

Abstract
Predisposition to myelodysplastic syndrome (MDS) and acute leukemia is a hallmark of Fanconi anemia (FA). Morphologic criteria for MDS in FA are not well established, nor is the significance of clonal chromosomal abnormalities. We reviewed bone marrow samples of 119 FA patients: 23 had MDS, with the most common subtype refractory cytopenia with multilineage dysplasia. The presence of MDS was highly correlated with the presence of clonal abnormalities. Neutrophil dysplasia and increased blasts were always associated with the presence of a clone, in contrast with dyserythropoiesis. The most frequent clones had gains of 1q and 3q and/or loss of 7. Karyotype complexity also correlated with MDS. One third of patients with 3q as a sole abnormality had no MDS; patients with 3q and an additional abnormality all had MDS. The data provide a rationale for integrating cytogenetic findings with independently evaluated morphologic findings for monitoring bone marrow status in FA.
Keywords: Fanconi, Hematopathology, Cytogenetics, Myelodysplastic syndrome, Clone, Chromosome, Dyserythropoiesis, Dysgranulopoiesis, Dysmegakaryopoiesis
Fanconi anemia (FA) is an inherited disorder associated with genomic instability, bone marrow failure (BMF), and predisposition to hematologic malignancies and/or solid tumors. Thirteen complementation groups have been identified,1 with 12 of the putative genes mapped to autosomal loci and the remaining gene to the X chromosome. The early life-threatening feature of FA is BMF, thought to result from excess apoptosis in stem and progenitor cells.2,3 The genetic instability and proapoptotic tendency then provide a selective force for the evolution of adapted hematopoietic stem cell clones that lead to acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS).4 In published literature reviews and studies of patients enrolled in the International Fanconi Anemia Registry, leukemia was reported in 8% to 9% and MDS in 7% of patients.5–7 Analysis of the data for 754 patients from International Fanconi Anemia Registry demonstrated a cumulative incidence of 90% for BMF, 33% for hematologic malignancy (AML or MDS), and 35% for solid tumors by age 50 years.7 The diagnosis of a malignancy preceded the diagnosis of FA in more than one third of the cases with cancer reported in the literature.3 MDS and AML are major predictors of clinical outcome in patients with FA.8,9
MDS is a particularly challenging diagnosis in FA owing to the frequent presence of dysplastic features, especially dyserythropoiesis, the complicating factor of hypocellularity, and the frequent administration of granulocyte colony-stimulating factor (G-CSF) resulting in hyposegmented neutrophils and circulating blasts.10,11 In addition, even among expert pathologists, it is difficult to reach consensus on certain morphologic features such as megaloblastosis in erythroid precursors or hypogranularity in neutrophils.11 Neither the current World Health Organization (WHO) nor the suggested pediatric classifications establish quantitative or qualitative criteria for MDS associated with constitutional/inherited abnormalities.
Relevant to the development of MDS in FA is the emergence of abnormal cytogenetic clones in the bone marrow (BM) of patients. Previous studies have documented acquired clonal abnormalities in FA.12–14 However, incomplete characterization of the aberrations owing to limitations in available techniques, the absence of the more common recurring abnormalities in AML, the presence of background chromosomal instability generating nonclonal abnormalities, and the occasional report of “transient” clones have raised the issue of the clinical significance of these clonal abnormalities.14,15 More recently, Tonnies et al16 have shown that a clone resulting in an extra copy of the distal long arm of chromosome 3 in FA is associated with a poor prognosis. Yet questions remain regarding the spectrum and clinical significance of clonal cytogenetic abnormalities. The present study was undertaken to characterize the morphologic findings of MDS in FA and their relationship to cytogenetic findings. These results have led to the generation of an integrated approach to the diagnosis of MDS in FA.
Materials and Methods
Eligibility criteria for the study were met by 119 consecutive patients with confirmed diagnoses of FA referred to the University of Minnesota Hospital, Minneapolis, between 1991 and 2006, for whom both hematopathology and cytogenetics data were available for review. All had a positive test for chromosomal hypersensitivity to diepoxybutane and/or mitomycin C in a College of American Pathologists–certified laboratory. In these 119 pre–BM transplant patients with FA, 173 BM biopsies and concurrent cytogenetic analyses were performed. For each patient, 1 to 4 BM biopsy specimens were evaluated. For patients with sequential biopsies, the most recent BM biopsy specimen or the biopsy specimen closest to the time of transplantation was selected for correlative statistical analysis. Hemogram data, peripheral blood (PB) and BM smears, touch imprints, biopsy and clot sections, and iron stains were reviewed by 2 experienced hematopathologists without knowledge of cytogenetic results. Cytogenetic studies were performed at the University of Minnesota and reviewed by 1 cytogeneticist without knowledge of the hematopathology findings. Clinical and demographic information were obtained from the patients’ medical records. Informed consent for study was obtained for all patients in accordance with University of Minnesota Medical School Institutional Review Board requirements.
