

Abstract
Age-related changes in the niche have long been postulated to impair the function of somatic stem cells. Here we demonstrate that the aged stem cell niche in skeletal muscle contains substantially reduced levels of fibronectin (FN), leading to detrimental consequences for the function and maintenance of muscle stem cells (MuSCs). Deletion of the gene encoding FN from young regenerating muscles replicates the aging phenotype and leads to a loss of MuSC numbers. By using an extracellular matrix (ECM) library screen and pathway profiling, we characterize FN as a preferred adhesion substrate for MuSCs and demonstrate that integrin-mediated signaling through focal adhesion kinase and the p38 mitogen-activated protein kinase pathway is strongly de-regulated in MuSCs from aged mice because of insufficient attachment to the niche. Reconstitution of FN levels in the aged niche remobilizes stem cells and restores youth-like muscle regeneration. Taken together, we identify the loss of stem cell adhesion to FN in the niche ECM as a previously unknown aging mechanism.
Extrinsic signals that originate in the immediate cellular environment, commonly known as the stem cell niche, are critical for the regulation of MuSCs1. Following injury, the stem cell niche in muscle is subject to a coordinated flux of various cell types that interact directly with MuSCs or that release regulatory growth factors and ECM. These niche interactions regulate the activation, self-renewal, differentiation and return to quiescence of MuSCs. Recent work has revealed a fundamental role of structural elements in the niche. Tissue stiffness, which is largely dependent on the composition of the ECM, is a critical fate determinant for MuSCs2–4. Moreover, the ECM molecules collagen VI and FN have been shown to provide signals that are essential for MuSC self-renewal during the regeneration of adult muscle5–7.
The MuSC niche can be severely perturbed by chronic degenerative diseases of skeletal muscle that are accompanied by aberrant deposition of ECM and altered support cell dynamics8. De-regulated niche signals eventually lead to stem cell dysfunction and inefficient tissue repair. Of note, a number of multisystemic conditions—such as aging, diabetes, obesity and cancer cachexia—are also accompanied by a loss of MuSC function and consequently by a decline of the regenerative capacity of skeletal muscle tissue9–12. In the elderly, this problem is also paralleled by a loss of MuSC numbers, resulting in dramatically delayed or incomplete healing of muscle following injury or surgery13–15. Impaired musculoskeletal recovery leads to prolonged immobility that in turn exacerbates the loss of muscle mass that often accompanies aging. Thus, inefficient muscle healing in the elderly is a major clinical problem, and therapeutic approaches for restoring MuSC function are needed.
It remains controversial whether intrinsic or extrinsic signals are the causative mediators of MuSC aging16. Changes in the niche may lead to long-lasting or irreversible cellular effects that could ultimately be interpreted as intrinsic MuSC aging. Notably, several studies have shown that a number of pathways are constitutively activated in aged MuSCs. This includes the p38 mitogen-activated protein (MAP) kinase and fibroblast growth factor (FGF)–ERK MAP kinase cascades, as well as signaling through the Janus kinase (JAK)–STAT transcription factor pathway17–21. Reduction of signaling through these pathways by using pharmaceutical inhibitors can restore MuSC self-renewal and promote muscle healing in aged mice. These observations raise the question of whether changes in the stem cell niche lead to an upstream induction of these signaling cascades.
Here we describe that loss of FN from the aged-niche ECM in regenerating muscles impairs MuSC function by affecting integrin signaling through PTK2 protein tyrosine kinase 2 (PTK2; also known as FAK) and MAP kinase pathways. Restoration of FN levels in muscle from old mice (aged muscle) rescues MuSC function and improves muscle healing. Thus, loss of stem cell adhesion to niche-derived FN is a root cause for MuSC aging that can be targeted to restore the regenerative capacity of muscle tissue in the elderly.
Results
Loss of fibronectin from the aged niche
To interrogate the effect of age-induced changes on MuSCs in the stem cell niche, we performed microarray profiling on freshly isolated cells from 9- to 10-week-old young animals and 20-month-old aged animals 3 d following muscle injury. The viability between young and aged cell populations using our flow cytometry isolation protocol was comparable (Supplementary Fig. 1a,b). As previously reported, we observed significant enrichment of components of the JAK–STAT and MAP kinase pathways in aged MuSCs, as compared to those in young cells, whereas expression of genes involved in cell cycle regulation was lower in aged cells17–22 (Fig. 1a–c and Supplementary Table 1). Notably, we also found that expression of components of the ECM– receptor pathway, including integrins and syndecans, was de-regulated in aged MuSCs, as compared to that in the young MuSCs (Fig. 1d and Supplementary Table 1). This observation suggested that the ECM composition of the niche is affected by aging. To test this hypothesis we used a spectrum of ECM-protein-specific slow-off-rate-modified aptamers23 on homogenates from uninjured muscles (uninj.) and from muscles that were collected 3, 7 and 14 d post injury (d.p.i.) (Fig. 1e). Consistent with increased baseline fibrosis in aged muscles8, the majority of ECM molecules were present at higher levels in the uninjured aged mice than in the young control mice. Notably, as compared to muscles from young mice (young muscle), aged muscle showed an impaired ability to globally upregulate ECM molecules over a 14-d regeneration time course following injury (Fig. 1e,f). As previously reported5, FN levels were strongly increased following injury in young muscles and, in contrast to the other ECM components, declined sharply at 14 d. Moreover, we observed that old regenerating muscles contained only 48% of FN protein, as compared to that in young muscles, at 3 d.p.i. and 43% of FN at 7 d.p.i. (Fig. 1g). Quantitative PCR (qPCR) analysis revealed lower levels of Fn1 mRNA at 3 d.p.i. but not at 7 d.p.i. in aged muscles, as compared to that in young muscles (Supplementary Fig. 1c), suggesting a substantial delay between Fn1 gene expression and the accumulation of protein. In summary, young regenerating muscle shows a sharp peak in FN levels following injury, whereas this response is blunted in aged muscle.
Figure 1.
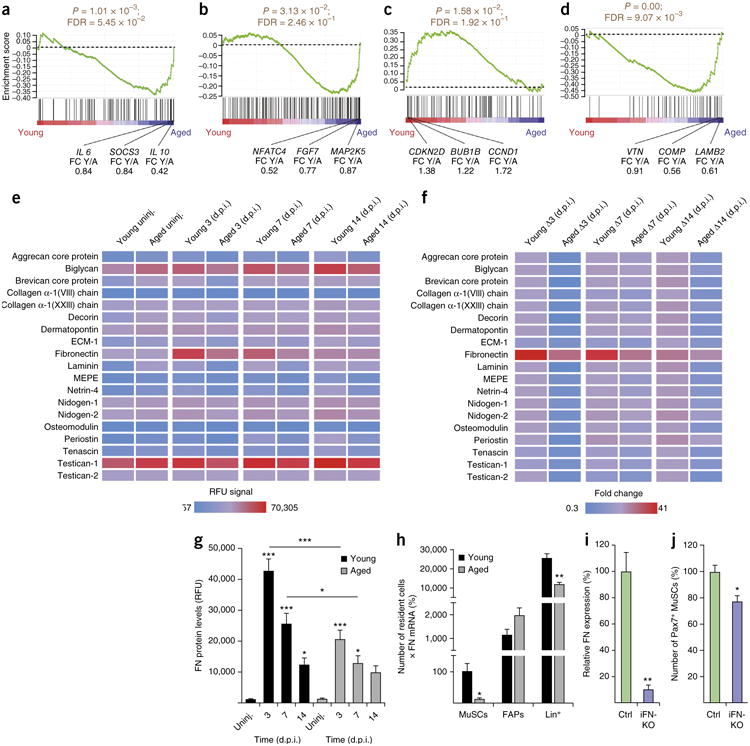
Aging affects FN levels during skeletal muscle regeneration. (a–d) Gene set enrichment analysis for KEGG-derived JAK–STAT (a), MAPK (b), cell cycle (c) and ECM–receptor interaction (d) pathways of freshly isolated young versus aged MuSCs at 3 d.p.i. Nominal P values and false discovery rate (FDR) Q values are reported. FC Y/A, fold change young/aged. (e) Detection of ECM proteins in young and aged muscles under uninjured (uninj.) conditions or at 3, 7 and 14 d.p.i. RFU, relative fluorescence units; ECM-1, extracellular matrix protein 1; MEPE, matrix extracellular phosphoglycoprotein. Data represent means. (f) Changes (Δ) of the RFU signal shown in e relative to the uninjured condition in young and aged muscles. Data represent means. (g) RFU signal for FN in young and aged muscles. (h) FN expression by different cell populations in young and aged muscles at 3 d.p.i. (i) qPCR for FN expression in muscles of control (Ctrl) or iFN-KO mice at 5 d.p.i. (j) Number of Pax7+ MuSCs per unit area in control and iFN–KO mice at 5 d.p.i. Unless otherwise specified, data are means + s.e.m. In a–d, n = 6 mice for young and n = 5 for aged. In e–g, n = 8 mice for young, aged uninj. and aged at 3 d.p.i.; n = 5 for aged at 7 d.p.i.; and n = 7 for aged at 14 d.p.i. In h, n = 5 mice for young and n = 6 for aged. In i,j, n = 3 mice per group for Ctrl and iFN–KO. ***P < 0.001, **P < 0.01, *P < 0.05; by one-way analysis of variance (ANOVA) followed by Bonferroni post hoc test (g) or by Student's t-test (h–j).
