

SUMMARY
Pyroptosis is a lytic form of programmed cell death mediated by the inflammatory caspases-1, -4, and -5. We recently discovered that small molecule inhibitors of the serine peptidases DPP8 and DPP9 (DPP8/9) induce pro-caspase-1 dependent pyroptosis in monocytes and macrophages. Notably, DPP8/9 inhibitors, unlike microbial agents, absolutely require caspase-1 to induce cell death. Therefore, DPP8/9 inhibitors are useful probes to study caspase-1 in cells. Here we show that, in the absence the pyroptosis-mediating substrate gasdermin D (GSDMD), caspase-1 activates caspases-3 and -7 and induces apoptosis, demonstrating that GSDMD is the only caspase-1 substrate that induces pyroptosis. Conversely, we found that, during apoptosis, caspases-3/7 specifically block pyroptosis by cleaving GSDMD at a distinct site from the inflammatory caspases that inactivates the protein. Overall, this work reveals bidirectional crosstalk between apoptosis and pyroptosis in monocytes and macrophages, further illuminating the complex interplay between cell death pathways in the innate immune system.
Keywords: pyroptosis, apoptosis, cell death, monocytes, macrophages, DPP8/9 inhibitors, gasdermin D, caspase-1, caspase-3, caspase-7
Graphical Abstract
INTRODUCTION
The inflammatory caspases-1, -4, and -5 are key effectors of the innate immune response against infectious organisms (Broz and Dixit, 2016; Guo, et al., 2015; Lamkanfi and Dixit, 2014). When activated, each of these enzymes is able to induce a form of programmed cell death known as pyroptosis. Pyroptosis, unlike apoptosis, involves cell swelling and pore formation in the plasma membrane, ultimately resulting in rupture of the membrane and the release of cytosolic contents into the extracellular space (Bergsbaken, et al., 2009). Pyroptosis is thought to play a key role in the clearance of infectious agents by releasing surviving intracellular bacteria for neutrophil-mediated killing, exposing pathogenic antigens to the adaptive immune system, and secreting cytokines and eicosanoids to promote inflammatory and repair responses and further stimulate adaptive immunity (Aachoui, et al., 2013; Broz and Dixit, 2016; Jorgensen, et al., 2016; Miao, et al., 2010).
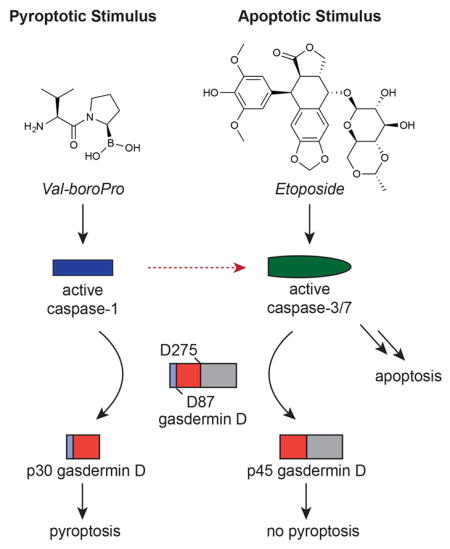
Pyroptosis mediated by caspase-1, often referred to as canonical pyroptosis, occurs in monocytes, macrophages, and dendritic cells. A remarkable variety of microbial structures and activities, including bacterial flagellin, double stranded DNA, anthrax lethal toxin, and bacterial modification of host Rho GTPases, trigger canonical pyroptosis (Broz and Dixit, 2016; Lamkanfi and Dixit, 2014). In each case, a central scaffold, for example NAIP-NLRP4 (Zhao, et al., 2011), AIM2 (Hornung, et al., 2009), NLPR1 (Boyden and Dietrich, 2006), and Pyrin (Xu, et al., 2014), respectively, for the stimuli listed above, detects its cognate ligand and then oligomerizes with the adapter protein ASC to form large multiprotein complexes called canonical inflammasomes. These inflammasomes then recruit and induce the autoproteolytic maturation and activation of caspase-1, which in turn cleaves and activates the pyroptotic substrate gasdermin D (GSDMD) and the inflammatory cytokines IL-1β and IL-18 (Kayagaki, et al., 2015; Shi, et al., 2015; Thornberry, et al., 1992). Although caspase-1 is usually activated by this general mechanism, two notable exceptions exist. First, in the absence of ASC, the NAIP-NLRC4 and Nlrp1b scaffolds can directly activate pro-caspase-1 without inflammasome complex formation or autoproteolysis (Broz, et al., 2010; Guey, et al., 2014; Van Opdenbosch, et al., 2014). Second, small molecule inhibitors of the cytosolic host serine proteases DPP8 and DPP9, including Val-boroPro (Figure 1A) and L-allo-Ile-isoindoline, can induce pyroptosis via activation of pro-caspase-1 without autoproteolysis through a still unknown mechanism (Okondo, et al., 2017). In both cases, activated pro-caspase-1 cleaves GSDMD and induces pyroptosis, but does not efficiently cleave IL-1β or IL-18.
Figure 1. Val-boroPro induces apoptosis in Gsdmd−/− macrophages. See also Fig. S1 and Movies S1–4.