Hematopathology
Bone Marrow Failure
BMF was classified into 3 categories based on hemoglobin level, absolute neutrophil count, and platelet count9 Table 1. BMF grade 1 or 2 had at least 1 cytopenia present; BMF grade 3 had 2 of the 3 cytopenias present. BM cellularity was graded based on comparison with expectation for age, as follows17: 0, at least 50% less cellular than expected (markedly hypocellular); 1, 30% to 40% less than expected (moderately hypocellular); 2, 10% to 20% less than expected (slightly hypocellular); 3, normocellular for age; and 4, cellularity of 10% or higher than expected (hypercellular).
Table 1.
Severity of Bone Marrow Failure
| Mild* (Grade 1) | Moderate (Grade 2) | Severe (Grade 3) | |
|---|---|---|---|
| Absolute neutrophil count,/μL (× 109/L) | ≥1,000 (1.0) | <1,000 (1.0) | <500 (0.5) |
| Platelet count, × 103/μL (× 109/L) | ≥50 (50) | <50 (50) | <30 (30) |
| Hemoglobin, g/dL (g/L) | ≥8 (80) | <8 (80) | <8 (80) |
The absolute neutrophil count, platelet count, or hemoglobin concentration was decreased for age.
RBC Abnormalities
The principal abnormalities documented on review of PB smears were the degree of anisopoikilocytosis and the presence of numerous macro-ovalocytes. Three grades of anisopoikilocytosis were established, based on the red cell distribution width (RDW) and visual inspection of the smear: 1, slight (RDW = 15–16); 2, moderate (RDW = 17–20); and 3, marked (RDW > 20). Macro-ovalocytes were quantified and coded as follows: 0, rare or occasional (1 to 3 per high-power field); or 1, numerous (at least 4 per high-power field).
BM Dysplastic Features
Dyserythropoietic features evaluated included nuclear lobulation, budding, karyorrhexis and fragmentation, internuclear chromatin bridging, multinuclearity, megaloblastoid nuclei, nuclear/cytoplasmic dyssynchrony, and vacuolated erythroblasts. Ringed sideroblasts (RS) were identified based on WHO recommendations10 and graded as follows: 0, absent; 1, 1% to 14% of sideroblasts; and 3, 15% or more of sideroblasts. Dysgranulopoietic features evaluated included deficient or aberrant secondary granule production, nuclear hyposegmentation (pseudo–Pelger-Huët changes), and hypersegmentation. Owing to the frequent administration of G-CSF in patients with FA, the morphologic abnormalities usually associated with this agent, such as nuclear segmentation defects in association with cytoplasmic hypergranularity, were documented but not included as features of granulocytic dysplasia. Features of dysmegakaryopoiesis evaluated included small size with nonlobulated or bilobed (hypolobulated) nuclei, nonlobulated nuclei in megakaryocytes of all sizes, and multiple, widely separated nuclei. The BM blast percentage was graded as follows: 0, up to 1%; 1, more than 1% but less than 5%; 2, at least 5% but less than 10%; and 3, at least 10% but less than 20%. The identification of increased circulating blasts without an increase in BM blasts was not construed as evidence of MDS if the patient was receiving G-CSF at the time of the BM biopsy.
Grading of Dysplasia
Modified WHO criteria were used on BM examination to grade dysplasia.10 Dysplastic features were graded and coded for the granulocytic and megakaryocytic lineage as follows: 0, no dysplastic cells; 1, fewer than 5% dysplastic cells; 2, 5% to 9% dysplastic cells; and 3, 10% or more dysplastic cells. For erythroids, grades 0, 1, and 2 followed the same cutoffs. Dyserythropoiesis grade 3 was defined by 10% to 49% dysplastic cells, and a grade 4 was established based on the presence of 50% or more dysplastic cells. For grading dysplasia, at least 200 erythroid precursors, at least 100 neutrophils and precursors, and all available megakaryocytes were evaluated.
Diagnostic Categories
Diagnostic categories were established as follows: non-MDS, borderline MDS, and MDS. MDS was classified according to WHO criteria including at least 10% dysplastic cells in a myeloid cell line, at least 15% RS, at least 2% PB blasts, or at least 5% BM blasts.10 Dyserythropoiesis alone was considered diagnostic of MDS only if grade 4. No diagnosis of MDS was based exclusively on the presence of dyserythropoiesis. However, the presence of grade 3 dyserythropoiesis was used as part of the criteria for classification of MDS. The category of borderline MDS was used for cases in which the BM morphologic findings were considered equivocal or marginal: borderline MDS was defined as grade 1 to 3 dyserythropoiesis accompanied by either grade 1 or 2 dysgranulopoiesis or grade 2 dysmegakaryopoiesis, but no increase in blasts. A higher grade of dysmegakaryopoiesis vs dysgranulopoiesis was used owing to the low numbers of megakaryocytes commonly found in FA.