To determine in which muscle-resident cell populations the expression of FN is affected by aging, we isolated MuSCs, fibro-adipogenic progenitors (FAPs) and lineage-positive (Lin+) cells (including immune, hematopoietic and endothelial cells) from muscles at 3 d.p.i. using flow cytometry. Of note, we observed 45% lower numbers of Lin+ cells in aged muscles than in young muscle controls (Supplementary Fig. 1d). qPCR analysis revealed a 77% reduction of Fn1 mRNA expression in aged MuSCs and a trend toward lower levels in aged Lin+ cells, as compared to those in young cells (Supplementary Fig. 1e). To determine the overall contribution of the different cells populations to FN expression in young and aged muscles, we adjusted Fn1 mRNA expression by cell abundance (Fig. 1h). This revealed that the contribution of Lin+ cells to FN expression was orders of magnitude higher than that for all of the other cell types. Notably, aging lowered these levels by 55%. These observations demonstrate that Lin+ cells are major contributors to FN expression and that they are lost from aged regenerating muscles.
To directly address the effect of loss of FN from the niche, we generated FN-knockout (KO) mice by crossing animals carrying a tamoxifen-inducible Cre allele under the ROSA26 promoter to animals with a loxP-flanked (‘floxed’) Fn1 allele24,25 (which we hereafter refer to as iFN-KO mice). 4 weeks after the tamoxifen injection, the iFN-KO and control (Ctrl) mice were injured. 5 d later the muscles were collected, and Fn1 mRNA levels were assessed by qPCR (Fig. 1i). Fn1 transcripts were found to be 89.9% lower in iFN-KO mice than in control mice. Immunostaining tissue sections revealed a strong reduction in FN protein in iFN-KO mice as compared to that in control mice at 5 d.p.i. (Supplementary Fig. 2). Notably, the lower FN concentration in iFN-KO mice was sufficient to phenocopy the age-related loss of MuSCs and was associated with a significantly lower number of paired box 7 (Pax7)-positive cells (to 77% of that measured in muscles from control mice) (Fig. 1j). Thus, FN is essential for the maintenance of MuSCs during muscle healing.
Fibronectin is a preferred adhesion substrate for MuSCs
MuSCs can adhere to FN through integrins and the syndecan-4– frizzled-7 (Sdc4–Fzd7) co-receptor complex5,26. This led us to investigate how FN compares to other ECM molecules with respect to cell adhesion. For this purpose we used an array containing 36 different ECM conditions spotted onto a layer hydrogel (Fig. 2a)27,28. Primary mouse-MuSC-derived myoblasts were seeded on these arrays, and the percentage of cells adhering to the different ECMs was determined after 3 h, 6 h and 24 h. Myoblasts could adhere most efficiently on spots that contained either FN or FN in combination with other ECMs at all of the time points tested (Fig. 2b). The best conditions at 3 h and 6 h were spots with FN and laminin or with FN and collagen I at a 1:1 ratio. At the 24 h mark, myoblasts were able to adhere more broadly over the array, but FN-containing spots were still preferred.
Figure 2.
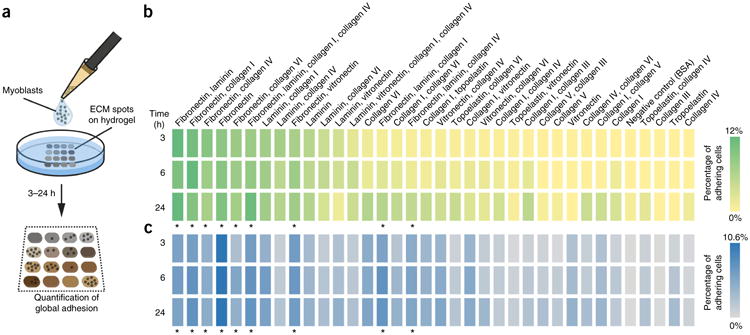
Fibronectin is a preferred adhesion substrate for mouse and human muscle progenitors. (a) Graphical overview of the experiment. (b,c) Quantification of the adhesion of mouse MuSC-derived myoblasts (b) and human myoblasts (c) on ECM hydrogel arrays 3 h, 6 h and 24 h after seeding. The total number of cells adhering to the array was set to 100%. Shades of green or blue color indicate changes in adherence. Asterisks indicate conditions containing FN. Values are means from n = 3 ECM hydrogel arrays per time point from independent experiments.
With the best ECM-protein-binding condition being FN alone, human myoblasts also showed a pronounced preference for adhesion to ECM components that were mixed with FN at all of the time points tested (Fig. 2c). Taken together, these results demonstrate that FN is a preferred binding substrate for mouse and human muscle progenitors and that its loss from the aged stem cell niche could result in a de-regulation of signaling pathways that are modulated by cell adhesion.
Adhesion to fibronectin modulates MuSC aging pathways
To get a comprehensive understanding of the signaling pathways that are affected by adhesion of MuSCs to FN, we undertook a phosphorylation-profiling approach. To this end, we used an antibody array consisting of 1,318 site-specific antibodies covering a wide spectrum of signaling molecules. Mouse MuSC-derived myoblasts were grown on FN for 72 h and compared to cells grown on collagen I (Col), an excellent adhesion substrate that is commonly used in primary myoblast culture. Following background subtraction, phosphospecific signals were normalized to the signals of the antibodies that were raised against the unphosphorylated antigen. A total of 64 proteins were found to be more than 10% changed in their phosphorylation levels after comparing myoblasts that were grown on FN to those grown on Col (Supplementary Table 2). Notably, among the factors affected by growth on FN were the cell matrix receptor β1-integrin and several components involved in the ERK and p38 mitogen-activated protein kinase (MAPK) and cell cycle regulatory pathways, which are also altered in MuSCs as a consequence of aging17,20,21 (Fig. 1b,c). Other factors whose phosphorylation levels was altered in response to growth on FN mapped to classes such as cytokine– cytokine receptor, phosphatidylinositol, Wnt, regulation of actin cytoskeleton, phosphatidylinositol 3-kinase (PI3K)–protein kinase B (Akt) pathway and cell cycle pathway.
To further verify that FN modulates the p38 and ERK MAPK aging pathways, we performed western blot analysis using extracts from myoblasts that were grown on Col, FN or laminin (LAM) for 72 h (Fig. 3a and Supplementary Fig. 3). Phosphorylation and total levels of p38 were lower in cells that were grown on FN than on Col or LAM. Growth on LAM led to increased ERK phosphorylation and total ERK levels. In contrast, total levels of ERK were reduced by growth on FN. These experiments demonstrate that sustained exposure to FN is able to curb signaling through two of the major aging-related pathways in MuSCs.
Figure 3.
