8.36 | 8.34 |
| 80 | 8.63 | 8.39 |
| 81 | 6.19 | 6.8 |
| 82 | 8.52 | 7.97 |
| 83 | 8.05 | 8.04 |
| 85 | 7.1 | 7.16 |
| 86 | 7.5 | 7.57 |
| 87 | 7.26 | 7.52 |
| 88 | 6.04 | 6.06 |
| 90 | 4.33 | 4.35 |
| 91 | 4.66 | 4.62 |
| 92 | 5 | 5.52 |
| 94 | 7.19 | 7.17 |
| 95 | 6.23 | 5.89 |
| 97 | 4.14 | 3.98 |
| 98 | 8.05 | 7.54 |
| 99 | 6.97 | 6.79 |
|
| ||
| Test set | ||
| 1 | 6.46 | 5.58 |
| 3 | 7.57 | 7.72 |
| 6 | 6.45 | 6.16 |
| 9 | 7.25 | 6.44 |
| 12 | 6.24 | 7.24 |
| 14 | 5.21 | 5.87 |
| 19 | 9.05 | 9.14 |
| 21 | 7.07 | 7.41 |
| 28 | 6.79 | 7.33 |
| 35 | 7.46 | 7.23 |
| 39 | 8.5 | 8.53 |
| 44 | 7.4 | 8.31 |
| 49 | 5.43 | 6.4 |
| 52 | 5.27 | 6.36 |
| 59 | 7.39 | 7.25 |
| 62 | 8.63 | 8.41 |
| 68 | 7.88 | 7.81 |
| 74 | 9.09 | 8.98 |
| 84 | 6.72 | 7.69 |
| 89 | 5.94 | 5.67 |
| 93 | 7.17 | 6.68 |
| 96 | 5.01 | 6.82 |
| 100 | 6.2 | 6.18 |
Figure 2.
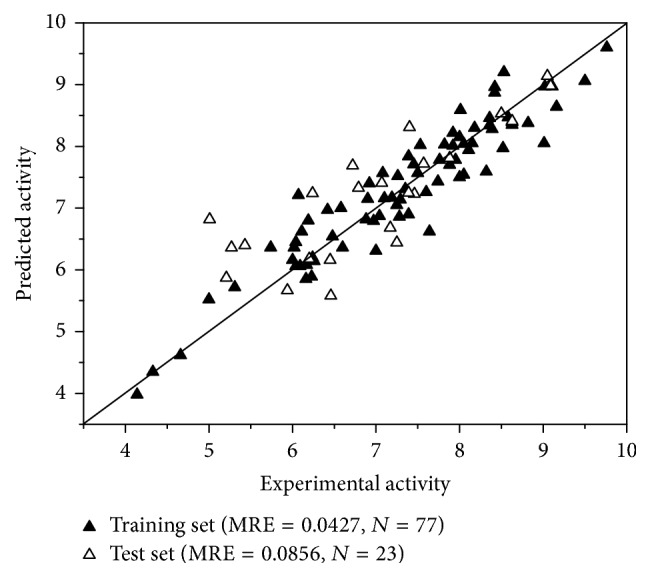
Scatterplot of experimental data versus predicted data from topomer CoMFA model 2.
Additionally, steric and electrostatic contour maps of R1 and R2 groups were obtained. Compound 33 was selected to study how to redesign EGFR inhibitors due to the highly activity (see Figure 3). From Figure 3, it could be concluded that large volume and positively charged groups were added, which can increase compound activity.
Figure 3.

3D contour maps of topomer CoMFA model for R1 and R2 of compound 33. (a) and (c) present steric contour map. (b) and (d) present electrostatic field map. Green, yellow, blue, and red represent large volume, small volume, positively charged, and negatively charged groups, respectively.
3.3. Molecular Docking
Compounds 27, 28, 30, 31, 32, and 33 were used for molecular docking with EGFR. As a result, these compounds have hydrogen bonds at Thr766 and Met769 which were in ATP binding sites (see Figure 4). These compounds interact with EGFR kinase at binding sites and the quinolone ring bound to the hydrophobic pocket of EGFR, instead of the purine ring of ATP.
Figure 4.