Cytogenetics
For cytogenetics studies, 20 or more metaphase cells from the BM aspirates of each patient were analyzed by G-banding. Metaphase fluorescence in situ hybridization (FISH) with single whole chromosome or multichromosome paint probes (Abbott Molecular, Des Plaines, IL) were applied as necessary to further characterize structural abnormalities. For small supernumerary marker chromosomes, array comparative genomic hybridization (a-CGH) with a 1-Mb spaced BAC (bacterial artificial chromosome) array (Spectral Genomics, Houston, TX) was applied. Abnormalities were characterized according to the 2005 International System for Human Cytogenetic Nomenclature.18 Almost all of the detected chromosomal abnormalities involved a gain or loss of a relatively large region of a chromosome. Therefore, for purposes of data analysis, each abnormality was described by a 3 character code: the chromosome number, “p” (short arm) or “q” (long arm), and “L” or “G” to designate a loss or gain of chromosomal material. For example, 1qG represents gain of a region of the long arm of chromosome 1.
Statistics
The χ2 test was used for statistical comparison of categorical data, and a Fisher exact test was applied to categories with small numbers. The general Wilcoxon test was used for comparison of continuous data and the κ statistic for measurement of the correlation between clonal abnormalities.
Results
Patient Characteristics
Of the 119 patients evaluated, 8 had AML. Because the focus of the present study was MDS and because these AML cases represent a unique subgroup, they were not included in any further analyses. Of the remaining 111 patients, 61 were male and 50 were female, with a median age of 9 years (range, 2–44 years). Eighty-eight patients subsequently underwent BM transplantation.
BM Findings
Frequent Dyserythropoiesis
Dyserythropoiesis was identified in 93.7% of patients. The most common morphologic abnormalities included irregular nuclear contours, budding nuclei, and karyorrhexis. In 72 (69.2%) of these 104 patients, the erythroid line was the sole cell lineage with dysplasia; 18 patients (17.3%) had grade 3 dyserythropoiesis. Grade 4 dyserythropoiesis was detected in 2 patients; both had more than 15% RS, and 1 patient also had dysmegakaryopoiesis.
RS, Dysmegakaryopoiesis, Dysgranulopoiesis, and Increased Blasts
Increased RS were identified in 21 patients (18.9%): 15 patients had 1% to 6% RS, and 6 patients had 17% to 42% RS. In 23 patients (20.7%), dysmegakaryopoiesis was present and characterized predominantly by small megakaryocytes with hypolobulated or nonlobulated nuclei. Dysmegakaryopoiesis grade 1 was diagnosed in 5 patients, grade 2 in 5 patients, and grade 3 in 13 patients. Dysgranulopoiesis was detected in 15 patients (13.5%): grade 1 in 4 patients, grade 2 in 2 patients, and grade 3 in 9 patients. The most common features of neutrophil dysplasia were hypogranular or agranular neutrophils and precursors and hyposegmented (pseudo–Pelger-Huët) neutrophils. Of 9 patients with increased BM blasts, 5 had 1.5% to 3% blasts, 3 had 5% to 7% blasts, and 1 had 18% blasts. One patient who had not received G-CSF had 4% circulating blasts and grade 3 dysgranulopoiesis, but no increase in BM blasts.
Multiple Subtypes of MDS
Of the patients, 23 (20.7%) had MDS, 5 (4.5%) had borderline MDS, and 83 (74.8%) showed insufficient morphologic abnormalities for MDS or borderline MDS and were assigned to the non-MDS category. MDS was subclassified as refractory cytopenia with multilineage dysplasia (RCMD; 13 patients, of whom 3 had more than 15% RS), refractory anemia with excess blasts (RAEB)-1 (4 patients), RAEB-2 (1 patient), refractory anemia with RS (3 patients), and MDS, unclassifiable (2 patients). Morphologic abnormalities identified in non-MDS, borderline MDS, and subclasses of MDS in FA BM samples are illustrated in Image 1.
Image 1.