(A) The structure of Val-boroPro. (B) Confirmation of Gsdmd knockout (KO) in RAW 264.7 macrophages by immunoblotting. An asterisk (*) indicates a background band. (C) LDH release from Gsdmd−/− RAW 264.7 cells treated with Val-boroPro was delayed, but not prevented. Caspase-1 is required for Val-boroPro-induced cell death. Data are means ± SEM of 3 independent experiments. *** p < 0.001 for GFP versus Gsdmd KO cells. (D) Static brightfield images of control and Gsdmd KO1 RAW 264.7 cells treated with Val-boroPro or etoposide. Arrows indicate dying cells. The images are representative of four randomly selected fields. (E) Immunoblotting reveals Parp cleavage in Gsdmd−/−, but not in control or Casp−/−, RAW 264.7 cells treated with Val-boroPro. As expected, etoposide induced Parp cleavage in all cells tested here. VbP, Val-boroPro; Etop., etoposide; FL, full-length; CL, cleaved.
In contrast to canonical pyroptosis, a non-canonical form of pyroptosis mediated by caspases -4 and -5, occurs in a variety of cell types (Kayagaki, et al., 2011; Shi, et al., 2014). In non-canonical pyroptosis, the direct recognition of bacterial lipopolysaccharide (LPS) by caspases-4 or -5 induces their activation (Hagar, et al., 2013; Kayagaki, et al., 2013; Shi, et al., 2014). Like activated pro-caspase-1 induced by DPP8/9 inhibitors, LPS-activated caspases-4/5 directly cleave GSDMD to induce pyroptosis, but do not cleave the inflammatory cytokines IL-1β and IL-18.
The N-terminal p30 fragment of GSDMD released by the inflammatory caspases forms pores in the plasma membrane, thereby mediating pyroptotic cell death (Ding, et al., 2016; Liu, et al., 2016; Sborgi, et al., 2016). In the absence of GSDMD, cells are completely resistant to intracellular LPS and caspase-4/5-mediated non-canonical pyroptosis. Whether GSDMD is required in canonical caspase-1-dependent cell death, however, is less clear, as the initial studies reporting the discovery of GSDMD differed as to whether canonical pyroptosis was entirely blocked (Shi, et al., 2015) or merely delayed (Kayagaki, et al., 2015) in Gsdmd−/− macrophages. A subsequent report showed that Gsdmd−/− macrophages treated with LPS plus nigericin or S. typhimurium do indeed undergo cell death, but the death was delayed and more closely resembled apoptosis than pyroptosis (He, et al., 2015). Intriguingly, caspase-1 appeared to not be required for this delayed cell death, as the delayed apoptotic-like response was also observed in Casp1−/− macrophages.
Unlike the cell death responses to these microbial stimuli, programmed cell death induced by DPP8/9 inhibitors is entirely dependent on caspase-1 (Okondo, et al., 2017). As such, DPP8/9 inhibitors can serve as useful tools to exclusively activate caspase-1 in cells. We previously showed that GSDMD−/− human THP-1 monocytes also undergo delayed cell death after treatment with the inhibitor Val-boroPro, indicating that GSDMD is not required for this delayed, but caspase-1-dependent cell death. However, we did not establish whether this delayed cell death was pyroptotic or apoptotic. Here, we show that it is apoptotic and, moreover, that it is mediated via the activation of caspases-3/7. These results confirm that caspase-1 itself can indeed induce apoptosis and indicate that no additional pyroptosis-inducing substrate of caspase-1 exists. We also report that the communication between the apoptotic and pyroptotic caspases is bidirectional, as we discovered that the apoptotic caspases-3/7 cleave GSDMD at a distinct site from the pyroptotic caspases during apoptosis to eliminate the ability of cells to undergo pyroptosis. Overall, these data show new regulatory mechanisms in monocyte and macrophage cell death, highlighting the complex interplay between apoptotic and pyroptotic signaling pathways.
RESULTS
Val-boroPro induces apoptosis in GSDMD knockout monocytes and macrophages
We previously showed that knockout of GSDMD delayed, but did not entirely prevent, DPP8/9 inhibitor-induced cell death in human THP-1 macrophages (Okondo, et al., 2017). To confirm that Gsdmd is similarly not essential for DPP8/9 inhibitor-induced cell death in mouse macrophages, we generated several Gsdmd−/− RAW 264.7 macrophage cell lines using CRISPR/Cas9 gene editing technology (Fig. 1B). Consistent with the results observed using THP-1 monocytes, Val-boroPro-induced cell death in Gsdmd−/− RAW 264.7 mouse macrophages was significantly delayed relative to control cells (Fig. 1C). As expected, Casp1−/− RAW 264.7 macrophages were completely resistant to Val-boroPro (Fig. 1C, Fig. S1A). These results demonstrate that both human and mouse caspase-1 cleave a substrate (or substrates) in addition to GSDMD that mediates cell death, and that this process occurs with slower kinetics than GSDMD-mediated pyroptosis.
We next wanted to determine whether this delayed cell death exhibited pyroptotic-like or apoptotic-like features. For an initial assessment, we imaged control and Gsdmd−/− RAW 264.7 cells treated with Val-boroPro or etoposide, which induces apoptosis by inhibiting DNA topoisomerase II. As expected, Val-boroPro-treated control RAW 264.7 cells underwent pyroptosis, which is lytic and does not involve membrane blebbing (Fig. 1D, Movie S1). In stark contrast, Val-boroPro-treated Gsdmd−/− RAW 264.7 cells exhibited apoptotic-like morphological features, including membrane blebbing, closely resembling etoposide-treated apoptotic cells (Fig. 1D, Movies S2–4). We next probed lysates from these cells for cleavage of the apoptotic caspase substrate Parp (Fig. 1E). As expected, control and Casp1−/− RAW 264.7 cells treated with DMSO or Val-boroPro showed no Parp cleavage. However, Gsdmd−/− RAW 264.7 cells treated with Val-boroPro, like cells treated with etoposide, exhibited significant Parp cleavage (Fig. 1E), consistent with their apoptotic morphology.