The docking result of the EGFR inhibitors with EGFR. (a) The binding site of compound 27 with EGFR is Thr766. (b) The binding site of compound 28 with EGFR is Met769. (c) The binding site of compound 30 with EGFR is Met769. (d) The binding site of compound 31 with EGFR is Met769. (e) The binding site of compound 32 with EGFR is Met769. (f) The binding sites of compound 33 with EGFR is Thr766 and Met769.
4. Discussion
4.1. 2D-QSAR Model
Feature selection via removal of some unnecessary features is required for a precise prediction model [25, 54, 55]. A subset containing nine features was obtained to build the 2D-QSAR prediction model. The prediction accuracy of the model was well for the training set and independent test set. This result indicated that the original data contained some redundant features, and feature selection was a helpful step in building a prediction model.
Although the accuracy of the prediction model with a subset containing nine features (DPLL, H, HF, HOMO, MR, Pc, TIndx, VP, and WIndx) was reliable, it was difficult to analyze the relationship between these descriptors and the activity of EGFR inhibitors as the prediction model is nonlinear. Thus, sensitivity was further applied for this problem [56]. Figure 1(a) shows the relationship between the Dipole length and activity. When the Dipole length is approximately 2 and 6.5, the activity levels are at minimum and maximum, respectively. Figure 1(b) shows the relationship between Henry's law constant and activity. The activity increases along with Henry's law constant from 0 to 30. When Henry's law constant is more than 30, the activity has a rising trend. Figure 1(c) shows the relationship between the Heat of Formation and activity. When the Heat of Formation ranges from −700 to 600, the activity increases. When the Heat of Formation is more than 600, the activity has a rising trend. Figure 1(d) shows the relationship between the HOMO energy and activity. When the HOMO energy ranges from −9.25 to −8.25, the activity increases. When the HOMO energy is approximately −8.25, the activity peaks. When the HOMO energy is greater than −8.25, the activity decreases. When the HOMO energy is more than −7.25, the activity has a decreasing trend. Figure 1(e) shows the relationship between the Molar refractivity and activity. When the Molar refractivity is approximately 10 and 14, the activity levels are at minimum and maximum, respectively. Figure 1(f) shows the relationship between the critical pressure and activity. When the critical pressure ranges from 0 to 60, the activity increases. When the critical pressure is more than 60, the activity has a rising trend. Figure 1(g) shows the relationship between the molecular topological index and activity. When the molecular topological index ranges from 0 to 60,000, the activity decreases. When the molecular topological index is more than 60,000, the activity has a decreasing trend. Figure 1(h) shows the relationship between the Vapor pressure and activity. When the Vapor pressure ranges from 0 to 1.4, the activity decreases. When the Vapor pressure was more than 1.4, the activity had a decreasing trend. Figure 1(i) shows the relationship between the Wiener index and activity. When the Wiener index and activity range from 0 to 9,000, the activity decreases. When the Wiener index is more than 9,000, the activity has a decreasing trend.
4.2. 3D-QSAR Model
Molecules in the topomer CoMFA models can be split into two, three, four, and more groups as needed [51, 57]. In this study, compounds were divided into two groups (R1 and R2). EGFR inhibitors' activity was related to the completeness of the pharmacophore. In topomer CoMFA models, the pharmacophore is related to cutting [44, 48, 58], which plays an important role in the model's predictive performance of the model [58]. In the topomer CoMFA analysis, all molecules of the training set are cut into two fragments. While the fragmentation was complete, the input structures were standardized and the topomers were generated. They all shared the same identical substructure. If the same identical substructure was recognized by the test set, the model's predictive ability was promising.