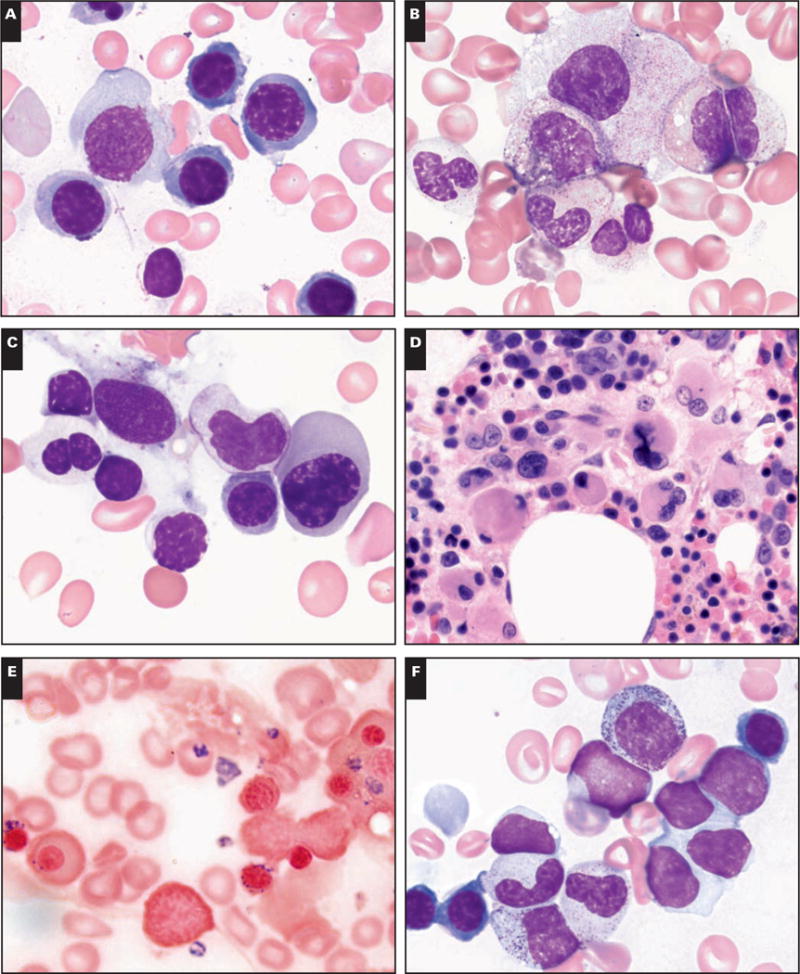
Morphologic abnormalities identified in the bone marrow of patients with Fanconi anemia. A, Rare erythroid precursor with irregular nucleus in non–myelodysplastic syndrome (MDS). B, Rare hypogranular neutrophil and rare megakaryocyte with nonlobated nucleus in borderline MDS. C, Pseudo–Pelger-Huët neutrophil and megaloblastic erythroid precursor with irregular nucleus in refractory cytopenia with multilineage dysplasia (RCMD). D, Numerous small bilobed megakaryocytes in RCMD. E, Increased ringed sideroblasts in refractory anemia with ringed sideroblasts. F, Increased blasts in refractory anemia with excess blasts-1 (A, B, C, E, and F, Wright-Giemsa, ×2,000; D, H&E, ×1,000).
Cytogenetics
Nonrandom Clonal Chromosomal Abnormalities
Clonal abnormalities were present in 36 patients (32.4%). A total of 73 abnormalities were observed, with all but 1 resulting in an unbalanced karyotype with a net loss and/or gain of chromosomal material. The distribution of abnormalities was nonrandom across the genome. Of the patients with abnormal clones, 77.8% had clones with 1qG, 3qG, and/or −7/7qL. These 3 abnormalities accounted for 56% of the total aberrations. Other abnormalities seen in at least 3 patients were 6pL, 11qL, 1pL, 6qL, and 17pL. 3qG most commonly formed part of a derivative chromosome, eg, the der(16) Image 2A or a supernumerary marker Image 2B. FISH or a-CGH was critical in defining the 3qG. 1qG also formed derivative and supernumerary chromosomes; in addition, it presented as an intrachromosomal duplication. 1qG, 3qG, and −7/7qL frequently occurred together in clones.
Image 2.

A, 3qG presenting as part of a derivative chromosome. Left panel, G-banding demonstrates additional material added to the short arm of chromosome 16. Right panel, Fluorescence in situ hybridization (FISH) with a whole chromosome 3 paint probe shows hybridization to the 2 normal No. 3 chromosomes and to the short arm of the abnormal chromosome 16. The abnormal chromosome 16 is designated as der(16)t(3;16)(q25;p13.3). B, G-banding, M-FISH, and array comparative genomic hybridization (CGH) resolve a complex karyotype with 3qG. Left panel, G-banding reveals gain of 2 extra copies of chromosome 1q resulting from a supernumerary t(1;1), an abnormal chromosome 18 with material of unknown origin added to its distal long arm, and a small supernumerary chromosome. Middle panel, Multicolor FISH confirms the gain of chromosome 1q material and further shows that the material added to chromosome 18 is derived from chromosome 9, and the small supernumerary chromosome is derived from chromosome 3. Right panel, CGH reveals the region of 3q in the marker extends frp, 3q21 to the 3q telomere (2 extra copies of this region are present).