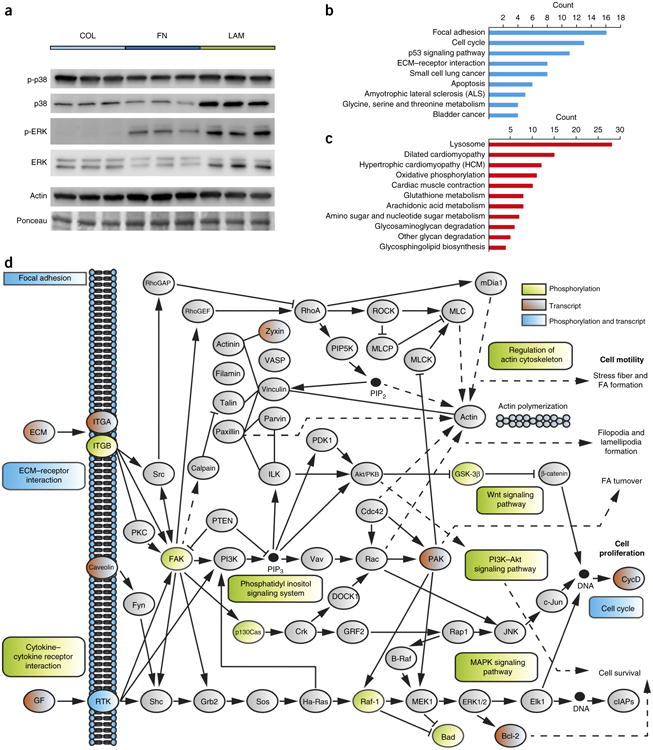
MuSC aging pathways are modulated by fibronectin. (a) Representative western blot analysis for the ERK and p38 MAP kinases from myoblasts grown on collagen I (COL), FN or laminin (LAM) for 72 h. Actin and a Ponceau-stained dominant band at the same molecular weight are shown as loading controls. (b,c) Gene Ontology analysis of statistically differentially regulated genes in microarray data from cells grown on FN versus those grown on Col for 72 h (n = 3 microarrays per condition). Graphs represent counts of up- (b) or downregulated (c) genes that are annotated to the indicated terms. (d) Mapping of proteins and genes that are differentially affected by FN to the KEGG adhesion–signaling system and its associated pathways. Light green indicates factors that are affected by FN at the proteomic level (phosphorylation), red denotes factors whose expression is changed at the transcript level, and blue indicates changes to factors at both levels.
Adhesion signaling is known to coordinate the activity of a number of transcription factors29. Because we observed strong changes in several signaling pathways after MuSCs were grown on FN for sustained periods of time, we decided to interrogate their transcriptional signature in comparison to that of cells grown on Col, using micro-array analysis. Genes with a log2(fold change) ≥0.5 and an adjusted P value of 0.05 (Fig. 3b,c and Supplementary Table 3) were tested for enrichment by using the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database. The highest numbers of genes that were upregulated by FN were found in pathways related to focal adhesion, cell cycle, p53 signaling, ECM–receptor interaction and small-cell lung cancer, whereas the highest numbers of genes downregulated by FN were associated with the lysosome pathway. Taken together, our data suggests that growth of cells on FN alters the activity of a number of signaling systems involved in cell adhesion and cell–matrix interaction on the proteomic and transcriptomic levels (Fig. 3d). These pathways involve integrin-mediated adhesion signaling, the p38 and ERK MAPKs and cell cycle regulatory pathways, which have all previously been implicated in MuSC aging17–22.
Lower adhesive capacity of aged MuSCs
Given the effects of FN on the adhesion signaling and aging pathways, we speculated that its prolonged loss from the aged regenerative niche might alter the adherence and survival capacity of MuSCs. To address this hypothesis we used flow cytometry to sort young and aged MuSCs, which we then plated on Col for 3 h, 6 h and 36 h directly after isolation. At the 3 h and 6 h time points, young and aged MuSCs were able to adhere equally well to the plates (Fig. 4a). After 36 h, however, the number of attached cells was reduced by 56% for the aged cells as compared to that for the young cells. To assess whether the mechanism leading to this difference involves anchorage-dependent programmed cell death (anoikis), we analyzed the cells by terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) (Fig. 4b). This experiment revealed 40% higher numbers of TUNEL+ aged cells at 36 h than young cells. Therefore, owing to their reduced adherence capacity, aged cells are more prone to anoikis.
Figure 4.
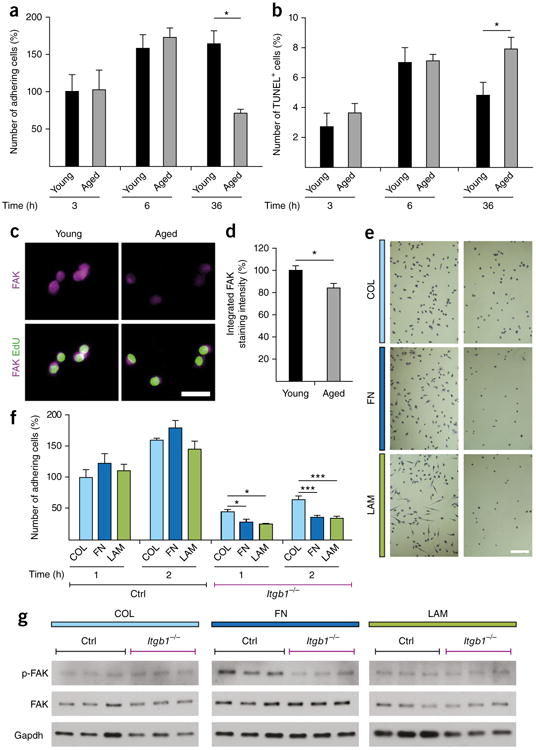
Impaired FN-mediated adhesion signaling in aged MuSCs. (a) Adhesion of freshly isolated young and aged MuSCs 3 h, 6 h and 36 h after seeding on Col. (b) Quantification of freshly isolated TUNEL+ apoptotic young and aged MuSCs at 3 h, 6 h and 36 h following adherence. (c,d) Representative images (c) and quantification (d) of freshly isolated MuSCs from young and aged mice that were seeded on Col and were subsequently stained for FAK. Scale bar, 25 μm. (e) Representative images of crystal violet–stained β1-integrin-knockout (Itgb1−/−) (right) and wild-type (Ctrl) (left) cells that were grown on Col (top), FN (middle) and LAM (bottom) 2 h after plating. Scale bar, 20 μm. (f) Adhesion capacity of Itgb1−/− and Ctrl cells on Col, FN and LAM at 1 h and 2 h following plating. (g) Representative western blots for FAK and phospho-FAK (pFAK) from Itgb1−/− and Ctrl cells that were grown for 72 h on Col, FN or LAM. Gapdh is shown as a loading control. Throughout, data are means + s.e.m. In a,b,d, n = 3 mice per group. In e,f, n = 4 independent cell culture replicates with n = 1 low-magnification image recorded per condition. In g, n = 3 myoblast lysates per condition, each from a different mouse. ***P < 0.001, *P < 0.05; by two-way ANOVA followed by Bonferroni post hoc test (a,b,f) or by Student's t-test (d).
The cytoplasmic nonreceptor tyrosine kinase FAK is a central hub in the adhesion signaling pathway that is activated by FN (Fig. 3d) and is a suppressor of anoikis30. To investigate whether FAK levels are affected by the aging process, we compared freshly isolated young and aged MuSCs by immunostaining. This revealed that aging leads to a downregulation of FAK expression (Fig. 4c,d). Thus, loss of FAK could be the reason for the reduced adhesive capacity and the increased amount of anoikis in aged MuSCs.
FN has been shown to activate FAK through integrins29. This indicates that a loss of integrin signaling would recapitulate aspects of the MuSC aging phenotype, including impaired adhesion and FAK signaling. Indeed, comparable to aged cells, myoblasts isolated from β1-integrin-knockout (Itgb1−/−) mice31 were severely defective in their adherence capacity as compared to those isolated from control mice (Fig. 4e,f). Of note, Itgb1−/− myoblasts were able to adhere better to Col than to FN or LAM. This indicates that ECM receptors other than β1-chain-containing integrin dimers are involved in the adhesion to Col. To determine whether β1-integrin (ITGB1) is required for FN-mediated FAK activation, we performed western blots from cells that were grown for 72 h on Col, FN or LAM. As expected, as compared to control cells, activating FAK phosphorylation by growth on FN was strongly perturbed in the Itgb1−/− cells (Fig. 4g and Supplementary Fig. 4). No differential FAK phosphorylation could be observed in cells that were grown on Col or Lam. These observations show that, similarly to aged cells, Itgb1−/− cells show impaired adhesive capacity and perturbed FAK signaling after exposure to FN.