It could be found that model 2 added an N element in R1 based on model 1, which contributed to the model's predictive ability (see Table 2). Thus, it is speculated that R1 and R2 in model 2 are the same identical substructures. The independent set test was used for evaluating model 2 (see Figure 2). It was observed that the predicted pIC50 of some compounds was poor, such as compound 9 and compound 34 (see Table 3). We guess this is because the same identical substructures of the two compounds (see Figure 5) were different from the other compounds. The poor predicted pIC50 of compounds may cause high MAE. According to Roy et al.'s report [46], the 3D-QSAR model in our study was reliable as the MAE of the external validation was both less than 0.1 × training set range and MAE (training set) + 3 × σ. It is well known that the presence of systematic error in predictions may easily be identified from the difference in mean error and mean absolute error. It is important to analyze prediction errors of compounds in test set in order to search any possible systematic error. In Roy et al.'s study [59], various metrics, including the number of positive prediction errors (NPE), the number of negative prediction errors (NNP), the absolute value for average of prediction errors (AE), the average of absolute prediction errors (AAE), the mean of positive prediction errors (MPE), and the absolute value for mean of negative prediction errors (MNE), were employed to analyze the prediction's error. If prediction error is complied with principles I–V defined by Roy, the results were recommended. In our study, the NPE, NNP, AE, AAE, MPE, and MNE were 12, 11, 0.219, 0.526, 0.713, and −0.321, respectively. ABS (MPE/MNE) and R2 (Y versus residuals) were 2.2 (threshold = 2) and 0.67 (threshold = 0.5), respectively. Hence, it was regarded that our 3D-QSAR model is reliable.
Figure 5.

Structures of compounds 9, 27, 28, 30, 31, 32, 33, and 34.
In addition, topomer CoMFA model provides opinions on modifying EGFR inhibitors in order to design potential highly selective and highly active EGFR inhibitors. Compound 33 (see Figure 5) was chosen to study the effect of R1 and R2 groups on activity due to its high activity. In R1 group, large group with a positive-charge in the yloxyethyl increases the compound's bioactivity (see Figure 3). In R2 group, small groups with a positive-charge in the benzene ring may also increase the compound's bioactivity.
4.3. Molecular Docking Analysis
Molecular docking was applied to predict the interaction sites between compounds and EGFR. As the structure of compound 33 is similar to erlotinib, EGFR also interacts with compound 33 at Thr766 and Met769 [50]. Interestingly, it is observed that the binding modes of compound 33-EGFR and erlotinib-EGFR were different despite the similar structure after calculation. Quinolone ring of erlotinib competitively binds to the hydrophobic pocket of EGFR kinase. For erlotinib, the aniline group reached into the pocket, and substituent groups of site 6 and site 7 were located outside of the hydrophobic pocket. For compound 33, it interacts with the EGFR by substituent groups of site 6 and site 7 in the hydrophobic pocket. In the steric and electrostatic fields, large volume group and positively charged group in site 6 and site 7 of compound 33 may increase inhibitor activity (see Figure 3). Then, the similar chemical series of compound 33 was selected to study the docking site. As a result, compounds 28, 30, 31, and 32 interact with EGFR at Met769, and compound 27 interacts with EGFR at Thr766. Thus, we considered that the Thr766 and Met769 played a crucial role in the EGFR activity.
Many studies performed the QSAR on kinase inhibitors, and the result was helpful for drugs design. In Farghaly et al.'s study [60], QSAR model was built, and the RMSE and r2 were applied to evaluate the model. After calculating, they selected out three predominant descriptors affecting the anticancer activity, and five anticancer agents were screened finally. Sharma showed the 2D-QSAR studies of c-Src tyrosine kinase inhibitors with q2 = 0.755 and r2 = 0.832 [61]. Sharma et al. reported QSAR studies of Aurora A kinase inhibitors [62]. q2 is 0.762 and r2 is 0.806. The difference in the number of samples causes the difference in q2 and r2. When q2 and r2 are more than 0.5 and 0.8, respectively, the model has statistical significance. In our QSAR study, q2 is 0.565 lower than these two studies, but r2 is higher (see Table 4). In addition, steric and electrostatic field and molecular docking analysis were applied in our study to explore the activity development and predict the interaction between inhibitors and protein, which is not showed in these studies. In conclusion, QSAR combined with molecular docking provides better insight into the future design of more potent EGFR inhibitors prior to synthesis.
Table 4.
The comparison of metrics between other studies and ours in QSAR study of the kinase inhibitors.
5. Conclusion
In this study, 2D-QSAR and 3D-QSAR prediction models were built to analyze EGFR inhibitors. Firstly, the 2D-QSAR model was built to predict whether a compound was an inhibitor or a noninhibitor. The accuracy of the 2D-QSAR model using the tenfold cross-validation test and independent set test was 98.99% and 97.67%, respectively. Then, the topomer CoMFA model was built based on EGFR inhibitors. Two models were obtained by cutting different molecular bonds. As a result, model 2 with higher q2 value and r2 values was selected to predict EGFR inhibitors. Finally, a series of similar chemical inhibitors were selected to study the interacting sites between EGFR and EGFR inhibitors using molecular docking tool. As a result, Thr766 and Met769 were received by studying the docking result. Thus, we considered that Thr766 and Met769 played a crucial role in the EGFR activity.