Non-MDS vs MDS Groups
The demographic and morphologic differences between the MDS/borderline MDS and non-MDS groups are listed in Table 2. The MDS/borderline MDS group included older patients with a lower average absolute neutrophil count, higher BM cellularity, and more significant dysplasia in each hematopoietic lineage compared with the non-MDS group. There were no significant differences in the male/female ratio and BMF grade (Table 2) or in the number of macro-ovalocytes, anisopoikilocytosis, hemoglobin level, or platelet count between the 2 groups (data not shown).
Table 2.
Demographic and Morphologic Differences Between the MDS/Borderline MDS and Non-MDS Patient Groups*
| MDS/Borderline MDS | Non-MDS | P | |
|---|---|---|---|
| Age range (median), y | 2–44 (12) | 2–21 (9) | <.01 |
| M/F ratio | 1.10 | 1.31 | .54 |
| Absolute neutrophil count | 1.75 | 1.05 | .01 |
| Bone marrow failure | 2.20 | 1.88 | .09 |
| Marrow cellularity | 1.22 | 0.29 | <.01 |
| Dyserythropoiesis | 2.25 | 1.41 | <.01 |
| Ringed sideroblasts | 0.74 | 0.07 | <.01 |
| Dysgranulopoiesis | 1.29 | 0 | <.01 |
| Dysmegakaryopoiesis | 2.08 | 0.05 | <.01 |
| Blasts | 0.50 | 0 | <.01 |
MDS, myelodysplastic syndrome.
Unless stated otherwise, the table includes mean values derived from the scoring systems delineated in “Materials and Methods.” The mean values of the dysplastic features are based on the grading of bone marrow dysplasia.
Clonal Abnormalities and the Presence of MDS
The presence of clonal abnormalities was highly correlated (P < .0001) with the presence of MDS. As shown in Table 3, 22 (95%) of 23 patients with MDS had a clonal abnormality, with only 1 patient with MDS (refractory anemia with RS) having normal cytogenetic findings. Of the 5 patients with borderline MDS, 4 had abnormal clones. The presence of a clone was associated with a 73-fold increase in risk of MDS and with a 26-fold increase in risk of borderline MDS. Of 83 non-MDS patients, cytogenetics was normal in 73 (88%). Among the remaining 10 non-MDS patients (12%), there was an abnormal clone, characterized by a single chromosomal abnormality.
Table 3.
Correlation of Morphologic and Cytogenetic Results in Patients With Fanconi Anemia
| Morphologic Diagnosis | Cytogenetics
|
|
|---|---|---|
| Abnormal | Normal | |
| MDS (n = 23) | 22 (96) | 1 (4) |
| Borderline MDS (n = 5) | 4 (80) 10 (12) | 1 (20) |
| Non-MDS (n = 83) | 73 (88) | |
MDS, myelodysplastic syndrome.
The relationships between MDS and the presence of chromosomal abnormalities differed significantly between different morphologic parameters: 100% of patients with any degree of dysgranulopoiesis or increased blasts had chromosomal abnormalities (P < .01) vs 83% of patients with dysmegakaryopoiesis (P < .01), 71% of patients with RS (P < .01), and 33% of patients with dyserythropoiesis (P = .97) Figure 1. There were no significant associations between a specific chromosomal abnormality (eg, 7qL) and a specific morphologic finding within the borderline MDS/MDS group. Higher karyotype complexity was associated with the presence of MDS. A karyotype with a single abnormality was found in 71 (86%) of 83 non-MDS cases compared with only 18.2% of cases with MDS (P < .001). Twenty patients had 3qG. Nine had 3qG as a sole abnormality: 4 had MDS, 2 had borderline MDS, and 3 were in the non-MDS group. All 11 patients with 3qG in combination with 1 or more other chromosomal abnormalities had MDS.
Figure 1.
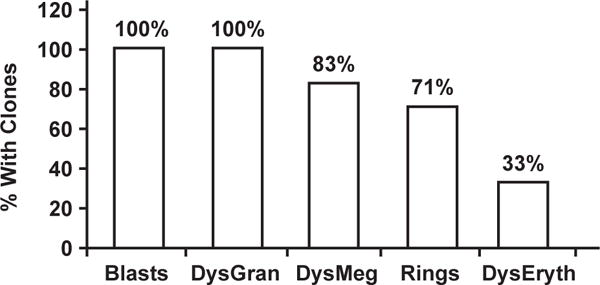
Relationship between different morphologic parameters and the presence of a clonal abnormality in Fanconi anemia (FA). The most reliable morphologic criteria for a diagnosis of myelodysplastic syndrome in FA are increased blasts and dysgranulopoiesis (DysGran), followed by dysmegakaryopoiesis (DysMeg) and increased ringed sideroblasts (Rings). Dyserythropoiesis (DysEryth) does not correlate with the presence of clonal abnormalities.