We next performed a closer analysis of the delayed cell death response in GSDMD−/− human THP-1 monocytes. Similar to the results in RAW 264.7 macrophages, we observed that Val-boroPro treatment of GSDMD−/− THP-1 cells (Okondo, et al., 2017), but not control or CASP1−/− cells, induced PARP cleavage (Fig. 2A, Fig. S1B) and an apoptotic morphology (Fig. S2A). Moreover, we observed a significant population of Val-boroPro-treated GSDMD−/− THP-1 cells, unlike control cells, stained positive for Annexin V and negative for propidium iodide (PI), consistent with apoptosis and not pyroptosis (Fig. 2B, Fig. S2B,C). Therefore, in monocytes and macrophages lacking GSDMD, Val-boroPro induces caspase-1-dependent apoptosis. It should be noted that, although LDH release is a hallmark of lytic cell death, LDH release in Val-boroPro-treated Gsdmd−/− cells (Fig. 1C) is likely due to secondary necrosis of apoptotic cells (Silva, 2010).
Figure 2. Val-boroPro activates caspases-3/7 in GSDMD knockout cells. See also Figs. S1-4.

(A) Immunoblotting reveals PARP cleavage in GSDMD−/−, but not in control or CASP−/−, THP-1 cells treated with Val-boroPro. In control cells, Val-boroPro induces the cleavage of GSDMD into the pyroptotic p30 fragment. Etoposide unexpectedly triggered the formation of a p43 fragment of GSDMD in control and CASP−/− cells. (B) Val-boroPro induces Annexin V positive/propidium iodide (PI) negative staining in GSDMD−/− THP-1 cells. Data are means ± SEM of 3 independent experiments. *** p < 0.001 compared to DMSO treated cells. (C–E) Effects of Val-boroPro, LPS plus nigericin, and etoposide on PARP and GSDMD in wild-type (C), GSDMD KO1 (D), and CASP1 KO1 (E) THP-1 cells as determined by immunoblotting. Nig, nigericin. (F) Val-boroPro induces cleavage of caspases-3 and -7 in GSDMD knockout cells. (G) Caspase-3/7 activity is elevated in GSMD KO THP-1 cells treated with Val-boroPro. Data are means ± SEM of 3 independent experiments. *** p < 0.001 compared to DMSO treated cells.
We next wanted to compare the cellular response induced by Val-boroPro to the response induced by LPS plus nigericin, which triggers pyroptosis by activating the NLRP3 inflammasome. As expected, treatment of THP-1 monocytes with Val-boroPro or LPS plus nigericin induced pyroptosis, as evidenced by the appearance of the p30 GSDMD fragment (Fig. 2C). It should be noted that LPS plus nigericin also induced some apoptosis in these cells, as evidenced by the appearance of cleaved PARP. In the absence of GSDMD, both LPS plus nigericin and Val-boroPro induced apoptosis (Fig. 2D). However, LPS plus nigericin, unlike Val-boroPro, also induced apoptosis in CASP1−/− THP-1 cells (Fig. 2E), consistent with previous reports that caspase-1 is not required for NLRP3 inflammasome-mediated cell death (He, et al., 2015; Lamkanfi, et al., 2008).
We next asked which apoptotic caspase (or caspases), if any, was activated in caspase-1-dependent-apoptosis induced by Val-boroPro. In GSDMD−/− knockout cell lines treated with Val-boroPro, we observed significant levels of the cleaved, active forms of caspases-3 and -7 (Fig. 2F,G, Fig. S3). Purified active caspase-1 was previously shown to directly cleave caspases-3/7 in vitro (Van de Craen, et al., 1999), and we confirmed that recombinant active caspase-1 indeed cleaves caspases-3 and 7 when added to THP-1 lysates (Fig. S4). We therefore speculate that active caspase-1 directly cleaves caspases-3/7 to trigger apoptosis, although direct in vivo cleavage remains to be definitively demonstrated. Collectively, these data suggest that caspases-3/7 mediate the caspase-1-dependent apoptotic cell death response.
GSDMD is cleaved at an alternate site during apoptosis
Intriguingly, we observed that a pronounced ~43 kDa fragment of GSDMD consistently appeared in THP-1 cells treated with etoposide (Fig. 2A,C). This p43 GSDMD fragment was not generated by caspase-1, as this band also appeared in caspase-1 knockout THP-1 cells treated with etoposide (Fig. 2A). Moreover, we observed this p43 GSDMD fragment in CASP1−/− THP-1 cells treated with LPS plus nigericin (Fig. 2E), which, like etoposide-treated cells, were undergoing at least some apoptosis. This data suggested that GSDMD is cleaved during apoptosis at a site distinct from the D275 site cleaved by the inflammatory caspases during pyroptosis that generates the p30 fragment.
To confirm that the generation of this p43 band was not specific to etoposide and LPS plus nigericin, we next treated THP-1 monocytes with three additional apoptosis-inducing agents, bortezomib, cycloheximide, and staurosporine, which induce cell death via proteasome inhibition, translation inhibition, and nonspecific kinase inhibition, respectively (Fig. 3A). In each case, we saw PARP cleavage, confirming that apoptosis was induced, as well as the appearance of the p43, but not the p30, GSDMD species. Intriguingly, LPS plus nigericin, which we showed induces both apoptosis and pyroptosis in monocytes (Fig. 2C), induces the formation of both the p30 and p43 GSDMD fragments (Fig. 3A). Collectively, these data indicate that cleavage of GSDMD into the p43 fragment is a general feature of apoptosis in cells expressing GSDMD.
Figure 3. Caspases-3/7 cleave GSDMD into p43 fragment during apoptosis.

