

Summary
Background
Secretion of Weibel-Palade body (WPB) contents is regulated, in part, by the phosphorylation of proteins that constitutes the endothelial exocytotic machinery. In comparison to protein kinases, a role for protein phosphatases in regulating endothelial exocytosis is undefined.
Results
We show that inhibition of protein phosphatase 2B (PP2B) activity by cyclosporine A (CsA), tacrolimus or a cell-permeable PP2B autoinhibitory peptide promotes the secretion of ultra-large von Willebrand factor (ULVWF) from human umbilical vein endothelial cells (HUVECs) in the absence of any other endothelial cell-stimulating agent. PP2B inhibitor-induced secretion and anchorage of ULVWF strings from HUVECs mediate platelet tethering. In support of a role for PP2B in VWF secretion, the catalytic subunit of PP2B interacts with the vesicle trafficking protein, Munc18c. Serine phosphorylation of Munc18c, which promotes granule exocytosis in other secretory cells, is increased in CsA-treated HUVECs, suggesting that this process may be involved in CsA-mediated WPB exocytosis. Furthermore, plasma VWF antigen level is also enhanced in CsA-treated mice, and siRNA-mediated knockdown of α and β isoforms of PP2B-A subunit in HUVECs enhanced VWF secretion.
Conclusions
These observations suggest that CsA promotes VWF release, in part by inhibition of PP2B activity and are compatible with the clinically observed association of CsA treatment and increased plasma VWF levels in humans.
Keywords: Protein phosphatase 2B, ULVWF, Secretion, HUVEC, Munc18c and Cyclosporine A
Introduction
Cyclosporine A (CsA), an immunosuppressive agent commonly used in organ transplantation, is also a potent inhibitor of protein phosphatase 2B (PP2B) activity [1]. A subset of patients treated chronically with CsA develops a type of thrombotic microangiopathy that clinically resembles thrombotic thrombocytopenic purpura (TTP) or hemolytic-uremic syndrome (HUS) [2]. CsA-treated patients also develop increased plasma levels of Von Willebrand factor (VWF) [3].
Ultra-large (UL) VWF multimers are produced and stored in the Weibel-Palade bodies (WPBs) of endothelial cells. ULVWF forms are released as long hyper-adhesive strings anchored to endothelial cells in response to different agonists (including histamine, thrombin, vasopressin, epinephrine, several cytokines, and Shiga toxins). Perfused platelets instantaneously attach to these ULVWF multimeric strings [4-5]. ULVWF-platelet strings are subsequently cleaved by the plasma metalloprotease, ADAMTS-13 (a disintegrin and metalloprotease with thrombospondin motifs), into smaller and less adhesive plasma VWF multimers.
The secretion of ULVWF is followed by the fusion of WPBs to the endothelial cell plasma membrane. Various signaling pathways can be involved. Thrombin and histamine increase intracellular Ca2+, whereas vasopressin and epinephrine increase intra-cellular cAMP [6-7]. Through the agonist-specific effectors, calcium/calmodulin (Ca2+/CaM) in thrombin-histamine signaling pathways, or protein kinase A (PKA) in vasopressin-epinephrine pathways, a small GTPase (Ral) is activated and promotes endothelial cell VWF secretion [6-7]. A guanine exchange factor for Ral, the Ral GDP dissociation stimulator (RalGDS), has been reported to regulate Ral-dependent exocytosis of WPBs in response to agonists that elevate either intra-cellular Ca2+ or cAMP [8]. In contrast to Ral, another GTPase, Rab3D may play a negative stimulatory role in VWF exocytosis as overexpression of Rab3D inhibits VWF secretion [9]. Recently, selective release of VWF but not tissue-type plasminogen activator (tPA) in response to histamine was shown to be mediated by phospholipase D1 [10]. Collectively, these signaling events could result in the re-organization of the cytoskeleton, particularly actin and microtubules, in order to achieve WPB redistribution and fusion to the endothelial cell plasma membrane. Microtubular “motors,” including dynein-dynactin, contribute to the contractile forces that facilitate the clustering of WPBs and secretion of their contents [11].