Discussion
The 10% cell line dysplasia threshold established by the WHO in 2001 for diagnosing MDS has been disputed by some investigators of pediatric MDS who suggest that marked dyspoiesis may be observed in children with non-MDS conditions, including infections, metabolic disorders, vitamin deficiencies, and inherited BM diseases.8,11,19 Consequently, the 2008 WHO classification of MDS was modified for pediatric patients by defining a subtype of refractory cytopenia (RC) for cases with at least 2 of the following: sustained cytopenia, clonal cytogenetic abnormality, and dysplastic changes present in 2 different myeloid cell lines or exceeding 10% in a single cell line.10,20 RC in children differs from that in adults by including predominantly cases of refractory neutropenia and thrombocytopenia rather than anemia and by the frequent presence of marked BM hypocellularity.20
Currently, it is suggested that inherited BMF syndromes cannot be separated from MDS subtypes that do not show an increase in blasts or a clonal abnormality, specifically from some cases of RC.20 Dyserythropoiesis characterized by nuclear/cytoplasmic dyssynchrony, binucleated or multinucleated erythroid cells, and fragmented erythroid nuclei have previously been described in the BM of patients with FA.9,21,22 Our study not only confirms the almost universal finding of dyserythropoiesis in FA BM samples but also identifies the most commonly encountered features and establishes that dyserythropoiesis as the sole abnormality should not be construed as evidence of MDS irrespective of degree. When dyserythropoiesis is accompanied by at least 10% dysplastic cells in 1 or 2 other myeloid cell lines, an unequivocal morphologic diagnosis of MDS (RCMD) can be made in FA BM samples by following the current WHO criteria. Unilineage dysplasia accounting for at least 10% of the line and restricted to the granulocytic or megakaryocytic lineage was a rare subtype of MDS in the current study.
The modified 2008 WHO classification for pediatric MDS provides criteria for the diagnosis of MDS in children with inherited BMF syndromes (“secondary” MDS), including BM hypercellularity in the presence of persistent PB cytopenia, increased BM blast count, or a persistent clonal abnormality.19,20 In the present study, BM cellularity appeared significantly increased in the MDS/borderline MDS group compared with FA BM samples without MDS. However, only 2 patients within the MDS/borderline MDS group showed hypercellular BM samples for age. Thus, BM hypercellularity per se does not appear to be a suitable criterion for MDS in these patients, especially because cytopenias are usually present at diagnosis of FA. Detection of increased BM blasts, although a reliable criterion,23 may be restrictive for diagnosing MDS in patients with FA. Similar to many non-Fanconi-associated cases of pediatric MDS,22 RAEB accounted for only a minority of patients with FA with MDS; RCMD was the most common subtype encountered in our series.
The morphologic evidence of MDS was highly correlated to the presence of clonal abnormalities. The most reliable criteria for diagnosing MDS were increased blasts and dysgranulopoiesis, which correlated with the presence of clonal abnormalities in all cases, followed by dysmegakaryopoiesis and increased RS. The cases defined as borderline MDS on morphologic review also showed high prevalence of clonal abnormalities. Thus, the presence of dysgranulopoiesis or dysmegakaryopoiesis in less than 10% of the line appears significant and supports the proposed criterion of less than 10% bilineage dysplasia for RC of childhood.20 In contrast with previous investigators who expressed concerns regarding the 10% threshold for dysplasia as being too sensitive in the context of an inherited BMF syndrome, we propose this may actually apply only to the erythroid lineage. Detection of an unequivocal but a lesser degree of dysplasia in the granulocytic or megakaryocytic lineage, as well as increased RS of less than 15%, should be correlated with the cytogenetic results and may have a significant impact on patient monitoring and therapeutic decisions.