(A) Immunoblotting of lysates from THP-1 cells treated with the indicated apoptotic stimuli reveals GSDMD cleavage into a p43 fragment. (B,C) Lysates from HEK 293T cells expressing C-terminally tagged human (B) or mouse (C) GSDMD were incubated (2 h, 37 °C) with the indicated recombinant caspases and cleavage was evaluated by immunoblotting.
Caspases-3/7 cleave GSDMD at distinct site from caspases-1/4/5
We speculated that one or more apoptotic caspase might be cleaving GSDMD into the p43 fragment during apoptotic cell death. To establish which caspase, if any, was responsible, we harvested lysates from HEK 293T cells ectopically expressing C-terminally V5-tagged human GSDMD and treated these lysates with various active, purified caspases. As expected, caspase-1 cleaved GSDMD at residue 275 into the p22 and p30 fragments (Fig. 3B) (Kayagaki, et al., 2015; Shi, et al., 2015). Importantly, we were able to visualize both fragments because the V5 antibody recognized the C-terminal V5-containing p22 fragment, and the GSDMD antibody, which was raised against an epitope containing residues 1–200, recognized the N-terminal p30 fragment. Strikingly, we observed that caspase-3, and to a lesser extent caspase-7, exclusively generated the p43 fragment (Fig. 3B). Caspases-8 and -9 did not significantly cleave GSDMD, consistent with previous results (Shi, et al., 2015). Moreover, because both the V5 and GSDMD antibodies detected the p43 fragment, this data indicated that the p43 cleavage site must be N-terminal to D275.
To confirm that this alternative GSDMD cleavage site was conserved in other species, we performed an analogous experiment with mouse Gsdmd expressed with a C-terminal Myc and Flag tags (Fig. 3C). In this analysis, we similarly observed that caspase-3 and -7, but not caspases-8 and -9, cleaved mouse Gsdmd at a site distinct from the canonical caspase-1 cleavage site.
Caspases-3/7 cleave GSDMD at Asp87
Only one apoptotic caspase cleavage motif (DXXD) is both N-terminal to D275 and evolutionarily conserved (Fig. 4A). In addition, cleavage at this site (D87 in humans) is predicted to produce a fragment ~43 kDa in size (Fig. 4B). We therefore hypothesized that caspases-3/7 likely cleave GSDMD after this residue. To determine if this is the specific cleavage site, the 43 kDa fragment was isolated and subjected to Edman degradation. As expected, this analysis revealed that the N-terminal residues of this fragment were indeed 88GQIQGSV (Table S1). We next mutated the aspartic acid at position 87 to alanine, and isolated lysates from HEK 293T cells expressing this mutant protein. While caspase-1 was still able to cleave GSDMD D87A at position 275 to generate the p30 and p22 fragments, caspases-3 and -7 now failed to generate the p43 fragment (Fig. 4C). Therefore, D87 is the caspase-3/7 cleavage site.
Figure 4. Apoptotic caspase cleavage at D87 inactivates GSDMD.

(A) A predicted caspase-3 cleavage site in GSDMD is conserved across the indicated species. (B) Schematic of GSDMD cleavage fragments. (C) Lysates from HEK 293T cells expressing C-terminally tagged human GSDMD D87A were incubated with recombinant caspases (2 h, 37 °C). While caspase-1 still cleaves this mutant protein at D275, caspases-3/7 cleavage was completely abolished. (D–G) HEK 293T cells were transiently transfected with the indicated GSDMD fragments. Each fragment possessed a C-terminal V5 tag. Expression of the fragment was evaluated by immunoblotting (D,F) and cell death was evaluated using an LDH assay (E,G). Data are means ± SEM of 3 independent experiments. *** p < 0.001 compared to GFP expressing cells.
Cleavage at D87 inactivates GSDMD
We next wanted to determine the effect of cleavage at position D87 on the function of GSDMD. Consistent with previous results, ectopic expression of GSDMD residues 1–275 in HEK 293T cells results in cell death (Fig. 4D,E) (Shi, et al., 2015). It should be noted that we found it difficult to see this species by immunoblotting (Fig. 4D), as others have previously (He, et al., 2015; Kayagaki, et al., 2015), likely because it is highly cytotoxic. In contrast, expression of GSDMD residues 1–87 or GSDMD 88–484 did not result in increased cell death relative to the GFP control (Fig. 4E), demonstrating that the fragments generated by D87 cleavage are not inherently cytotoxic. Moreover, expression of GSDMD 88–275, which would be formed if the p43 fragment were further processed by caspases-1/4/5, is also non-toxic (Fig. 4F,G). Taken together, our data indicates that cleavage of GSDMD at D87 inactivates its ability to induce pyroptosis, and suggests that cells specifically block pyroptotic cell death during apoptosis.
DISCUSSION
Before this study, a major unresolved question was whether or not GSDMD was an essential mediator of caspase-1-induced pyroptosis (Broz, 2015). Although GSDMD knockout completely eliminated caspase-4/5-induced pyroptosis, caspase-1-induced pyroptosis appeared to be merely delayed (Kayagaki, et al., 2015; Shi, et al., 2015). This result raised at least three, not necessarily mutually exclusive, possibilities: 1) that caspase-1 cleaves an additional substrate that mediates pyroptosis, 2) that caspase-1 activity can trigger an alternative cell death pathway, or 3) the activated inflammasomes (or some other upstream mediator) can directly activate an alternative cell death protease. In support of the latter two possibilities, previous studies had demonstrated that LPS plus ATP, S. typhimurium, and L. pneumophila activate caspases-3/7 in addition to caspase-1 in macrophages (Akhter, et al., 2009; Lamkanfi, et al., 2008), suggesting that these agents could quite likely also induce apoptosis. However, these studies did not confirm that these stimuli do in fact induce apoptosis in the absence of GSDMD, which had not yet been identified as the key pyroptotic substrate. Moreover, it appeared that caspase-1 was only required for the activation of caspase-7, but not caspase-3, leaving it unclear whether apoptosis, if it occurred, would depend on caspase-1.