Supplementary Material
The Supplementary Material includes: 1. A training set for building prediction model. 2. A test set for validation.
Acknowledgments
The authors would like to express gratitude towards scholarship from Natural Science Foundation of Shanghai Science and Technology Commission (no. 17ZR1422500), the Shanghai Jiao Tong University Medical Engineering Crossover Fund Project (no. YG2016MS26), the Shanghai University High Performance Computing, Shanghai Municipal Education Commission, the National Natural Science Foundation of China (81271384, 81371623, 31571171, and 31100838), Shanghai Key Laboratory of Bio-Energy Crops (13DZ2272100), the Shanghai Natural Science Foundation (Grant no. 15ZR1414900), the Key Laboratory of Medical Electrophysiology (Southwest Medical University) of Ministry of Education of China (Grant no. 201502), and the Young Teachers of Shanghai Universities Training Program. They also would like to thank Professor Mingyue Zheng from the State Laboratory of Drug Research of Chinese Academy of Science for helping calculate the molecular descriptors.
Conflicts of Interest
The authors declare that there are no conflicts of interest regarding the publication of this paper.
Authors' Contributions
Manman Zhao, Lin Wang, and Linfeng Zheng contributed equally to this work.
References
- 1.Ciardiello F., Tortora G. EGFR antagonists in cancer treatment. The New England Journal of Medicine. 2008;358(11):1160–1174. doi: 10.1056/nejmra0707704. [DOI] [PubMed] [Google Scholar]
- 2.He Y., Harrington B. S., Hooper J. D. New crossroads for potential therapeutic intervention in cancer—intersections between CDCP1, EGFR family members and downstream signaling pathways. Oncoscience. 2016;3(1):5–8. doi: 10.18632/oncoscience.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang R., Wang X., Wu J. Q., et al. Efficient porcine reproductive and respiratory syndrome virus entry in MARC-145 cells requires EGFR-PI3K-AKT-LIMK1-COFILIN signaling pathway. Virus Research. 2016;225:23–32. doi: 10.1016/j.virusres.2016.09.005. [DOI] [PubMed] [Google Scholar]
- 4.Imamura F., Uchida J., Kukita Y., et al. Monitoring of treatment responses and clonal evolution of tumor cells by circulating tumor DNA of heterogeneous mutant EGFR genes in lung cancer. Lung Cancer. 2016;94:68–73. doi: 10.1016/j.lungcan.2016.01.023. [DOI] [PubMed] [Google Scholar]
- 5.Wang K., Li D., Sun L. High levels of EGFR expression in tumor stroma are associated with aggressive clinical features in epithelial ovarian cancer. OncoTargets and Therapy. 2016;9:377–386. doi: 10.2147/OTT.S96309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cho A., Hur J., Moon Y. W., et al. Correlation between EGFR gene mutation, cytologic tumor markers, 18F-FDG uptake in non-small cell lung cancer. BMC Cancer. 2016;16(1, article 224) doi: 10.1186/s12885-016-2251-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holdman X. B., Welte T., Rajapakshe K., et al. Upregulation of EGFR signaling is correlated with tumor stroma remodeling and tumor recurrence in FGFR1-driven breast cancer. Breast Cancer Research. 2015;17(1, article 141) doi: 10.1186/s13058-015-0649-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sarkar C. Epidermal growth factor receptor (EGFR) gene amplification in high grade gliomas. Neurology India. 2016;64(1):27–28. doi: 10.4103/0028-3886.173635. [DOI] [PubMed] [Google Scholar]