Overall, gain of 1q, gain of 3q, and loss of 7 or 7q accounted for 56% of the chromosomal abnormalities detected. Although 1qG and −7/7qL have been recognized as frequent recurrent abnormalities in FA in the earlier investigations of FA,12–14 it is noteworthy that 3qG, albeit reported in a few of these earlier studies, has only recently been identified as a frequent and important recurrent cytogenetic finding.16 This is likely because the 3qG occurs most commonly as a small bit of chromosomal material translocated to another chromosome or as a relatively small supernumerary marker chromosome that cannot be definitively resolved by its G-banding pattern. Thus, these 3qG abnormalities were likely previously unrecognized, being cryptically embedded in karyotypes. It is probable that many of the chromosomal abnormalities designated as “add” (additional material of unknown origin) in the earlier French studies13,14 actually represented gains of 3q. The advent of FISH and a-CGH has enabled elucidation of the 3qG abnormalities, with current efforts aimed at identifying critical genes lying within the region of gain. All but 1 case of 3qG in our study encompassed the region 3q25 to the 3q telomere, thus including the EVI1 gene that has been shown to be overexpressed in FA-derived acute myeloid leukemia cell lines.24 The identification of the 3qG is also important because it appears to be an acquired abnormality that is associated with the diagnosis of FA. Although other abnormalities of 3q involving inversion or translocation are well documented in myeloid malignancies from non-FA patients, gain of 3q is only rarely seen in BM from a non-Fanconi patient. Thus, in our institution, on detection of 3qG in the BM in any pediatric patient referred for cytogenetic testing, diepoxybutane/mitomycin C testing is recommended to rule out FA.
Clones with a single abnormality were detected in some non-MDS cases. There were no morphologic indicators predicting a clone in this subgroup. Two critical questions are: Does the presence of a clone necessarily herald the onset of MDS? And if so, what is the latency from the time of appearance to the onset of MDS? Because most patients in this series were referred to our institution for transplantation, we do not have the longitudinal data necessary to address these questions. However, included among the patients with FA in this series were 8 patients showing changes in cytogenetics and/or morphologic status on multiple BM examinations spanning 1 to 5 years. Five patients’ initial samples showed normal cytogenetics and non-MDS but changed over time: 2 developed a clone (1 with 7pL, 1 with 1qG) but no evidence of MDS, 1 developed a clone and borderline MDS, and 2 developed a clone and MDS. Two patients whose initial studies showed a clone and borderline MDS showed expansion or evolution of the clone and progression to MDS. One patient who presented with borderline MDS and with 3qG in 30% of metaphases had a second study 2 years later with expansion of the 3qG clone to 100% of metaphases with persistent borderline MDS. Thus, in 5 of these 8 patients, the change in cytogenetic status was accompanied by a change in hematologic status. In no cases did a clone identified at initial presentation disappear over time.
Acknowledgments
We acknowledge all patients with Fanconi anemia and their families; Richard Brunning, MD, for hematopathology review and critical review of the manuscript; Phuong Nguyen, MD, for efforts in preliminary work on this project; Heather Zierhut for assistance with record review, and the Cytogenetics Laboratory staff for cytogenetic analyses.
Cytogenetic analyses were supported in part by grant P30CA077598-09 from the National Institutes of Health Comprehensive Cancer Center, Bethesda, MD.
Footnotes
Presented in abstract form at the 49th Annual Meeting of the American Society of Hematology; December 8-11, 2007; Atlanta, GA,
References
- 1.Ameziane N, Errami A, Leveille F, et al. Genetic subtyping of Fanconi anemia by comprehensive mutation screening. Hum Mutat. 2008;29:159–166. doi: 10.1002/humu.20625. [DOI] [PubMed] [Google Scholar]
- 2.Bagby GC, Alter BP. Fanconi anemia. Semin Hematol. 2006;43:147–156. doi: 10.1053/j.seminhematol.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 3.Alter BP. Diagnosis, genetics, and management of inherited bone marrow failure syndromes. Hematology Am Soc Hematol Educ Program. 2007:29–39. doi: 10.1182/asheducation-2007.1.29. [DOI] [PubMed] [Google Scholar]
- 4.Bagby GC, Lipton JM, Sloand EM, et al. Marrow failure. Hematology Am Soc Hematol Educ Program. 2004:318–336. doi: 10.1182/asheducation-2004.1.318. [DOI] [PubMed] [Google Scholar]