Consistent with these reports, a recent study showed that LPS plus nigericin or S. typhimurium-treated Gsdmd−/− mouse macrophages seemed to undergo apoptosis (He, et al., 2015). Here, we confirmed that LPS plus nigericin indeed induced apoptosis in the absence of GSDMD in THP-1 monocytes (Fig. 2D, Fig. S3A), and we even observed that LPS plus nigericin induced some apoptosis in wild-type cells (Fig. 2C). Interestingly, the apoptotic response to LPS plus nigericin does not require caspase-1 (Fig. 2E), but it does require the inflammasome sensor protein NLRP3 (He, et al., 2015). Thus, inflammasomes can activate apoptosis independent of caspase-1.
In striking contrast, Val-boroPro-induced cell death, whether pyroptotic or apoptotic, absolutely requires caspase-1. Here, we exploited this dependency to investigate the role of caspase-1 in GSDMD knockout cells, and discovered that Val-boroPro induces caspase-1-dependent apoptosis in these cells via the activation of caspases-3 and -7. As purified caspase-1 can cleave pro-caspase-3 and -7 (Lamkanfi, et al., 2008; Van de Craen, et al., 1999), it is likely caspase-1 is directly cleaving these executioner caspases. As noted above, a previous study found that the microbial agent activation of caspase-7, but not caspase-3, required caspase-1. Based on this data, these authors proposed that a distinct inflammasome/caspase-1/caspase-7 activation pathway might exist that does not activate caspase-3 (Lamkanfi, et al., 2008). However, because caspase-3 was activated in these cells by proteins upstream of caspase-1, that caspase-1 is also capable of activating caspase-3 could not be ruled out. Our data now prove that caspase-3 can be activated by both a caspase-1-dependent mechanism and a caspase-1-independent mechanism, and therefore the inflammasome/caspase-1/caspase-7 activation pathway is not specific to caspase-7. More generally, these data show that at least two distinct mechanisms, activated caspase-1 and activated inflammasomes, can induce apoptosis in monocytes and macrophages in the absence of GSDMD. As we found no evidence of any lytic, pyroptotic-like cell death occurring in GSDMD knockout monocytes and macrophages treated with Val-boroPro, it is unlikely an additional pyroptosis-inducing substrate of caspase-1 exists. We speculate that the inflammasomes and caspase-1 activate apoptosis in order to ensure that the infected cell is eliminated. On that note, the activation of these executioner caspases does appear to be important, as caspase-7 has been shown to play a role in restricting the growth on L. pneumophila in mice (Akhter, et al., 2009).
During the course of this study, we also observed the appearance of a new GSDMD fragment in monocytes treated with several apoptosis inducing small molecules, including etoposide, bortezomib, and cycloheximide. We found that caspase-3, and to a lesser degree caspase-7, cleaves GSDMD after D87. This ‘apoptotic cleavage site’ is evolutionarily conserved and lies within the p30 fragment of GSDMD that forms membrane pores. Not surprisingly, proteolysis at this site renders GSDMD incapable of activating pyroptosis. We speculate that GSDMD is inactivated by the executioner apoptotic caspases in order to ensure that an apoptotic cell does not accidentally trigger cell lysis. Consistent with this premise, caspases-3/7 are unable to cleave and activate the inflammatory caspases-1 and -4 (Van de Craen, et al., 1999). Therefore, not only do caspases-3/7 avoid triggering pyroptosis by not proteolytically activating the inflammatory caspases, but they also inactivate the key substrate of the inflammatory caspases that mediates cell lysis.
One particularly intriguing insight from this study is that the canonical inflammasomes and caspase-1 activate caspases-3 and -7, which are precisely the enzymes that inactivate GSDMD and block pyroptosis. It may seem counterintuitive for the pyroptotic machinery to activate a pathway that downregulates pyroptosis. However, the activation of caspases-3/7 occurs more slowly than the cleavage of GSDMD and activation of pyroptosis. We therefore hypothesize that the activation of caspase-3/7 by the inflammasomes and caspase-1 might not only serve the purpose of ensuring that the infected cell is eliminated, as suggested above, but it may also serve as a counter regulatory feedback mechanism to dampen pyroptosis and inflammation.
In summary, we now propose a bidirectional model of crosstalk between pyroptosis and apoptosis in monocytes and macrophages (Fig. 5). In particular, we have now unequivocally demonstrated that pyroptotic stimuli can also activate apoptosis by multiple mechanisms, but conversely that apoptotic stimuli specifically avoid and even inactivate pyroptosis. In addition to these biological insights, this work demonstrates the utility of Val-boroPro as a tool to activate caspase-1 in monocytes and macrophages. We expect that additional studies using this molecule will continue to uncover new and surprising aspects of the innate immune response to microbial pathogens.
Figure 5. Apoptosis and pyroptosis interplay in monocytes and macrophages.