In other secretory cells (e.g., mast cells, neurons and pancreatic islet cells), vesicular trafficking proteins of the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) superfamily is also involved in granule exocytosis. Endothelial cells contain SNARE proteins and there is evidence that SNARE and SNARE-associated proteins may control the exocytosis of WPBs and secretion of their contents [12-13].
Reversible post-translational modifications, including S-nitrosylation of N-ethylmaleimide-sensitive factor (NSF) [12] or phosphorylation of tyrosine and/or serine/threonine residues on vesicular trafficking proteins are capable of regulating granule exocytosis [14]. The phosphorylation status of any trafficking protein is determined by the action of both protein kinases and protein phosphatases, and alterations in the activity of either type of enzyme can alter exocytosis. A role for protein kinase A (PKA) [11] or protein kinase C (PKC δ [15] in endothelial cell VWF exocytosis has been investigated. In contrast, any contribution of serine/threonine phosphatases to the regulation of WPB exocytosis and secretion of their contents is unknown.
PP2B (or calcineurin) is a serine/threonine phosphatase that exists as a heterodimer composed of a 58-64 kDa catalytic subunit (A subunit) [PP2B-A] and a 19 kDa Ca2+ regulatory subunit (B subunit) [PP2B-B]. PP2B-A subunit has three isoforms (α, β and γ); α and β isoforms are ubiquitous, while γ is testis specific. PP2B is a Ca2+/CaM -activated enzyme involved in the regulation of a variety of cellular functions [16]. The PP2B-A subunit contains an autoinhibitory (AI) domain and the binding sites for the PP2B-B subunit and CaM. In the absence of CaM, the AI domain binds to the active cleft of PP2B and inhibits the phosphatase activity [17]. Peptides mimicking the AI domain are potent and specific inhibitors of PP2B activity [18]. The immunosuppressant's, cyclosporine (CsA) and tacrolimus (FK506) are inhibitors of PP2B. In order for this inhibition to occur, CsA must first form a complex with the resident cytoplasmic protein, cyclophilin, and tacrolimus must complex with FK506-binding protein 12 (FKBP12) [1]. This phosphatase inhibitory activity could provide in part a mechanism for the CsA-associated thrombotic microangiopathies found in a small subset of organ transplant patients.
Since PP2B is a serine/threonine phosphatase, we hypothesized that inhibition of PP2B activity may induce ULVWF secretion (at least in part) by increased serine phosphorylation of a vesicle trafficking protein that promotes WPB exocytosis and ULVWF secretion. We evaluated this possibility in vitro using human umbilical vein endothelial cells (HUVECs) and in vivo in mice.
Methods and Materials
Endothelial cell cultures
Human umbilical cord vein endothelial cells (HUVECs) were obtained as previously described [5]. Briefly, umbilical cord veins were washed in phosphate buffer, subjected to collagenase digestion and seeded onto 6-well or 35-mm culture dishes coated with 1 % gelatin. HUVECs were maintained in medium 199 supplemented with 20% FBS, 3% penicillin-streptomycin-neomycin (PSN) and 0.2 mM L-glutamine and grown to confluency (5-7 days). Most of the cells used in these studies were either primary or used after 1 passage.
Reverse transcription-polymerase chain reaction (RT-PCR) analysis
The presence of mRNA for the α and β isoforms of PP2B-A subunit was quantified using SYBR-Green–based real-time PCR. Total RNA was isolated from HUVEC's by RNeasy Mini Kit (Qiagen, Valencia CA) and cDNA was synthesized from 2 μg of total RNA using the Superscript III First RT-PCR system (Invitrogen, Carlsbad CA). PCR amplification of the cDNA samples was performed using 7 μl of 2X Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA), 1 μl of 100 pmol of primers, 13 μl of CDNA preparation to a final volume of 20 μl. The following primers were used 5′-ACGCCAACCTTAACTCCATCA-3 (forward) and 5′-TGCTGTCCGTGCCGTTAGT-3′ (reverse) for PP2B-Aα, and 5′TGGTGAAAGAAGGTCGAGTAGATG-3′ (forward) and 5′TGGCAGCACCCTCATTGA-3′ (reverse) for PP2B-Aβ and 5′-ATGGAAATCCCATCACCATCTT-3′ (forward) and 5′-CGCCCCACTTGATTTTGG-3′ (reverse) for glyceraldehyde 3-phosphate dehydrogenase (GAPDH). PCR cycling conditions included one cycle of initial activation at 50°C for 2 minutes, followed by 1 cycle of initial denaturation at 95°C for 10 minutes, 40 cycles of denaturation at 95°C for 15 minutes, 40 cycles of annealing at 60°C for 1 minute. The PCR products, their dissociation curves and absolute quantity of mRNA were detected with the 7300 Real-Time PCR System (Applied Biosystems, Foster City, CA).