- 5.Auerbach AD, Allen RG. Leukemia and preleukemia in Fanconi anemia patients: a review of the literature and report of the International Fanconi Anemia Registry. Cancer Genet Cytogenet. 1991;51:1–12. doi: 10.1016/0165-4608(91)90002-c. [DOI] [PubMed] [Google Scholar]
- 6.Alter BP. Cancer in Fanconi anemia, 1927–2001. Cancer. 2003;97:425–440. doi: 10.1002/cncr.11046. [DOI] [PubMed] [Google Scholar]
- 7.Kutler DI, Singh B, Satagopan J, et al. A 20-year perspective on the International Fanconi Anemia Registry (IFAR) Blood. 2003;101:1249–1256. doi: 10.1182/blood-2002-07-2170. [DOI] [PubMed] [Google Scholar]
- 8.Alter BP, Caruso JP, Drachtman RA, et al. Fanconi anemia: myelodysplasia as a predictor of outcome. Cancer Genet Cytogenet. 2000;117:125–131. doi: 10.1016/s0165-4608(99)00159-4. [DOI] [PubMed] [Google Scholar]
- 9.Owen J, Frohnmayer L, Eiler ME. Fanconi Anemia: Standards for Clinical Care. 2nd. Eugene, OR: Fanconi Anemia Research Fund; 2003. [Google Scholar]
- 10.Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th. Vol. 2 Lyon, France: IARC Press; 2008. WHO Classification of Tumours. [Google Scholar]
- 11.Cantu Rajnoldi A, Fenu S, Kerndrup G, et al. Evaluation of dysplastic features in myelodysplastic syndromes: experience from the morphology group of the European Working Group of MDS in Childhood (EWOG-MDS) Ann Hematol. 2005;84:429–433. doi: 10.1007/s00277-005-1034-4. [DOI] [PubMed] [Google Scholar]
- 12.Berger R, Bernheim A, Le Coniat M, et al. Chromosomal studies of leukemic and preleukemic Fanconi’s anemia patients: examples of acquired “chromosomal amplification”. Hum Genet. 1980;56:59–62. doi: 10.1007/BF00281569. [DOI] [PubMed] [Google Scholar]
- 13.Berger R, Jonveaux P. Clonal chromosome abnormalities in Fanconi anemia. Hematol Cell Ther. 1996;38:291–296. doi: 10.1007/s00282-996-0291-6. [DOI] [PubMed] [Google Scholar]
- 14.Maarek O, Jonveaux P, Le Coniat M, et al. Fanconi anemia and bone marrow clonal chromosome abnormalities. Leukemia. 1996;10:1700–1704. [PubMed] [Google Scholar]
- 15.Alter BP, Scalise A, McCombs J, et al. Clonal chromosomal abnormalities in Fanconi’s anaemia: what do they really mean? Br J Haematol. 1993;85:627–630. doi: 10.1111/j.1365-2141.1993.tb03362.x. [DOI] [PubMed] [Google Scholar]
- 16.Tonnies H, Huber S, Kuhl JS, et al. Clonal chromosomal aberrations in bone marrow cells of Fanconi anemia patients: gains of the chromosomal segment 3q26q29 as an adverse risk factor. Blood. 2003;101:3872–3874. doi: 10.1182/blood-2002-10-3243. [DOI] [PubMed] [Google Scholar]
- 17.Hartsock RJ, Smith EB, Petty CS. Normal variations with aging of the amount of hematopoietic tissue in bone marrow from the anterior iliac crest: a study made from 177 cases of sudden death examined by necropsy. Am J Clin Pathol. 1965;43:326–331. doi: 10.1093/ajcp/43.4.326. [DOI] [PubMed] [Google Scholar]
- 18.Shaffer LG, Tommerup N, editors. ISCN 2005: An International System for Human Cytogenetic Nomenclature (2005) Basel, Switzerland: Karger; 2005. [Google Scholar]
- 19.Hasle H, Niemeyer CM, Chessells JM, et al. A pediatric approach to the WHO classification of myelodysplastic and myeloproliferative diseases. Leukemia. 2003;17:277–282. doi: 10.1038/sj.leu.2402765. [DOI] [PubMed] [Google Scholar]
- 20.Niemeyer CM, Baumann I. Myelodysplastic syndrome in children and adolescents. Semin Hematol. 2008;45:60–70. doi: 10.1053/j.seminhematol.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 21.Guinan EC, Shimamura A. Acquired and inherited aplastic anemia syndromes. In: Greer JP, Foerster J, Lukens JN, editors. Wintrobe’s Clinical Hematology. 11th. Vol. 1. Philadelphia, PA: Lippincott Williams & Wilkins; 2003. pp. 1409–1412. [Google Scholar]
- 22.Elghetany MT. Myelodysplastic syndromes in children: a critical review of issues in the diagnosis and classification of 887 cases from 13 published series. Arch Pathol Lab Med. 2007;131:1110–1116. doi: 10.5858/2007-131-1110-MSICAC. [DOI] [PubMed] [Google Scholar]
- 23.Faivre L, Guardiola P, Lewis C, et al. for the European Fanconi Anemia Research Group Association of complementation group and mutation type with clinical outcome in Fanconi anemia. Blood. 2000;96:4064–4070. [PubMed] [Google Scholar]
- 24.Meyer S, Fergusson WD, Whetton AD, et al. Amplification and translocation of 3q26 with overexpression of EVI1 in Fanconi anemia–derived childhood acute myeloid leukemia with biallelic FANCD1/BRCA2 disruption. Genes Chromosomes Cancer. 2007;46:359–372. doi: 10.1002/gcc.20417. [DOI] [PubMed] [Google Scholar]