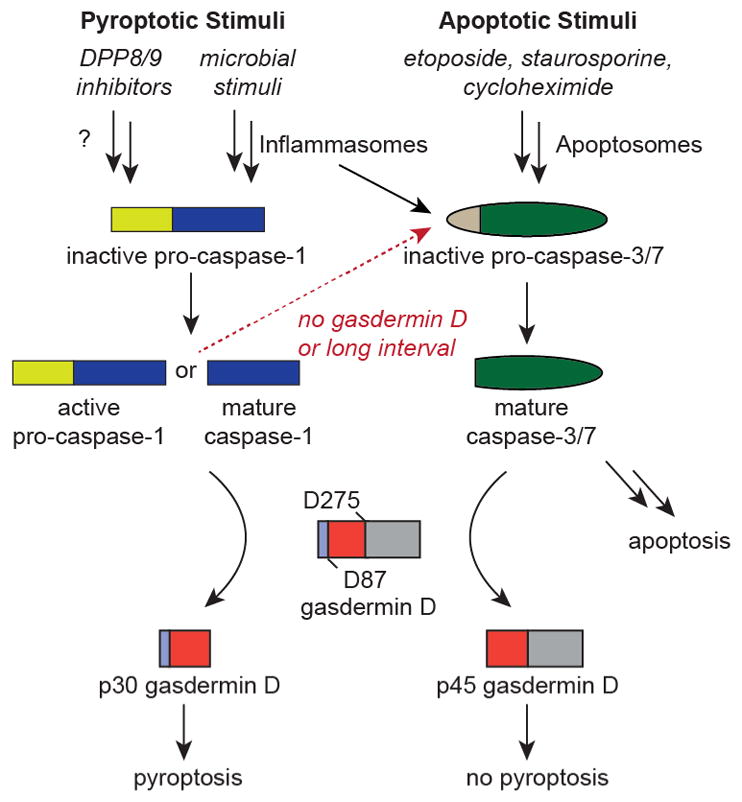
Schematic depicting the bidirectional crosstalk between the apoptotic and pyroptotic cell death pathways in monocytes and macrophages. The dashed red arrow indicates that caspase-1 activates caspase-3/7 in cells, but that it is not yet definitively established if this is due to direct or indirect cleavage in vivo.
SIGNIFICANCE
Caspase-1 plays a central role in the innate immune response to microbial infections by activating inflammatory cytokines and mediating a lytic form of cell death known as pyroptosis. We recently discovered that small molecule inhibitors of DPP8/9, including Val-boroPro L-allo-Ile-isoindoline, induce pro-caspase-1-dependent pyroptosis selectively in monocytes and macrophages. Unlike microbial agents that induce caspase-1 dependent pyroptosis, DPP8/9 inhibitors absolutely require caspase-1 to mediate cell death. Here, we demonstrated the utility of DPP8/9 inhibitors as tools to selectively activate caspase-1 in cells. We first showed that active caspase-1 induces apoptosis in the absence of GSDMD, confirming that GSDMD is indeed the only pyroptosis-inducing substrate of caspase-1 in these cells. Second, we demonstrated that caspase-1 activates both caspases-3 and -7 to mediate this apoptotic cell death. Finally, we also discovered that, during apoptosis, caspases-3 and -7 cleave GSDMD at a distinct site from the inflammatory caspases. Caspase-3/7 cleavage of GSDMD renders the protein inactive and unable to induce pyroptosis, showing that apoptotic cells specifically disable lytic cell death. Overall, our data unequivocally demonstrates a role of caspase-1 in apoptosis and reveals previously unknown bidirectional interplay between apoptotic and pyroptotic pathways in innate immune cells.
STAR METHODS
KEY RESOURCES TABLE
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Daniel Bachovchin (bachovcd@mskcc.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
HEK 293T and RAW 264.7 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS). THP-1 cells were grown in RPMI-1640 medium with 10% FBS. All cells were grown at 37 °C in a 5% CO2 incubator. Cell lines were tested for mycoplasma using the MycoAlertTM Mycoplasma Detection Kit (Lonza).
METHODS DETAILS
Cloning
cDNA coding for full length human GSDMD (Dharmacon), truncated human GSDMD variants, and GFP (Yang, et al., 2011) were cloned with a C-terminal V5 tag into pLEX_307 vector (Addgene) using Gateway technology (Thermo Fisher Scientific). The single amino acid point mutation D87A was generated with the QuickChange site-directed mutagenesis kit (Agilent). cDNA for mouse Gsdmd containing C-terminal Myc and Flag tags in the pCMV6-Entry vector was purchased from Origene. sgRNAs were designed using the Broad Institute’s web portal (Doench, et al., 2016) (http://www.broadinstitute.org/rnai/public/analysis-tools/sgrna-design) and cloned into the lentiGuide-Puro vector (Addgene #52963) as described previously (Sanjana, et al., 2014). The sgRNA sequences used were: hCASP1 5′-CTAAACAGACAAGGTCCTGA-3′, mCasp1 5′-TTAAACAGACAAGATCCTGA-3′, hGSDMD 5′-TGAGTGTGGACCCTAACACC-3′, mGsdmd 5′-AGGTTGACACATGAATAACG-3′, and GFP 5′-GGGCGAGGAGCTGTTCACCG-3′ All plasmids were verified by DNA sequencing (Genewiz).
Knockout cell lines
Constructs were packaged into lentivirus in HEK 293T cells using the Fugene HD transfection reagent (Promega) and 2 μg of the vector, 2 μg psPAX2, and 0.2 μg pMD2-G. THP-1 or RAW 264.7 cells were spinfected with virus for 2 h at 1000g at 30 °C supplemented 8 μg/mL polybrene. After 2 days, cells were selected for stable expression of S. pyogenes Cas9 (Addgene #52962) using blasticidin (5 μg/mL for THP-1, 1 μg/mL for RAW 264.7) and for stable expression of sgRNAs using puromycin (0.5 μg/mL for THP-1, 5 μg/mL for RAW 264.7) or hygromycin (100 μg/mL for THP-1). After 10 d, single cells were isolated by serial dilution and expanded. Cells were then harvested for immunoblotting or used in experiments.