Fibronectin rescues the adhesion capacity of aged MuSCs
Our data indicate that FN is a preferred adhesion substrate for MuSCs and that the concentration of this ECM component is decreased in the aged regenerative niche, leading to impaired FAK signaling and increased anoikis. Therefore, we decided to test whether aged MuSCs would show better attachment to FN, as compared to attachment in the presence of abundant adhesion substrates such as Col and LAM. Indeed, FN significantly improved the attachment of freshly isolated aged MuSCs at all of the time points following plating (Fig. 5a). Young MuSCs showed a similar preference for FN over Col or LAM (Supplementary Fig. 5). Growth on FN also reduced the num ber of TUNEL+ aged cells (Fig. 5b) and led to a minor but significantly higher number of proliferating cells that incorporated EdU (Fig. 5c). These data indicate that FN can overcome several age-related defects of MuSCs, including their impaired adhesion capacity, propensity for anoikis and reduced proliferative capacity.
Figure 5.
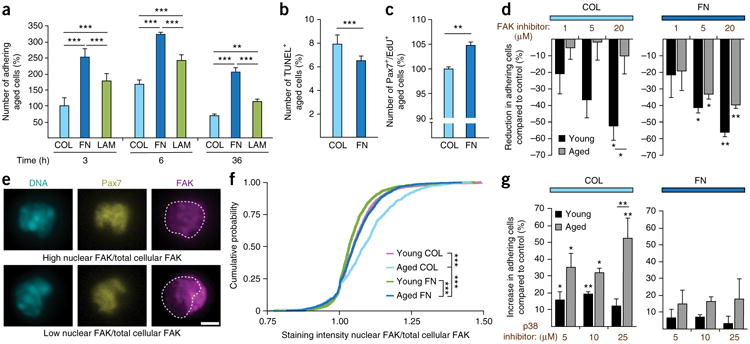
Exposure to FN rescues adhesion signaling in aged MSCs. (a) Adhesion of freshly isolated aged MuSCs on Col, FN and LAM at 3 h, 6 h and 36 h after seeding. (b) Quantification of freshly isolated TUNEL+ aged MuSCs 36 h after isolation and plating on Col or FN. (c) Proliferation of freshly isolated aged MuSCs grown on Col or FN for 96 h. (d) Percentage reduction in numbers of MuSCs that were seeded on Col (left) or FN (right) for 36 h and then exposed to a FAK inhibitor or to vehicle (control). (e) Representative images of MuSCs showing differential FAK subcellular localization after growth on Col (top) or FN (bottom). Scale bar, 2.5 μm. (f) Cumulative probability of the ratio of nuclear FAK over total FAK in young and aged MuSCs that were plated on Col or FN for 6 h. Empirical cumulative distribution functions were built on the basis of n = 3 mice per condition. Kolmogorov– Smirnov distance (D) in young MuSCs on FN (young FN) versus aged MuSCs on FN (aged FN), aged FN versus aged MuSCS on Col (aged COL), and young MuSCs on Col (young COL) versus aged COL are 0.13, 0.12 and 0.21, respectively, and ***P values are 1.05 × 10−6, 5.17 × 10−6 and 1.55 × 10−14, respectively. (g) Percentage increase in numbers of adhering MuSCs seeded on either Col or FN for 36 h and exposed to either the p38 inhibitor or vehicle. Throughout, bars represent means + s.e.m. For all experiments, unless otherwise noted, n = 3 mice. In e,f, n ≥ 60 cells were analyzed per mouse, n = 3 mice per condition. Unless otherwise noted, ***P < 0.001, **P < 0.01, *P < 0.05 versus vehicle-treated cells or as indicated; by two-way ANOVA followed by Bonferroni post hoc test (a,d,g) or by Student's t-test (b,c).
To get further insights into the down-stream mechanisms involved in the FN-mediated restoration of adhesive capacity of aged MuSCs, we plated cells on either Col or FN in the presence of a FAK inhibitor (Fig. 5d). Treatment with the FAK inhibitor, as compared to that with a vehicle (Veh) control, led to lower numbers of adherent young cells on both Col and FN. However, aged cells that were plated on Col lost their responsiveness to FAK inhibition. In contrast, growth on FN restored the sensitivity of aged cells to the FAK inhibitor to levels observed in the young cells. These data demonstrate that FAK activity is specifically lost in aged cells and restored by FN. Therefore, FAK signaling is required for the improved adherence of aged MuSCs to FN.
Under conditions of enhanced stress and cell detachment, FAK can translocate into the nucleus32. Given the critical role for FAK in MuSCs that we identified, we set out to determine whether there were any changes in the subcellular localization of FAK between the young and aged cells. When comparing the growth of freshly isolated young and aged MuSCs on Col, we observed a higher proportion of cells that had a high ratio of nuclear FAK to total cellular FAK (Fig. 5e,f). Notably, growth on FN was able to restore the subcellular localization of FAK to that found in young cells. Thus, aged MuSCs that are exposed to FN are under less stress, and their FAK signaling activity is restored to that observed in young cells.
Because our pathway-profiling assays (Fig. 3, Supplementary Fig. 3 and Supplementary Table 2) showed that the ERK and p38 MAPKs are affected by FN, we decided to also interrogate these pathways for a possible role in the age-induced anchorage deficit of MuSCs. Pharmacological inhibition of the ERK MAPK pathway did not alter the number of young and aged cells that adhered to Col or FN (data not shown). In contrast, treatment with a p38 MAPK inhibitor, as compared to treatment with vehicle, led to a significantly higher number of adherent aged cells after growth on Col (Fig. 5g). In line with the suppression of the p38 pathway after prolonged exposure to FN (Fig. 3b), we observed that, as compared to vehicle-treated cells, p38 inhibitor had no further beneficial effects on the adhesion of aged cells on this substrate. These results demonstrate that ERK signaling does not affect the adhesive capacity of MuSCs, whereas the age-related induction of the p38 pathway diminishes adhesion and cell survival.
FN treatment rejuvenates MuSCs and improves regeneration
To demonstrate that FN can also improve adhesion signaling and MuSC function in aged skeletal muscle, we injected mice with purified mouse FN at 2 d.p.i. and analyzed the regenerating tissue 3 d later (Fig. 6a). Injection of FN, as compared to injection with vehicle, led to a significantly higher abundance of FAK puncta in aged Pax7+ MuSCs (Fig. 6b). No significant effect of FN on total FAK levels was observed in muscles of young mice (Supplementary Fig. 6a). Similar to its effect on freshly isolated MuSCs in vitro (Fig. 5e,f), FN treatment also restored FAK subcellular localization in vivo in Pax7+ cells to that seen in young cells (Fig. 6c). Moreover, by using immunostaining to detect the cell proliferation marker Ki67, we observed that MuSCs from muscles of FN-treated aged mice are more proliferative, as compared to MuSCs from muscles of vehicle-treated mice (Fig. 6d). No effect of FN treatment on MuSC proliferation was observed in muscles from young mice (Supplementary Fig. 6b). To investigate whether the FN-mediated restoration of adhesion signaling and proliferation of aged MuSCs also results in a higher number of cells available for differentiation and muscle repair, we quantified the abundance of cells that were positive for the myogenic commitment marker MyoD1 (also known as MyoD). This revealed higher numbers of Pax7+MyoD+ and Pax7−MyoD+ cells in muscles from aged FN-treated mice, as compared to those from aged vehicle-treated mice (Fig. 6e,f). No positive effects on the abundance of Pax7+MyoD+ and Pax7−MyoD+ cells were observed as a consequence of FN injections, as compared to vehicle injections, in young muscles (Supplementary Fig. 6c,d). Taken together, these observations demonstrate that FN treatment of aged muscle is able to rescue FAK signaling in MuSCs and thereby restores their proliferative and myogenic potential.
Figure 6.