- 9.Oda K., Matsuoka Y., Funahashi A., Kitano H. A comprehensive pathway map of epidermal growth factor receptor signaling. Molecular Systems Biology. 2005;1:p. E1. doi: 10.1038/msb4100014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baer J. C., Freeman A. A., Newlands E. S., Watson A. J., Rafferty J. A., Margison G. P. Depletion of O6-alkylguanine-DNA alkyltransferase correlates with potentiation of temozolomide and CCNU toxicity in human tumour cells. British Journal of Cancer. 1993;67(6):1299–1302. doi: 10.1038/bjc.1993.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Minkovsky N., Berezov A. BIBW-2992, a dual receptor tyrosine kinase inhibitor for the treatment of solid tumors. Current Opinion in Investigational Drugs. 2008;9(12):1336–1346. [PubMed] [Google Scholar]
- 12.Dearden J. C. The history and development of quantitative structure-activity relationships (QSARs) International Journal of Quantitative Structure-Property Relationships. 2016;1(1):1–44. [Google Scholar]
- 13.Cherkasov A., Muratov E. N., Fourches D., et al. QSAR modeling: where have you been? Where are you going to? Journal of Medicinal Chemistry. 2014;57(12):4977–5010. doi: 10.1021/jm4004285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roy K., Kar S., Das R. N. Understanding the Basics of QSAR for Applications in Pharmaceutical Sciences and Risk Assessment. Academic Press; 2015. [Google Scholar]
- 15.Gadaleta D., Mangiatordi G. F., Catto M., Carotti A., Nicolotti O. Applicability domain for QSAR models: where theory meets reality. International Journal of Quantitative Structure-Property Relationships. 2016;1(1):45–63. doi: 10.4018/ijqspr.2016010102. [DOI] [Google Scholar]
- 16.Roy K., Kar S., Ambure P. On a simple approach for determining applicability domain of QSAR models. Chemometrics and Intelligent Laboratory Systems. 2015;145:22–29. doi: 10.1016/j.chemolab.2015.04.013. [DOI] [Google Scholar]
- 17.Lee S., Barron M. G. Development of 3D-QSAR model for acetylcholinesterase inhibitors using a combination of fingerprint, molecular docking, and structure-based pharmacophore approaches. Toxicological Sciences. 2015;148(1):60–70. doi: 10.1093/toxsci/kfv160. [DOI] [PubMed] [Google Scholar]
- 18.Tresadern G., Bemporad D. Modeling approaches for ligand-based 3D similarity. Future Medicinal Chemistry. 2010;2(10):1547–1561. doi: 10.4155/fmc.10.244. [DOI] [PubMed] [Google Scholar]
- 19.Chou K.-C. Structural bioinformatics and its impact to biomedical science. Current Medicinal Chemistry. 2004;11(16):2105–2134. doi: 10.2174/0929867043364667. [DOI] [PubMed] [Google Scholar]
- 20.Liang J.-W., Zhang T.-J., Li Z.-J., Chen Z.-X., Yan X.-L., Meng F.-H. Predicting potential antitumor targets of Aconitum alkaloids by molecular docking and protein–ligand interaction fingerprint. Medicinal Chemistry Research. 2016;25(6):1115–1124. doi: 10.1007/s00044-016-1553-7. [DOI] [Google Scholar]
- 21.Blake L., Soliman M. E. S. Identification of irreversible protein splicing inhibitors as potential anti-TB drugs: insight from hybrid non-covalent/covalent docking virtual screening and molecular dynamics simulations. Medicinal Chemistry Research. 2014;23(5):2312–2323. doi: 10.1007/s00044-013-0822-y. [DOI] [Google Scholar]
- 22.Ambure P., Kar S., Roy K. Pharmacophore mapping-based virtual screening followed by molecular docking studies in search of potential acetylcholinesterase inhibitors as anti-Alzheimer's agents. BioSystems. 2014;116(1):10–20. doi: 10.1016/j.biosystems.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 23.Lv J., Su J., Wang F., Qi Y., Liu H., Zhang Y. Detecting novel hypermethylated genes in Breast cancer benefiting from feature selection. Computers in Biology and Medicine. 2010;40(2):159–167. doi: 10.1016/j.compbiomed.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 24.Ajdadi F. R., Gilandeh Y. A., Mollazade K., Hasanzadeh R. P. Application of machine vision for classification of soil aggregate size. Soil and Tillage Research. 2016;162:8–17. doi: 10.1016/j.still.2016.04.012. [DOI] [Google Scholar]