LDH cytotoxicity assays
For experiments involving RAW 264.7 macrophages, RAW 264.7 cells were seeded at 1 × 106 cells/well in 6-well plates. After 24 h, cells were washed 2× with PBS and replenished with fresh media. The cells were then treated with Val-boroPro (2 μM) and supernatant was removed at the indicated time intervals. For cell culture experiments, Val-boroPro (Coutts, et al., 1996) was resuspended in DMSO containing 0.1% TFA to prevent compound cyclization. For experiments involving HEK 293T cells, HEK 293T cells were seeded at 0.5 × 106 cells/well in 6-well plates in DMEM containing 10% FBS one day prior to transfection. Cells were then transiently transfected with 2 μg of plasmid DNA using the Fugene HD transfection reagent (Promega), and supernatants and cell lyates were collected after 24 h. Supernatants were analyzed for LDH activity using the Pierce LDH Cytotoxicity Assay Kit (Life Technologies) according to the manufacturer’s instructions. The HEK 293T lysates were also used for immunoblotting experiments. A two-tailed t-test was performed on the cytotoxicity data.
Immunoblotting cytotoxicity experiments
THP-1 cells (7 × 106 cells/plate) were seeded in 10 cm dishes and treated with DMSO, Val-boroPro (2 μM), etoposide (50 μM), bortezomib (2 μM), cycloheximide (2 μM), and staurosporine (100 nM) for 48 hours. THP-1 cells were also treated with LPS (10 μg/mL) for 48 h. After 48 h, LPS-primed cells were treated with nigericin (20 μM) for 30 minutes. RAW 264.7 cells (7 × 106 cells/plate) were seeded overnight in 10 cm dishes then treated with DMSO, Val-boroPro (2 μM), and etoposide (50 μM) for 24 h. Cells were washed 2x in PBS (pH = 7.4), resuspended in PBS, and lysed by sonication. Protein concentrations were determined using the DCA Protein Assay kit (Bio-Rad). The samples were separated by SDS-PAGE, immunoblotted, and visualized using the Odyssey Imaging System (Li-Cor).
GSDMD cleavage assays
HEK 293T cells transiently expressing Gsdmd-Myc-Flag, GSDMD-V5 or GSDMD D87A-V5 were prepared as described above. After 24 h, the cells washed 2x with PBS, resuspended in PBS, and lysed by sonication. Protein concentrations were determined DCA Protein Assay kit (Bio-Rad). Lysate (2 mgs/mL) was mixed at a ratio of 1:1 with 2x caspase reaction buffer (100 mM HEPES, 2 mM EDTA, 200 mM NaCl, 20 mM DTT, 20% Glycerol, pH 7.5). Caspases-1, -3, -7, -8, and -9 were reconstituted according to the manufacturer protocols. 1 μL of recombinant mature caspase was added to lysates and incubated at 37°C for 2 hours. The final concentrations of caspases were 7 ng/mL for caspase-3 and 1 Unit for caspases-1, -7, -8, and 9. Cleavage of GSDMD was assessed by immunoblotting. For identification of the GSDMD caspase-3 cleavage site, recombinant GSDMD was incubated with caspase-3 as described above. Products were transferred to PVDF membranes and analyzed by Edman degradation (UC Davis Genome Center).
Caspase-3/7 cleavage and activation assays
To assess the ability of caspase-1 to cleave caspases-3/7, THP-1 lysates were incubated with 1 Unit of caspase 1 (1 hour, 37°C) before immunoblotting for caspase-3 and -7. To determine whether caspase-3/7 were activated in cells, 1 × 106 THP-1 cells were treated with DMSO or Val-boroPro (2 μM). After 24 hours, 10 μL was diluted with 10 μL of RPMI in a white 384-well clear bottom plate (Corning), and then mixed with 20 μL of the Caspase-3/7 Glo Reagent according to the manufacturer’s protocol. The mixture was incubated in the dark for 3 hours at room temperature. Luminescence was then read using a Cytation 5 Cell Imaging Multi-Mode Reader (BioTek).
Annexin V and PI staining
1 × 106 THP-1 cells of the indicated genotype were treated with DMSO or Val-boroPro (2 μM) for 24 hours. The cells were then analyzed using the Annexin V-FITC Apoptosis Kit (Clontech) according to the manufacturer’s protocol.
Imaging of cell death by microscopy
To examine cell death morphology, RAW 264.7 or THP-1 cells were plated at 1.5 × 104 cells on 8 chambered slides (Nunc Lab-Tek II Chambered Coverglass) and treated with DMSO, Val-boroPro (2 μM), or etoposide (50 μM). For RAW 264.7 cells, static bright field images were captured using Zeiss AxioObserver inverted microscope with a 10×/0.45NA objective every 5 minutes over 48 hours. For THP-1 cells, the cells were treated 12 hours before imaging for 24 hours. Images were processed using the Zeiss ZEN 2 Blue Edition software. The data shown is representative of 4 randomly selected fields.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical values including the exact n and statistical significance are also reported in the Figure Legends.
Supplementary Material
Taabazuing et al Highlights file.
DPP8/9 inhibitors induce apoptosis instead of pyroptosis in the absence of GSDMD
Active caspase-1 can cleave and activate both caspases-3 and -7
Caspases-3 and -7 cleave GSDMD at Asp87 during apoptosis
Cleavage of GSDMD at Asp87 inactivates its pyroptotic activity
Acknowledgments
We thank S. Fujisawa for microscopy assistance and R. Gardner for flow cytometry assistance. This work was supported by Josie Robertson Foundation (D.A.B) and the MSKCC Core Grant (P30 CA008748).