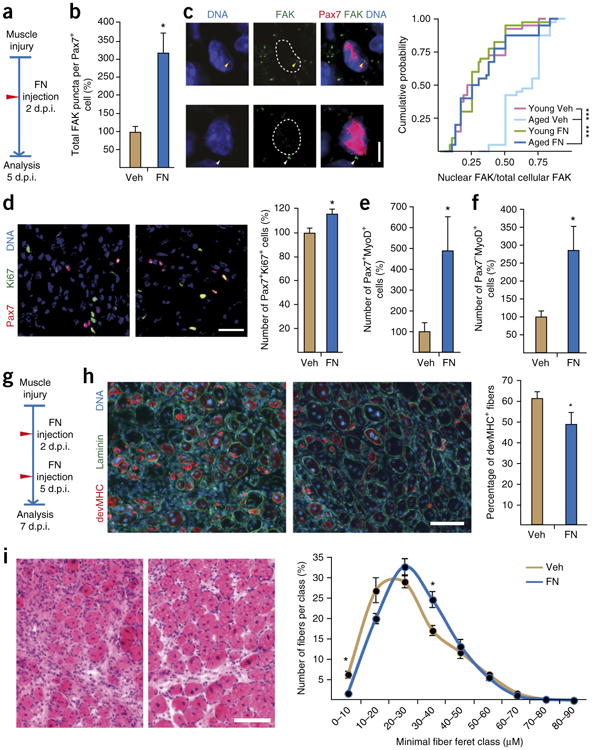
Fibronectin treatment restores the regenerative capacity of aged muscles. (a) Experimental protocol used for b–f. (b) Quantification of FAK levels in Pax7+ MuSCs in tissue sections of vehicle (Veh)- or FN-treated muscles. (c) Representative images of nuclear (yellow arrowheads) (top) or cytosolic (white arrowheads) (bottom) FAK puncta in Pax7+ cells (left) and quantification of the cumulative probability of the ratio (per cell) of nuclear FAK puncta/total FAK puncta. Kolmogorov– Smirnov distance (D) in aged FN versus aged COL, and in young COL versus aged COL, are 0.725 and 0.7, respectively. ***P values are 1.48 × 10−9 and 6.15 × 10−9, respectively. (d) Representative images of Pax7 and Ki67 staining in tissue sections from vehicle- (left) or FN-treated (middle) mice, and quantification of Ki67+ cells within the Pax7+ cell population (Pax7+Ki67+) (right). (e,f) Quantification of Pax7+MyoD+ (e) and Pax7−MyoD+ (f) cells per unit area in muscles of vehicle- or FN-treated muscles. (g) Experimental protocol used for h,i. (h) Representative images of muscle sections stained for developmental myosin heavy chain (devMHC) and laminin in vehicle- (left) or FN-treated (middle) aged mice, and quantification of the percentage of devMHC+ fibers in muscles after the indicated treatments (right). (i) Representative H&E-stained images of muscle cross-sections from vehicle- (left) or FN-treated (middle) mice and quantification of fiber size on the basis of laminin staining (right). Throughout, bars and data points represent means + s.e.m. and ± s.e.m., respectively. Mice used were n = 3 (e, FN; f, FN; i) or n = 4 (b–e, Veh; f, Veh; h) per condition. In c, n = 10 cells were analyzed per mouse. In d,h,i, quantification was performed on stitched images covering the entire cross section of the tibialis anterior muscle of each mouse. **P < 0.01, *P < 0.05; by Student's t-test (b,d–f,h,i). Scale bars, 5 μm (c), 25 μm (d) and 100 μm (h,i).
To examine whether FN treatment can improve the function of MuSCs sufficiently to restore tissue healing in the aged, we injected muscles twice over a longer period of regeneration (Fig. 6g). Staining for developmental myosin heavy chain (devMHC), a marker of the earliest stage of muscle fiber formation, revealed that aged muscles injected with FN contained less number of immature fibers at 7 d.p.i. than those injected with vehicle (Fig. 6h). No changes in the abundance of devMHC+ fibers were observed between young FN- or vehicle-treated muscles (Supplementary Fig. 6e). Finally, in contrast to injection with vehicle, injection of aged mice with FN accelerated muscle regeneration and led to significantly larger muscle fibers at 7 d.p.i. (Fig. 6i). No effect on muscle fiber size was observed between young FN-injected or vehicle-treated mice (Supplementary Fig. 6f). Taken together, these results demonstrate that restoration of FN levels during muscle regeneration to those seen in young animals is able to rescue the niche-dependent loss of MuSC function that is associated with aging.
Discussion
Old age in mammals is accompanied by a loss of MuSC function and number, leading to impaired healing of skeletal muscle following injury13–16. Aging is a multisystemic process, and it has been suggested that changes in circulating factors could be the principal mediators of MuSC dysfunction33. However, it has remained unclear whether such alterations in the systemic environment act directly on the stem cells or whether they lead to local changes in the niche that indirectly affect MuSC function. In support of the latter hypothesis, our study revealed that structural regulatory elements in the stem cell niche in skeletal muscle are profoundly changed as a consequence of the aging process and that these alterations de-regulate the majority of pathways that have previously been associated with MuSC aging17–22.
In contrast to the higher baseline fibrosis seen in uninjured aged muscles, we observed that following injury, old muscles failed to upregulate a transitional regenerative FN-rich extracellular matrix. Concomitantly, ubiquitous deletion of FN in iFN-KO mice leads to a reduction of MuSC numbers that reiterates the aging phenotype. We found that FN is a preferred adhesion substrate for MuSCs that regulates the p38 and ERK MAPK aging pathways through ITGB1 and FAK. Restoration of attachment to FN in the aged niche reactivates FAK signaling in MuSCs and thereby restores the regenerative capacity of old skeletal muscle.
Our results revealed that Lin+ cells—which include immune, hematopoietic and endothelial cells—express high levels of FN and are extremely abundant in young regenerating muscles early after injury. Coinciding with the notably lower overall FN content in aged muscles, the contribution of Lin+ cells to Fn1 mRNA expression is much lower as compared to that in young tissue. In addition, our results show lower levels of Fn1 mRNA expression in aged MuSCs than in young MuSCs. It has been shown that self-renewal in the muscle lineage is dependent on autologous regulation by FN that is derived from MuSCs themselves5,7. Therefore, decreased local FN production by MuSCs further exacerbates the age-associated decline in niche FN and is likely to affect the maintenance of MuSCs in the tissue during regeneration. It is also possible that aging affects FN expression in muscle fibroblasts. However, there are no suitable surface markers available for the flow cytometric isolation of muscle fibroblasts34. Thus future studies will have to address the spatiotemporal contribution of individual FN-secreting cell populations to the MuSC niche using specific Cre drivers and reporter alleles.
Aged muscles have been demonstrated to contain higher levels of FGF2, which induces a break in MuSC quiescence, leading to regenerative failure17. FGF signaling is known to activate the JAK–STAT, phosphatidylinositol, PI3K–Akt and MAPK pathways35. Notably, we found that most of these pathways are also affected by FN. FGF signaling is regulated by integrins that can serve as FN receptors36. Our results demonstrate that signaling pathways downstream of integrins are de-regulated as a consequence of loss of environmental FN in aged cells. Thus, these observations indicate that changes in the content of FN in the MuSC niche are closely connected to age-related alterations in FGF signaling37.
At a very advanced age in mice, MuSCs enter a senescent state that is marked by high expression of the cell cycle inhibitor p16INK4A and failure of autophagy22,38. Of note, p16INK4A has also been shown to induce anoikis in several different cell types39. In line with our results, this study demonstrated that p16INK4A-mediated anoikis is strongly inhibited by FN but not by LAM. On the basis of these observations we speculate that restoration of the FN content of the niche could also be a strategy to overcome MuSC senescence in geriatric individuals.
Taken together, we discovered FN as a structural element in the stem cell niche that is critical for the maintenance and function of MuSCs during muscle regeneration. Loss of FN from the niche affects a substantial number of pathways and cellular mechanisms that have been implicated in MuSC aging17–22. Our work reveals that alterations in the ability of stem cells to adhere to the niche are a root cause of MuSC aging and a promising target for the rejuvenation of skeletal muscle tissue.
Methods
Methods and any associated references are available in the online version of the paper.