PP2B inhibitor–induced stimulation
Cells were stimulated with either CsA, FK506 (1 nM and 10 nM diluted in 0.001% DMSO) (Sigma), vehicle control DMSO (0.001%), cell permeable peptides containing the scrambled or PP2B autoinhibitory (AI) domain (1 μM or 2 μM) or 100 μM histamine as positive controls for 15 minutes. PP2B AI cell-permeable peptide was composed of the calcineurin (CaN) autoinhibitory domain fused to a poly-arginine-based protein transduction domain (11R) Ac-RRRRRRRRRRRGGGRMAPPRRDAMPSDA-NH2 (Calbiochem, SanDiego, CA). (Ac-RRRRRRRRRRRGGGRMRDRPAPAMDPSA-NH2) was the scrambled peptide synthesized by Gene Script Corporation, (Piscataway, NJ). Presence of any cellular debris in the supernatant was removed by centrifugation at 150 × g for 3 minutes. VWF antigen levels were measured in cell supernatants using a standard ELISA technique. Samples were incubated for 1 hr on microtiter plates pre-coated with 1 μg/ml rabbit anti-human-VWF antibody (Ramco Laboratories, Stafford, TX). After several washes, samples were incubated with 0.5 μg/ml of goat anti-human-VWF and rabbit anti-goat HRP antibodies (Bethyl Laboratories, Montgomery, TX) and color-developed with the addition of TMB (3, 3′, 5, 5′-tetramethylbenzidine; Invitrogen, Carlsbad, CA). The reaction was stopped with 1 M HCl and the absorbance was read at 450 nm. Human plasma was used as a standard in the VWF antigen ELISA. Fold differences in VWF antigen levels were determined after normalizing the absorbance values obtained for test samples to that obtained from media only. In some experiments, equal volume of samples (cell supernatants) obtained from equivalent number of HUVECs treated with various agents were separated by sodium dodecyl sulfate (SDS)-1% agarose gel electrophoresis and VWF multimers detected by immunoblotting with polyclonal anti-human-VWF antibody and secondary anti-IgG-horseradish peroxidase (HRP) conjugate (Bethyl Laboratories).
Phosphatase activity
PP2B activities were measured in HUVEC lysate obtained after treatment with either: DMSO, CsA, FK506, AI and scrambled peptides, control siRNA and PP2B-Aα or PP2B-Aβ depleted cells using the Calcineurin Cellular Assay Kit Plus (Biomol International, Plymouth Meeting, PA). Free phosphate within the lysates was removed by gel filtration using desalting resin. PP2B activity in the lysate was measured by removal of phosphate from the PP2B substrate (RII phosphopeptide) provided. The released phosphate was detected by addition of Biomol green reagent and absorbance measurements at 620 nm were converted into released phosphate using a generated phosphate standard curve.
Quantification of ULVWF-platelet strings induced by PP2B inhibitors
Isolation and washing of platelets from healthy voluntary donors was performed as before [5]. Human subjects gave written informed consent and the study was approved by the Baylor College of Medicine and Rice University Review Boards. The number of ULVWF strings (with adherent platelets) secreted by PP2B inhibitor-induced stimulated endothelial cells was quantified under flowing conditions (2.5 dynes/cm2) in a parallel-plate flow chamber system as previously described [5]. HUVECs seeded onto 35-mm culture dishes were treated with either: 100 μM histamine (positive control); 0.001% DMSO (solvent control); 1 nM and 10 nM CsA and FK506; 1 and 2 μM AI peptide or Tyrode's buffer (basal) for 2 minutes. Washed platelets re-suspended in Ca+2, Mg+2-free Tyrode's buffer were perfused over the treated HUVECs for 2 min and the number of ULVWF strings observed by adherent platelets and phase-contrast microscopy was counted in 20 separate fields at 200× for each experimental condition.