- 25.Sadeghia R., Zarkami R., Sabetraftar K., Van Damme P. Application of genetic algorithm and greedy stepwise to select input variables in classification tree models for the prediction of habitat requirements of Azolla filiculoides (Lam.) in Anzali wetland, Iran. Ecological Modelling. 2013;251:44–53. doi: 10.1016/j.ecolmodel.2012.12.010. [DOI] [Google Scholar]
- 26.Burges C. J. C. A tutorial on support vector machines for pattern recognition. Data Mining and Knowledge Discovery. 1998;2(2):121–167. doi: 10.1023/A:1009715923555. [DOI] [Google Scholar]
- 27.Imani F., Boada F. E., Lieberman F. S., Davis D. K., Mountz J. M. Molecular and metabolic pattern classification for detection of brain glioma progression. European Journal of Radiology. 2014;83(2):e100–e105. doi: 10.1016/j.ejrad.2013.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Emblem K. E., Zoellner F. G., Tennoe B., et al. Predictive modeling in glioma grading from MR perfusion images using support vector machines. Magnetic Resonance in Medicine. 2008;60(4):945–952. doi: 10.1002/mrm.21736. [DOI] [PubMed] [Google Scholar]
- 29.Cramer R. D. Topomer CoMFA: a design methodology for rapid lead optimization. Journal of Medicinal Chemistry. 2003;46(3):374–388. doi: 10.1021/jm020194o. [DOI] [PubMed] [Google Scholar]
- 30.Myint K. Z., Xie X.-Q. Recent advances in fragment-based QSAR and multi-dimensional QSAR methods. International Journal of Molecular Sciences. 2010;11(10):3846–3866. doi: 10.3390/ijms11103846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gadhe C. G. CoMFA vs. Topomer CoMFA, which one is better a case study with 5-lipoxygenase inhibitors. Journal of the Chosun Natural Science. 2011;4(2):91–98. [Google Scholar]
- 32.Rewcastle G. W., Denny W. A., Bridges A. J., et al. Tyrosine kinase inhibitors. 5. Synthesis and structure-activity relationships for 4-[(phenylmethyl)amino]- and 4-(phenylamino)quinazolines as potent adenosine 5′-triphosphate binding site inhibitors of the tyrosine kinase domain of the epidermal growth factor receptor. Journal of Medicinal Chemistry. 1995;38(18):3482–3487. doi: 10.1021/jm00018a008. [DOI] [PubMed] [Google Scholar]
- 33.Thompson A. M., Bridges A. J., Fry D. W., Kraker A. J., Denny W. A. Tyrosine kinase inhibitors. 7. 7-Amino-4-(phenylamino)- and 7-amino-4-[(phenylmethyl)amino]pyrido[4,3-d]pyrimidines: a new class of inhibitors of the tyrosine kinase activity of the epidermal growth factor receptor. Journal of Medicinal Chemistry. 1995;38(19):3780–3788. doi: 10.1021/jm00019a007. [DOI] [PubMed] [Google Scholar]
- 34.Rewcastle G. W., Bridges A. J., Fry D. W., Rubin J. R., Denny W. A. Tyrosine kinase inhibitors. 12. Synthesis and structure-activity relationships for 6-substituted 4-(phenylamino)pyrimido[5,4-d]pyrimidines designed as inhibitors of the epidermal growth factor receptor. Journal of Medicinal Chemistry. 1997;40(12):1820–1826. doi: 10.1021/jm960879m. [DOI] [PubMed] [Google Scholar]
- 35.Thompson A. M., Murray D. K., Elliott W. L., et al. Tyrosine kinase inhibitors. 13. Structure—activity relationships for soluble 7-substituted 4-[(3-bromophenyl)amino]pyrido[4,3-d]pyrimidines designed as inhibitors of the tyrosine kinase activity of the epidermal growth factor receptor. Journal of Medicinal Chemistry. 1997;40(24):3915–3925. doi: 10.1021/jm970366v. [DOI] [PubMed] [Google Scholar]