Online Methods
Mice
All mice were housed under standard conditions and allowed access to food and water ad libitum. Itgb1−/− myoblasts and controls were isolated from both genders of mice, otherwise only male mice were used. Unless otherwise indicated, the strain C57BL/6JRj (Janvier) was used. Young mice were between 9 and15 weeks of age, and aged mice were 20–24 months old. For isolation of cells from regenerating muscle, tibialis anterior, gastrocnemius and quadriceps muscles were injected with 50 μl, 100 μl or 50 μl of 50% vol/vol glycerol in phosphate-buffered saline (PBS), respectively40. Animal experiments were approved by the Vaud Cantonal Commission (Switzerland) for animal experimentation under licenses VD2620 and VD2947, the Institutional Animal Care and Use Committee (IACUC) of the Carnegie Institution for Science (USA) under the permit number A3861-01 and the Landesamt für Verbraucherschutz Abteilung Gesundheitlicher und technischer Verbraucherschutz (Germany) under Reg.-Nr 03-010/15. Fibronectin-floxed mice were provided by R.F., and the ROSA26–CreERT2 mice were provided by F. Stewart (Technische Universität Dresden, Germany)24,25. Experiments with iFN-KO and the respective control animals were performed in accordance with University of Ottawa guidelines for animal handling and animal care, as determined by the University of Ottawa Animal Care Committee. iFN-KO and Ctrl mice were treated with four daily intraperitoneal injections of 100 mg per kg body weight (mg/kg) tamoxifen (Sigma) in corn oil at 2–3 weeks of age. Tibialis anterior muscle injury in iFN-KO mice was induced at 6–7 weeks of age by a single injection of 50 μl of 10 μM cardiotoxin (Sigma) solution in 0.9% saline. For in vivo FN treatment, tibialis anterior muscles of young and aged mice were injured with a single injection of 50 μl of 20 μM cardiotoxin solution in 0.9% saline. Mice were then injected with 0.5 mg/ml mouse FN solution (Biopur) or vehicle (Veh) at 2 and 5 d.p.i. Veh was 50 mM Tris and 0.5 M NaCl at pH 7.5 in water. Muscles were isolated and analyzed at 5 or 7 d.p.i.
Slow-off-rate-modified aptamer assay
Muscles samples were pulverized using the cryoPREP impactor system (Covaris). The muscle powder was then subjected to mechanical lysis using a Polytron homogenizer, and the proteins were extracted in 50 mM Tris (pH 7.5), 150 mM NaCl, 50 mM NaF, 1 mM EDTA, 0.5% Triton X-100. Protein concentration was determined by a bicinchoninic acid (BCA) assay assay (Pierce), and samples were diluted at 250 ug/ml. Protein extracts were analyzed by using DNA-aptamer-based recognition on the SOMAscan platform (Somalogic, Boulder, CO, USA), as described23. Median normalized relative fluorescence units (RFUs) were log2-transformed before applying principal component analysis and linear models. Statistical analyses were performed in R 3.1.3 (R Foundation for Statistical Computing).
ECM arrays and coating
Mouse primary myoblasts and human primary myoblasts (HSMM, Lonza) between passage 4 and 10 were seeded on MicroMatrix 36 arrays (Microstem) containing ECMs spotted onto a 10% polyacrylamide hydrogel of 10 kPa in stiffness. After 3 h, 6 h and 24 h, the number of adhering cells was determined by visual counting. For ECM coating, dishes were covered with collagen (C7774, Sigma), fibronectin (F2006, Sigma) or laminin (L6274, Sigma).
Antibody arrays and western blot
Mouse primary myoblasts were grown on FN- or Col-coated dishes for 72 h, trypsinized, collected by centrifugation and frozen in dry ice. Phospho Explorer Antibody Arrays were analyzed by Full Moon BioSystems. Briefly, as described in the manufacturer's instructions, proteins were extracted by using nondenaturing lysis buffer, concentration was adjusted between samples, and protein extracts were biotinylated, coupled to the antibodies on the array and detected by dye-conjugated streptavidin using a microarray scanner. Cells for western blot analysis were grown for 3 h or 72 h on Col-, FN- or LAM-coated dishes and lysed in RIPA buffer (Sigma) following collection. After adjustment of protein concentrations (as determined by BCA assays) samples were boiled in Laemmli buffer and interrogated by standard western blot procedures. Antibodies used were: rabbit p44/42 MAPK (Cell Signaling #9102), rabbit phospho-p44/42 MAPK (Cell Signaling #4370), rabbit p38 MAPK (Cell Signaling#9212), rabbit phospho-p38 MAPK (Cell Signaling #9211), rabbit FAK (Cell Signaling #13009), rabbit pFAK (Cell Signaling #3284), mouse β-actin (Sigma A5441) and mouse Gapdh (RDI/Fitzgerald, RDI–TRK5G4–6C5). Antibodies for western blots were diluted 1/1,000, except for β-actin, which was used at 1/5,000. Antibody validation is provided on the manufacturers' websites.
Quantitative PCR
RNA was extracted from frozen muscles or freshly sorted cells by using miRNeasy Mini Kit or RNeasy Micro Kit (Qiagen), respectively. RNA samples were subjected to reverse transcription using random primers (High Capacity cDNA Reverse Transcription Kit, ABI). SYBR and Taqman quantitative PCR was performed on a LightCycler 480. The following primers were used: Fn1 sense: GGCCACACCTACAACCAGTA, Fn1 antisense: TCGTCTCTGTCAGCTTGCAC Primers for housekeeping genes were: Actb sense: CAGCTTCTTTGCAGCTCCTT, Actb antisense: GCAGCGATATCGTCATCCA. Taqman probe for Fn1 was Mm01256744_m1 (Applied Biosystems). For qPCR using whole regenerating muscles, reference genes were selected based on their stability across time points of regeneration from microarray data: Atp5b sense: ACCTCGGTGCAGGCTATCTA, Atp5b antisense: AATAGCCCGGGACAACACAG, Eif2a sense: CACGGTGCTTCCCAGAGAAT, Eif2a antisense: TGCAGTAGTCCCTTGTTAGCG, Psmb4 sense: GCGAGT CAACGACAGCACTA, Psmb4 antisense: TCATCAATCACCATCTGGCCG.
Flow cytometry, MuSC in vitro assays and myoblast culture
For isolation of cell populations, muscles were collected and digested with Dispase II (2.5 U/ml; Roche), Collagenase B (0.2% Roche) and MgCl2 (5 mM) at 37 °C. Cells were then incubated at 4 °C for 30 min with antibodies against CD45 (Invitrogen, MCD4501 or MCD4528; dilution for both 1/25), CD31 (Invitrogen, RM5201 or RM5228; dilution for both 1/25), CD11b (Invitrogen, RM2801 or RM2828; dilution for both 1/25), CD34 (BD Biosciences, 560230 or 560238; dilution for both 1/60), Ly-6A–Ly-6E (Sca1) (BD Biosciences, 561021; dilution 1/150), α7-integrin (R&D, FAB3518N; dilution 1/30) and CD140a (eBioscience, 12–1401–81 or 17–1401–81; dilution for both 1/30). Antibody validation is provided on the manufacturer's website. FACS isolation was performed on a Beckman-Coulter Astrios Cell sorter. MuSCs were CD45−CD31−CD11b−Sca1−CD34+Itga7+; FAPs (fibro-adipogenic progenitors) were CD45−CD31−CD11b−Sca1+CD34+PDGF Ra+; and Lin+ cells were CD45+CD31+CD11b+. For high-throughput imaging, MuSCs were distributed into 96-well plates at a density of 1,765 cells/cm2. Freshly sorted MuSCs were maintained in high-glucose Dulbecco's modified Eagle's medium (DMEM), 20% heat-inactivated FBS, 10% inactivated horse serum, 2.5 ng/ml basic FGF (bFGF; Invitrogen), 1% penicillin–streptomycin (P–S), 1% l-glutamine, 1% Na pyruvate (Invitrogen). EdU was added to the medium at 10 μM for 6 h before fixation. For long-term culture, myoblasts were maintained in Ham's F-10 (Gibco), 20% heat-inactivated FBS, 2.5 ng/ml bFGF (Invitrogen) and 1% P–S. Small molecular inhibitors were: FAK inhibitor (F14, CAS 4506–66–5, #sc–203950) and p38 inhibitor (SB 203580 hydrochloride, R&D systems TOCRIS #1402). Itgb1-KO myoblasts and controls were isolated from Pax7CE/+;Itgb1f/f;R26RYFP/YFP mice31,41,42 and Pax7CE/+;R26RYFP/YFP, respectively37. Primary human myoblasts from adult donors were obtained from Lonza (HSMM) after the supplier received informed consent from the donors and after consent was obtained from the Vaud ethics commission for human research (CER–VD) under protocol 281/14.