Treatment of mice with CsA
Balb/C mice (8-12 weeks) were intraperitoneally injected with either 20 mg/kg of CsA, or control vehicle DMSO, or mock injected with an equal volume of 0.9% saline (NaCl) solution under a protocol approved by the Institutional Animal Care and Use Committee at Baylor College of Medicine. Blood was collected after 15 minutes after injection. VWF levels in the plasma were measured using a sandwich ELISA described above; with the exception that the antibodies used were rabbit anti-human VWF (Dako, Glostrup, Denmark). This antibody cross reacts with mouse VWF and has been previously used to detect mouse VWF antigen levels [19]. Mouse plasma was used as a standard in the VWF antigen ELISA. Fold differences in mouse VWF antigen levels was determined after normalizing the absorbance values obtained for test samples (CsA or DMSO treatment) to that obtained from control (mock NaCl treatment).
siRNA construct, transfection and secretion
A pre-formed mix of 4 independent siRNAs targeting the α isoform of the A subunit of PP2B [PPP3CA] (M-008300-02), and the β isoform of A subunit of PP2B [PPP3CB] (M-009704-01) were purchased from Dharmacon (Lafayette, CO). A non-specific control siRNA pool with no sequence homology to any human or mouse sequence (D-001206-13-05) was used as control in this study. HUVECs were transfected with 100 nM siRNA oligos using DharmaFect transfecting reagent (Dharmacon) according to the manufacturer's instruction. After 48 hours, the supernatant was collected and VWF antigen levels were measured by VWF ELISA assay kit (Ramco Laboratories). The % of VWF antigen was determined using an internal prediluted human VWF calibrator solution. Fold differences in VWF antigen levels was determined after normalizing the % of VWF antigen obtained for PP2B-Aα or PP2B-Aβ siRNA treated cell to that obtained from control siRNA treated cells. Silencing of the α and β isoforms of PP2B-A subunit were confirmed by RT-PCR and immunoblotting with isoform specific antibodies (Santa Cruz Biotechnology, Santa Cruz, CA). The blots were stripped and reprobed with an antibody to actin (loading control).
Protein interaction studies
The interaction of the catalytic subunit of PP2B (PP2B-Aα subunit) with vesicle trafficking protein Munc18c was determined by a PP2B pull down assay. PPP3CA truncated at residue 347 and tagged to glutathione S-transferase in pGEX-6P plasmid was obtained from Dr. Anjana Rao (Harvard Medical School, Boston. MA). This cDNA lacks the autoinhibitory domain but retains the catalytic and much of the PP2B-B binding subunits that are necessary to sustain interaction with any potential substrates. GST-tagged protein was expressed in E.coli after isopropyl-beta-D-thiogalactopyranoside (IPTG) induction, and purified using glutathione beads. The GST and GST-PP2B-Aα proteins were characterized by Coomassie blue staining. Purified GST-PP2B-Aα and GST proteins (3 μg) were precoupled with glutathione beads and mixed overnight at 4° C with 100 μg of lysates obtained from resting HUVECs. Beads were washed three times and analyzed on a 10 % SDS-PAGE gel followed by immunoblotting with anti-Munc18c antibody (gift from Dr. Pressin, State University of New York at Stony Brook, NY).
Immunoprecipitation and Immunoblotting
Lysates obtained from HUVECs (750 μg) following treatment with DMSO or CsA (1 and 10 nM) for 15 minutes were immunoprecipitated using 3 μg of anti-phosphoserine antibody (Invitrogen) and protein A-sepharose. Proteins were separated by 10% SDS-PAGE, transferred to nitrocellulose, probed with polyclonal antibody to Munc18c, and developed using the ECL system (Amersham Bioscience). The signals were scanned using photoshop version 6 software (Adobe), and densitometric quantification was performed using a BioRad Gel documentation system.