- 36.Bridges A. J., Zhou H., Cody D. R., et al. Tyrosine kinase inhibitors. 8. An unusually steep structure-activity relationship for analogues of 4-(3-bromoanilino)-6,7-dimethoxyquinazoline (PD 153035), a potent inhibitor of the epidermal growth factor receptor. Journal of Medicinal Chemistry. 1996;39(1):267–276. doi: 10.1021/jm9503613. [DOI] [PubMed] [Google Scholar]
- 37.Rewcastle G. W., Palmer B. D., Thompson A. M., et al. Tyrosine kinase inhibitors. 10. Isomeric 4-[(3-bromophenyl)amino]pyrido[d]-pyrimidines are potent ATP binding site inhibitors of the tyrosine kinase function of the epidermal growth factor receptor. Journal of Medicinal Chemistry. 1996;39(9):1823–1835. doi: 10.1021/jm9508651. [DOI] [PubMed] [Google Scholar]
- 38.Li S., Guo C., Zhao H., Tang Y., Lan M. Synthesis and biological evaluation of 4-[3-chloro-4-(3-fluorobenzyloxy) anilino]-6-(3-substituted-phenoxy)pyrimidines as dual EGFR/ErbB-2 kinase inhibitors. Bioorganic and Medicinal Chemistry. 2012;20(2):877–885. doi: 10.1016/j.bmc.2011.11.056. [DOI] [PubMed] [Google Scholar]
- 39.Waterson A. G., Petrov K. G., Hornberger K. R., et al. Synthesis and evaluation of aniline headgroups for alkynyl thienopyrimidine dual EGFR/ErbB-2 kinase inhibitors. Bioorganic and Medicinal Chemistry Letters. 2009;19(5):1332–1336. doi: 10.1016/j.bmcl.2009.01.080. [DOI] [PubMed] [Google Scholar]
- 40.Suzuki N., Shiota T., Watanabe F., et al. Synthesis and evaluation of novel pyrimidine-based dual EGFR/Her-2 inhibitors. Bioorganic and Medicinal Chemistry Letters. 2011;21(6):1601–1606. doi: 10.1016/j.bmcl.2011.01.119. [DOI] [PubMed] [Google Scholar]
- 41.Suzuki N., Shiota T., Watanabe F., et al. Discovery of novel 5-alkynyl-4-anilinopyrimidines as potent, orally active dual inhibitors of EGFR and Her-2 tyrosine kinases. Bioorganic and Medicinal Chemistry Letters. 2012;22(1):456–460. doi: 10.1016/j.bmcl.2011.10.103. [DOI] [PubMed] [Google Scholar]
- 42.Irwin J. J. Software review: ChemOffice 2005 Pro by Cambridgesoft. Journal of Chemical Information and Modeling. 2005;45(5):1468–1469. doi: 10.1021/ci050286x. [DOI] [Google Scholar]
- 43.Varma S., Simon R. Bias in error estimation when using cross-validation for model selection. BMC Bioinformatics. 2006;7(supplement 5):91–98. doi: 10.1186/1471-2105-7-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang L., Shen H., Li B., Hu D. Classification of schizophrenic patients and healthy controls using multiple spatially independent components of structural MRI data. Frontiers of Electrical and Electronic Engineering in China. 2011;6(2):353–362. doi: 10.1007/s11460-011-0142-2. [DOI] [Google Scholar]
- 45.Zhang Y. P., Sussman N., Klopman G., Rosenkranz H. S. Development of methods to ascertain the predictivity and consistency of SAR models: application to the U.S. National toxicology program rodent carcinogenicity bioassays. Quantitative Structure-Activity Relationships. 1997;16(4):290–295. doi: 10.1002/qsar.19970160403. [DOI] [Google Scholar]
- 46.Roy K., Das R. N., Ambure P., Aher R. B. Be aware of error measures. Further studies on validation of predictive QSAR models. Chemometrics and Intelligent Laboratory Systems. 2016;152:18–33. doi: 10.1016/j.chemolab.2016.01.008. [DOI] [Google Scholar]
- 47.Yu S., Yuan J., Shi J., et al. HQSAR and topomer CoMFA for predicting melanocortin-4 receptor binding affinities of trans-4-(4-chlorophenyl) pyrrolidine-3-carboxamides. Chemometrics and Intelligent Laboratory Systems. 2015;146:34–41. doi: 10.1016/j.chemolab.2015.04.017. [DOI] [Google Scholar]
- 48.Kumar S., Tiwari M. Topomer-CoMFA-based predictive modelling on 2,3-diaryl-substituted-1,3-thiazolidin-4-ones as non-nucleoside reverse transcriptase inhibitors. Medicinal Chemistry Research. 2015;24(1):245–257. doi: 10.1007/s00044-014-1105-y. [DOI] [Google Scholar]