Footnotes
AUTHOR CONTRIBUTIONS
C.Y.T. performed experiments, analyzed data, and wrote the paper. M.C.O. performed experiments and analyzed data. D.A.B. conceived and directed the project, analyzed data, and wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, Tan MH, Cotter PA, Vance RE, Aderem A, et al. Caspase-11 protects against bacteria that escape the vacuole. Science. 2013;339:975–978. doi: 10.1126/science.1230751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhter A, Gavrilin MA, Frantz L, Washington S, Ditty C, Limoli D, Day C, Sarkar A, Newland C, Butchar J, et al. Caspase-7 activation by the Nlrc4/Ipaf inflammasome restricts Legionella pneumophila infection. PLoS Pathog. 2009;5:e1000361. doi: 10.1371/journal.ppat.1000361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7:99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet. 2006;38:240–244. doi: 10.1038/ng1724. [DOI] [PubMed] [Google Scholar]
- Broz P. Immunology: Caspase target drives pyroptosis. Nature. 2015;526:642–643. doi: 10.1038/nature15632. [DOI] [PubMed] [Google Scholar]
- Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16:407–420. doi: 10.1038/nri.2016.58. [DOI] [PubMed] [Google Scholar]
- Broz P, von Moltke J, Jones JW, Vance RE, Monack DM. Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe. 2010;8:471–483. doi: 10.1016/j.chom.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutts SJ, Kelly TA, Snow RJ, Kennedy CA, Barton RW, Adams J, Krolikowski DA, Freeman DM, Campbell SJ, Ksiazek JF, et al. Structure-activity relationships of boronic acid inhibitors of dipeptidyl peptidase IV. 1. Variation of the P2 position of Xaa-boroPro dipeptides. J Med Chem. 1996;39:2087–2094. doi: 10.1021/jm950732f. [DOI] [PubMed] [Google Scholar]
- Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, Shao F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–116. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guey B, Bodnar M, Manie SN, Tardivel A, Petrilli V. Caspase-1 autoproteolysis is differentially required for NLRP1b and NLRP3 inflammasome function. Proc Natl Acad Sci U S A. 2014;111:17254–17259. doi: 10.1073/pnas.1415756111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21:677–687. doi: 10.1038/nm.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013;341:1250–1253. doi: 10.1126/science.1240988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang ZH, Zhong CQ, Han J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015;25:1285–1298. doi: 10.1038/cr.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen I, Lopez JP, Laufer SA, Miao EA. IL-1beta, IL-18, and eicosanoids promote neutrophil recruitment to pore-induced intracellular traps following pyroptosis. Eur J Immunol. 2016 doi: 10.1002/eji.201646647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–671. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszynski A, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341:1246–1249. doi: 10.1126/science.1240248. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–1022. doi: 10.1016/j.cell.2014.04.007. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, Kanneganti TD, Van Damme P, Vanden Berghe T, Vanoverberghe I, Vandekerckhove J, Vandenabeele P, Gevaert K, Nunez G. Targeted peptidecentric proteomics reveals caspase-7 as a substrate of the caspase-1 inflammasomes. Mol Cell Proteomics. 2008;7:2350–2363. doi: 10.1074/mcp.M800132-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–158. doi: 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD, Aderem A. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010;11:1136–1142. doi: 10.1038/ni.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okondo MC, Johnson DC, Sridharan R, Go EB, Chui AJ, Wang MS, Poplawski SE, Wu W, Liu Y, Lai JH, et al. DPP8 and DPP9 inhibition induces pro-caspase-1-dependent monocyte and macrophage pyroptosis. Nat Chem Biol. 2017;13:46–53. doi: 10.1038/nchembio.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sborgi L, Ruhl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, Farady CJ, Muller DJ, Broz P, Hiller S. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016;35:1766–1778. doi: 10.15252/embj.201694696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187–192. doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- Silva MT. Secondary necrosis: the natural outcome of the complete apoptotic program. FEBS Lett. 2010;584:4491–4499. doi: 10.1016/j.febslet.2010.10.046. [DOI] [PubMed] [Google Scholar]
- Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J, et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature. 1992;356:768–774. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- Van de Craen M, Declercq W, Van den brande I, Fiers W, Vandenabeele P. The proteolytic procaspase activation network: an in vitro analysis. Cell Death Differ. 1999;6:1117–1124. doi: 10.1038/sj.cdd.4400589. [DOI] [PubMed] [Google Scholar]
- Van Opdenbosch N, Gurung P, Vande Walle L, Fossoul A, Kanneganti TD, Lamkanfi M. Activation of the NLRP1b inflammasome independently of ASC-mediated caspase-1 autoproteolysis and speck formation. Nat Commun. 2014;5:3209. doi: 10.1038/ncomms4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Yang J, Gao W, Li L, Li P, Zhang L, Gong YN, Peng X, Xi JJ, Chen S, et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature. 2014;513:237–241. doi: 10.1038/nature13449. [DOI] [PubMed] [Google Scholar]
- Yang X, Boehm JS, Yang X, Salehi-Ashtiani K, Hao T, Shen Y, Lubonja R, Thomas SR, Alkan O, Bhimdi T, et al. A public genome-scale lentiviral expression library of human ORFs. Nat Methods. 2011;8:659–661. doi: 10.1038/nmeth.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, Liu L, Shao F. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature. 2011;477:596–600. doi: 10.1038/nature10510. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.