Immunostaining and image analysis
Adhesion of freshly isolated MuSCs was assessed at different time points by fixation in 4% paraformaldehyde (PFA) followed and counterstaining with the nuclear dye DAPI. TUNEL+ cells were quantified using the In situ Cell Death Detection Kit, TMR red (Roche #12 156 792 910). EdU incorporation was revealed by using the Click-0iT assay (Molecular Probes) according to manufacturer's instructions. Briefly, cells were fixed for 15 min in 4% PFA, permeabilized for 20 min in PBS containing 0.5% (vol/vol) Triton X-100 (PBTX), stained with the Click-iT reaction mix and counterstained with DAPI. For immunostaining, cells were blocked for 1–2 h in 5% goat serum, 1% BSA and 0.2% PBTX, before incubation with primary and secondary antibodies. Image acquisition was performed using the ImageXpress (Molecular Devices) platform. Quantifications were done using the MetaXpress software. The number of Pax7-, MyoD- and Ki67-positive cells was determined by counting of immunostainings in muscle sections, as previously described5. In-situ quantifications were done using the ImageJ software43 (U. S. National Institutes of Health) over the entire cross-sectional area of the muscle. For minimal fiber feret measurements, only centralized fibers on LAM-stained sections were considered. Antibodies were: mouse Pax7 (DHSB; for tissue sections, undiluted hybridoma culture supernatant and purified at 2.5 μg/ml)5, mouse devMHC (DSHB, F1.652; for tissue sections, dilution 1/500), rabbit FAK antibody (Abcam, ab40794; for tissue sections, dilution 1/200; for isolated cells, dilution 1/40), rabbit Ki67 (Abcam, ab833; for tissue sections, dilution 1/200), rabbit laminin (L9393 Sigma; for tissue sections, dilution 1/1,000), rabbit fibronectin (Abcam, ab23750; for tissue sections, dilution 1/1,000)5. Where no citation is provided, antibody validation and references can be found on the manufacturer's website.
Microarrays
For isolation of RNA from freshly sorted activated MuSCs, the RNeasy Micro Kit (Qiagen) was used. RNA samples were then subjected to 3′ microarray analysis on Illumina MouseRef–8_V2 chips. 3 ng of total RNA were used to produce cRNA in a two-round amplification protocol, using first Messageamp II aRNA amplification kit (AM1751, Life Technologies, Inc.) followed by Messageamp II–biotin enhanced aRNA amplification kit (AM1791, Life Technologies, Inc.). 750 ng of cRNA were hybridized for 16 h at 55 °C on Illumina MouseRef–8 v2 microarrays. Quality of total RNA was checked by using the Bioanalyzer 2100 with Total RNA Pico kit, and quality of cRNA was checked by using the Bioanalyzer 2100 with the Total RNA Nano kit (Agilent Technologies). Quantifications were done using the Quant-iT RiboGreen RNA Assay Kit (Life Technologies, Inc.). For microarrays comparing FN and Col, cells were grown for 72 h on FN- or Col-coated dishes. RNA was extracted with the Agencourt RNAdvance Tissue Kit (Beckman Coulter, Inc). cRNA was produced with the Illumina TotalPrep-96 Kit (Life Technologies Inc). 15 μg of cRNA were fragmented prior to a 16-h hybridization at 45 °C on an Affymetrix Mouse Genome 430 2.0 Array. Quality of total RNA and cRNA was checked by using the Bioanalyzer 2100 with the Total RNA Nano kit (Agilent Technologies). Quantification was done by using the Quant-iT RiboGreen RNA Assay Kit assay (Life Technologies Inc). Microarray data have been deposited to Gene Expression Omnibus under accession number GSE81096 and GSE81225.
Transcriptomic analysis
The robust-multiarray-average (RMA) approach was used for the creation and normalization of the summarized Affymetrix probe set signals. We applied a nonspecific filter to discard probe sets with low variability; we retained 19,050 Affymetrix probe sets whose s.d. was greater than the median of the s.d. of all of the probe sets. Illumina expression signals were quantile-normalized. We applied a nonspecific filter to discard probe sets with low variability and retained 12,848 Illumina probe sets whose s.d. was greater than the median of the s.d. of all of the probe sets. For differential expression analysis and pathway analyses, genes (represented by probe sets) were tested for differential expression using the moderated t-statistic as implemented in LIMMA44 for both data sets. We exploited DAVID Bioinformatics Resources 6.7 (ref. 45) to assess whether the differentially expressed genes were related to specific KEGG pathways. Gene Set Enrichment Analysis (GSEA), using the Broad Institute algorithm v2.2.0 (ref. 46), was conducted on the preranked gene lists defined according to the LIMMA results and the gene sets derived from the KEGG pathway database (c2.cp.kegg.v5.0.symbols.gmt) were tested.
Statistical analysis
All wild-type mice were randomized according to body weight before interventions, and no mouse was excluded from the study, except for those that died a natural death during the course of the experiment. Sample size determination was based on the expected effect size and variability that was previously observed for similar readouts in the investigators' labs. In vivo treatments were not blinded, but imaging readouts were analyzed in a blinded manner. Genome-wide statistical analyses and Kolmogorov–Smirnov tests were performed using R version 3.1.3 and relevant Bioconductor packages as described in the sections above. Gene Set Enrichment Analyses were performed using the Broad Institute algorithm v2.2.0. All other statistical analyses were performed using GraphPad Prism (GraphPad Software) assuming normal distribution of the variables measured. Statistical significance for binary comparisons was assessed by a Student's t-test after checking that variances do not differ between groups or by a Welch correction when variances differed between groups. All exploratory and signaling experiments were analyzed by using two-tailed tests, and in vivo phenotypic rescue experiments were tested using a one-tailed test. For comparison of more than two groups, one-way or two-way ANOVAs were used, according to the experimental design, and followed by Bonferroni multiple-comparison testing. All measurements that were fully independent were analyzed using unpaired statistics, whereas experiments in which treatments were performed with primary cells isolated from the same animal(s) were analyzed by using paired statistics. All data are expressed as mean + s.e.m. or mean ± s.e.m. For plotting the cumulative probability of the ratio of nuclear FAK puncta over total FAK puncta per cell in tissue resident MuSCs, 0.5 pseudo puncta were added to the entire data set before performing statistics.
Supplementary Material
Acknowledgments
We thank J. Ritchie for help with mouse husbandry. We are grateful to the NIHS community for fruitful discussions and support, in particular to the Aging and Stem Cells groups, and to E. Rolland and E. Baetge. We thank C. Poser for excellent technical assistance. ROSA26–CreERT2 mice were provided by F. Stewart (Technische Universität Dresden, Germany) as a kind gift to M.A.R. J.V.M. and M.J.J. were supported by a grant from the German Research Foundation (DFG) (grant MA–3975/2–1). M.A.R. holds the Canada Research Chair in Molecular Genetics and is supported by a grant from the Government of the Ontario Ministry of Research and Innovation (MRI) (grant ORF–RE05–084). M.R. and C.M.F. are supported by the National Institutes of Health grants (grants HD075345 and AR060042). C.F.B. is supported by the Fondation Suisse de Recherche sur les Maladies Musculaires (FSRMM).
Footnotes
Accession codes: Gene Expression Omnibus: microarray data have been deposited under accession number GSE81096 and GSE81225.
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
Author Contributions: M.J.J., N.H., C.P.-C., M.R., S.K., M.S., L. Li, S.M., U.L., F.S., N.A.D., A.P., C.-M.F. and J.v.M., designed and conducted experiments, and analyzed results; E.M. and F.R. performed data analysis; G.J. provided support with high-content image acquisition and analysis; M.A.R. provided critical reagents, edited the manuscript and helped with data interpretation; R.F. provided critical reagents; D.H.W. helped with data acquisition and analysis; P.S. and P.D. helped with experimental design, data interpretation and editing of the manuscript; and L. Lukjanenko, J.N.F. and C.F.B. designed and conducted experiments, analyzed results and wrote the manuscript.
Competing Financial Interests: The authors declare competing financial interests: details are available in the online version of the paper.