- 49.Tian Y., Shen Y., Zhang X., et al. Design some new type-I c-met inhibitors based on molecular docking and topomer comfa research. Molecular Informatics. 2014;33(8):536–543. doi: 10.1002/minf.201300118. [DOI] [PubMed] [Google Scholar]
- 50.Tresadern G., Cid J.-M., Trabanco A. A. QSAR design of triazolopyridine mGlu2 receptor positive allosteric modulators. Journal of Molecular Graphics and Modelling. 2014;53:82–91. doi: 10.1016/j.jmgm.2014.07.006. [DOI] [PubMed] [Google Scholar]
- 51.Tang H., Yang L., Li J., Chen J. Molecular modelling studies of 3,5-dipyridyl-1,2,4-triazole derivatives as xanthine oxidoreductase inhibitors using 3D-QSAR, Topomer CoMFA, molecular docking and molecular dynamic simulations. Journal of the Taiwan Institute of Chemical Engineers. 2016;68:64–73. doi: 10.1016/j.jtice.2016.09.018. [DOI] [Google Scholar]
- 52.Joshi S. D., More U. A., Koli D., Kulkarni M. S., Nadagouda M. N., Aminabhavi T. M. Synthesis, evaluation and in silico molecular modeling of pyrroyl-1,3,4-thiadiazole inhibitors of InhA. Bioorganic Chemistry. 2015;59:151–167. doi: 10.1016/j.bioorg.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 53.Stamos J., Sliwkowski M. X., Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. Journal of Biological Chemistry. 2002;277(48):46265–46272. doi: 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- 54.Miao J., Niu L. A survey on feature selection. Procedia Computer Science. 2016;91:919–926. doi: 10.1016/j.procs.2016.07.111. [DOI] [Google Scholar]
- 55.Bolón-Canedo V., Sánchez-Maroño N., Alonso-Betanzos A. Feature selection for high-dimensional data. Progress in Artificial Intelligence. 2016;5(2):65–75. doi: 10.1007/s13748-015-0080-y. [DOI] [Google Scholar]
- 56.Niu B., Su Q., Yuan X., Lu W., Ding J. QSAR study on 5-lipoxygenase inhibitors based on support vector machine. Medicinal Chemistry. 2012;8(6):1108–1116. doi: 10.2174/1573406411208061108. [DOI] [PubMed] [Google Scholar]
- 57.Ding W., Sun M., Luo S., et al. A 3D QSAR study of betulinic acid derivatives as anti-tumor agents using topomer CoMFA: model building studies and experimental verification. Molecules. 2013;18(9):10228–10241. doi: 10.3390/molecules180910228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xiang Y., Song J., Zhang Z. Topomer CoMFA and virtual screening studies of azaindole class renin inhibitors. Combinatorial Chemistry and High Throughput Screening. 2014;17(5):458–472. doi: 10.2174/1386207317666140107094708. [DOI] [PubMed] [Google Scholar]
- 59.Roy K., Ambure P., Aher R. B. How important is to detect systematic error in predictions and understand statistical applicability domain of QSAR models? Chemometrics and Intelligent Laboratory Systems. 2017;162:44–54. doi: 10.1016/j.chemolab.2017.01.010. [DOI] [Google Scholar]
- 60.Farghaly T. A., Hassaneen H. M. E., Elzahabi H. S. A. Eco-friendly synthesis and 2D-QSAR study of novel pyrazolines as potential anticolon cancer agents. Medicinal Chemistry Research. 2015;24(2):652–668. doi: 10.1007/s00044-014-1175-x. [DOI] [Google Scholar]
- 61.Sharma M. C. 2D QSAR studies of the inhibitory activity of a series of substituted purine derivatives against c-Src tyrosine kinase. Journal of Taibah University for Science. 2016;10(4):563–570. doi: 10.1016/j.jtusci.2015.11.002. [DOI] [Google Scholar]
- 62.Sharma M. C., Sharma S., Bhadoriya K. QSAR studies on pyrazole-4-carboxamide derivatives as Aurora A kinase inhibitors. Journal of Taibah University for Science. 2016;10(1):107–114. doi: 10.1016/j.jtusci.2015.06.003. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The Supplementary Material includes: 1. A training set for building prediction model. 2. A test set for validation.