Statistical analysis and presentation of VWF antigen data
Experimental conditions were compared by using paired student's t test. p value at 0.05 was considered significant. The absolute amount of VWF secreted from HUVECs showed some variation from experiment to experiment but there was consistency in the relative ratio of control to PP2B-inhibited samples. Therefore, the VWF antigen were primarily expressed as a fold difference in PP2B inhibited samples compared to control or mock treated samples.
Results
Figure 1A demonstrates the presence of mRNA for α and β isoforms of PP2B-A subunit in HUVECs. Immunoblotting of the lysates from HUVECs with isoform specific antibodies confirm the presence of PP2B-Aα and PP2B-Aβ proteins (Figures 1B and 1C). Furthermore, the regulatory subunit of PP2B (PP2B-B subunit) was also expressed in HUVECs (Figure 1D).
Figure 1. Inhibition of PP2B was associated with VWF secretion from HUVECs.
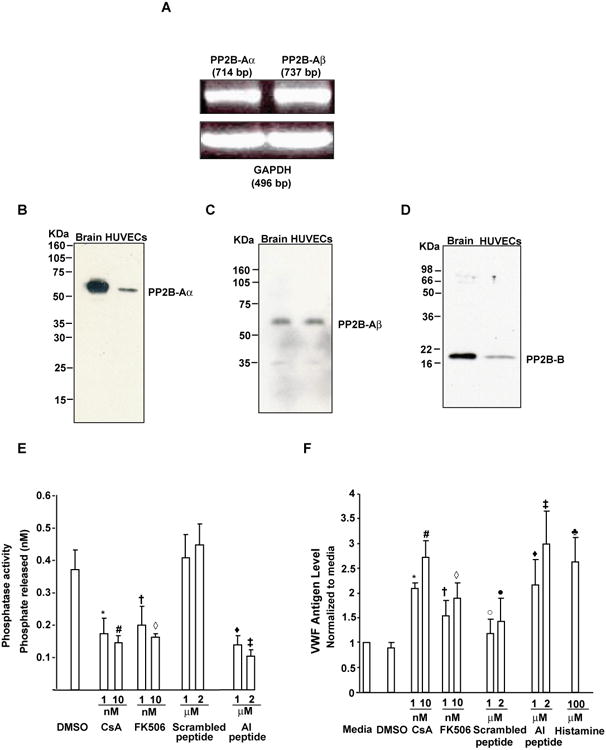
(A) Expression of PP2B-Aa and PP2B-Aβ mRNA by RT-PCR. An amplification product of 714 base pair (bp) specific for the α isoform of PP2B-A subunit and a 737 bp specific for the β isoform of PP2B-a subunit was identified by RT-PCR. GAPDH (496 bp) was used as a loading control. (B-D) Expression of PP2B-Aα, PP2B-Aβ and PP2B-B proteins. HUVEC lysate (40 μg) were separated by SDS-PAGE and immunoblotted with antibodies specific to for PP2B-Aα (B) or PP2B-Aβ (C) or the regulatory subunit of PP2B (PP2B-B) (D). Lysate obtained from mouse brain (brain) are also shown as a positive control (E) CsA, FK506 and AI peptide inhibited PP2B activity. Data are the mean +/- SEM of 3-8 experiments. Compared to DMSO, *P=0.0043 (for 1 nM CsA), #P=0.0028 (for 10 nM CsA), †P=0.056 (for 1nM FK506) and ◊P=0.04 (for 10 nM FK506). Compared to the scrambled peptide, ŨP=0.03 and ‡P=0.02 for 1 and 2 μM AI peptide, respectively. (F) PP2B inhibitors promoted VWF release. HUVECs were incubated with the indicated compounds, supernatant collected and the released VWF measured by ELISA. Data are mean +/-SE from 5-11 experiments. Compared to media, *P<0.001 (for 1 nM CsA), #P=0.004 (for 10 nM CsA), †P=0.05 (for 1nM FK506), ◊P=0.01 (for 10 nM FK506), °P=0.40 (for 1 μM scrambled peptide), (x000183)P=0.069 (for 2 μM scrambled peptide) and ♣p=0.005 (for 100 μM histamine). Compared to scrambled peptide, ŨP=0.044 and ‡P=0.01 for 1 and 2 μM AI peptide, respectively.