References
- 1.Bentzinger CF, Wang YX, Dumont NA, Rudnicki MA. Cellular dynamics in the muscle satellite cell niche. EMBO Rep. 2013;14:1062–1072. doi: 10.1038/embor.2013.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gilbert PM, et al. Substrate elasticity regulates skeletal muscle stem cell self-renewal in culture. Science. 2010;329:1078–1081. doi: 10.1126/science.1191035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lv H, et al. Mechanism of regulation of stem cell differentiation by matrix stiffness. Stem Cell Res Ther. 2015;6:103. doi: 10.1186/s13287-015-0083-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bonaldo P, et al. Collagen VI deficiency induces early onset myopathy in the mouse: an animal model for Bethlem myopathy. Hum Mol Genet. 1998;7:2135–2140. doi: 10.1093/hmg/7.13.2135. [DOI] [PubMed] [Google Scholar]
- 5.Bentzinger CF, et al. Fibronectin regulates Wnt7a signaling and satellite cell expansion. Cell Stem Cell. 2013;12:75–87. doi: 10.1016/j.stem.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Urciuolo A, et al. Collagen VI regulates satellite cell self-renewal and muscle regeneration. Nat Commun. 2013;4:1964. doi: 10.1038/ncomms2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tierney MT, et al. Autonomous extracellular matrix remodeling controls a progressive adaptation in muscle stem cell regenerative capacity during development. Cell Rep. 2016;14:1940–1952. doi: 10.1016/j.celrep.2016.01.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mann CJ, et al. Aberrant repair and fibrosis development in skeletal muscle. Skelet Muscle. 2011;1:21. doi: 10.1186/2044-5040-1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.D'Souza DM, et al. Diet-induced obesity impairs muscle satellite cell activation and muscle repair through alterations in hepatocyte growth factor signaling. Physiol Rep. 2015;3:e12506. doi: 10.14814/phy2.12506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sousa-Victor P, García-Prat L, Serrano AL, Perdiguero E, Muñoz-Cánoves P. Muscle stem cell aging: regulation and rejuvenation. TEM. 2015;26:287–296. doi: 10.1016/j.tem.2015.03.006. [DOI] [PubMed] [Google Scholar]
- 11.He WA, et al. NF-κB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J Clin Invest. 2013;123:4821–4835. doi: 10.1172/JCI68523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fujimaki S, Wakabayashi T, Takemasa T, Asashima M, Kuwabara T. Diabetes and stem cell function. BioMed Res Int. 2015;2015:592915. doi: 10.1155/2015/592915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carlson ME, et al. Molecular aging and rejuvenation of human muscle stem cells. EMBO Mol Med. 2009;1:381–391. doi: 10.1002/emmm.200900045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Watters JM, Clancey SM, Moulton SB, Briere KM, Zhu JM. Impaired recovery of strength in older patients after major abdominal surgery. Ann Surg. 1993;218:380–390. doi: 10.1097/00000658-199309000-00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Müller M, Tohtz S, Dewey M, Springer I, Perka C. Age-related appearance of muscle trauma in primary total hip arthroplasty and the benefit of a minimally invasive approach for patients older than 70 years. Int Orthop. 2011;35:165–171. doi: 10.1007/s00264-010-1166-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blau HM, Cosgrove BD, Ho AT. The central role of muscle stem cells in regenerative failure with aging. Nat Med. 2015;21:854–862. doi: 10.1038/nm.3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chakkalakal JV, Jones KM, Basson MA, Brack AS. The aged niche disrupts muscle stem cell quiescence. Nature. 2012;490:355–360. doi: 10.1038/nature11438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Price FD, et al. Inhibition of JAK-STAT signaling stimulates adult satellite cell function. Nat Med. 2014;20:1174–1181. doi: 10.1038/nm.3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tierney MT, et al. STAT3 signaling controls satellite cell expansion and skeletal muscle repair. Nat Med. 2014;20:1182–1186. doi: 10.1038/nm.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cosgrove BD, et al. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat Med. 2014;20:255–264. doi: 10.1038/nm.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bernet JD, et al. MAPK signaling underlies a cell-autonomous loss of stem cell self-renewal in skeletal muscle of aged mice. Nat Med. 2014;20:38. 265–271. doi: 10.1038/nm.3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sousa-Victor P, et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature. 2014;506:316–321. doi: 10.1038/nature13013. [DOI] [PubMed] [Google Scholar]
- 23.Gold L, et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS One. 2010;5:e15004. doi: 10.1371/journal.pone.0015004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sakai T, et al. Plasma fibronectin supports neuronal survival and reduces brain injury following transient focal cerebral ischemia but is not essential for skin-wound healing and hemostasis. Nat Med. 2001;7:324–330. doi: 10.1038/85471. [DOI] [PubMed] [Google Scholar]
- 25.Seibler J, et al. Rapid generation of inducible mouse mutants. Nucleic Acids Res. 2003;31:e12. doi: 10.1093/nar/gng012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siegel AL, Atchison K, Fisher KE, Davis GE, Cornelison DD. 3D time-lapse analysis of muscle satellite cell motility. Stem Cells. 2009;27:2527–2538. doi: 10.1002/stem.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brafman DA, Shah KD, Fellner T, Chien S, Willert K. Defning long-term maintenance conditions of human embryonic stem cells with arrayed cellular microenvironment technology. Stem Cells Dev. 2009;18:1141–1154. doi: 10.1089/scd.2008.0410. [DOI] [PubMed] [Google Scholar]
- 28.Brafman DA, et al. Investigating the role of the extracellular environment in modulating hepatic stellate cell biology with arrayed combinatorial microenvironments. Integr Biol (Camb) 2009;1:513–524. doi: 10.1039/b912926j. [DOI] [PubMed] [Google Scholar]
- 29.Moreno-Layseca P, Streuli CH. Signaling pathways linking integrins with cell cycle progression. Matrix Biol. 2014;34:144–153. doi: 10.1016/j.matbio.2013.10.011. [DOI] [PubMed] [Google Scholar]
- 30.Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer. 2014;14:598–610. doi: 10.1038/nrc3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raghavan S, Bauer C, Mundschau G, Li Q, Fuchs E. Conditional ablation of β1-integrin in skin. Severe defects in epidermal proliferation, basement membrane formation and hair follicle invagination. J Cell Biol. 2000;150:1149–1160. doi: 10.1083/jcb.150.5.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lim ST, et al. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol Cell. 2008;29:9–22. doi: 10.1016/j.molcel.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Conboy IM, et al. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 2005;433:760–764. doi: 10.1038/nature03260. [DOI] [PubMed] [Google Scholar]
- 34.Tigges J, et al. The hallmarks of fibroblast aging. Mech Ageing Dev. 2014;138:26–44. doi: 10.1016/j.mad.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 35.Lanner F, Rossant J. The role of FGF–Erk signaling in pluripotent cells. Development. 2010;137:3351–3360. doi: 10.1242/dev.050146. [DOI] [PubMed] [Google Scholar]
- 36.Kim SH, Turnbull J, Guimond S. Extracellular matrix and cell signaling: the dynamic cooperation of integrin, proteoglycan and growth factor receptor. J Endocrinol. 2011;209:139–151. doi: 10.1530/JOE-10-0377. [DOI] [PubMed] [Google Scholar]
- 37.Rozo M, Li L, Fan CM. Targeting β1-integrin signaling enhances regeneration in aged and dystrophic mice. Nat Med. 2016 doi: 10.1038/nm.4116. http://dx.doi.org/10.1038/nm.4116. [DOI] [PMC free article] [PubMed]
- 38.García-Prat L, et al. Autophagy maintains stemness by preventing senescence. Nature. 2016;529:37–42. doi: 10.1038/nature16187. [DOI] [PubMed] [Google Scholar]
- 39.Plath T, et al. A novel function for the tumor suppressor p16INK4a: induction of anoikis via upregulation of the α5β1 fibronectin receptor. J Cell Biol. 2000;150:1467–1478. doi: 10.1083/jcb.150.6.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lukjanenko L, Brachat S, Pierrel E, Lach-Triflieff E, Feige JN. Genomic profiling reveals that transient adipogenic activation is a hallmark of mouse models of skeletal muscle regeneration. PLoS One. 2013;8:e71084. doi: 10.1371/journal.pone.0071084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Srinivas S, et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lepper C, Conway SJ, Fan CM. Adult satellite cells and embryonic muscle progenitors have distinct genetic requirements. Nature. 2009;460:627–631. doi: 10.1038/nature08209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:e3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 45.Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 46.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.