In order to evaluate the possible functional role for PP2B in VWF secretion from WPBs, we treated primary HUVECs in culture with PP2B inhibitors and measured VWF antigen release using an ELISA assay. Compared to the controls (DMSO or cell permeable scrambled peptide), exposure of HUVECs with CsA, FK506 or a cell-permeable peptide containing the PP2B autoinhibitory (AI) domain [20] showed a maximal of ∼48-56 % inhibition of PP2B activity, respectively (Figure 1E). The inhibition of PP2B activity by these agents was associated with the rapid release of VWF antigen from HUVECs in a dose-dependent manner (Figure 1F). Since, AI but not the scrambled peptide significantly enhanced VWF secretion, only AI peptide was used for the rest of the studies. CsA, FK506 and AI peptide were about as effective as histamine in promoting the release of VWF antigen from HUVECs.
HUVECs secrete ULVWF multimers from the WPBs rapidly in response to histamine [4]. We conducted experiments to determine if inhibition of PP2B activity also promotes the secretion of ULVWF multimeric strings from the HUVECs. Compared to normal pooled plasma (reference), HUVECs exposed either to CsA, AI peptide or FK506 revealed VWF multimers enriched in ULVWF forms (Figure 2A). In complementary experiments, PP2B inhibitor-induced ULVWF strings were visualized using microscopy by the adherence of washed platelets under flowing conditions (∼2.5 dynes/cm2 shear stress) to the hyper-adhesive ULVWF strings. Compared to the DMSO treated endothelial cells (ECs), HUVECs secreted ∼4-5-fold or ∼2.5-3 fold more ULVWF-platelet strings in response to CsA, and FK506, respectively (Figure 2B). Moreover, AI peptide or histamine (positive control) treated HUVECs also supported ∼3.5 fold increased ULVWF strings compared to cells in Tyrode's buffer (basal conditions) (Figure 2B). These studies suggest that inhibition of PP2B provokes the secretion of ULVWF strings from endothelial cells in vitro. Exposure to the PP2B inhibitors or vehicle control did not damage HUVECs, as demonstrated by the failure of either agents to diminish cell metabolic/reductive capacity using 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl-tetrazolium bromide (MTT) (Data not shown). These results indicated that HUVEC release of VWF multimers enriched in ULVWF forms is not the consequence of cell damage induced by the PP2B inhibitors.
Figure 2. PP2B inhibitors induced release of ULVWF/VWF multimers from HUVECs.
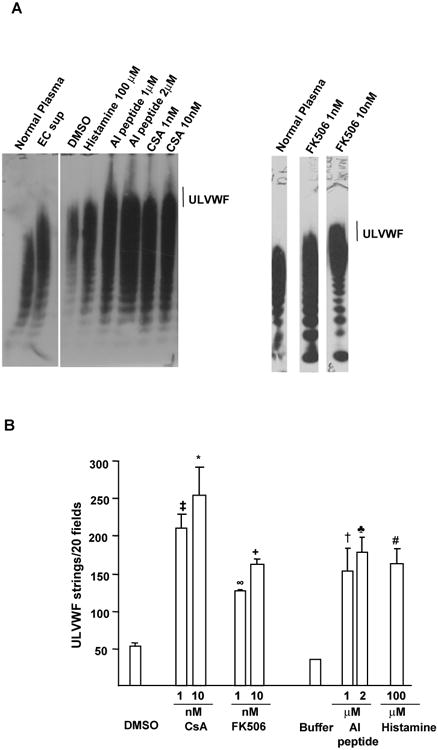
(A) ULVWF/VWF multimeric patterns. HUVECs were treated with PP2B inhibitors and ULVWF/VWF multimers from the cell supernatants analyzed by unreduced 1% agarose/SDS electrophoresis/anti-VWF antibody immunoblotting. VWF multimeric patterns from normal platelet-poor plasma or endothelial cell supernatant (EC sup) from histamine-treated cells are shown for comparison. ULVWF multimers are indicated by a vertical line. Compared to the normal platelet-poor plasma lane, ULVWF multimers were prominently enhanced in lane containing the PP2B inhibitor-treated samples. Occasionally, DMSO treated lane also showed some VWF and ULVWF forms. (N=3). (B) Perfused platelets adhered instantaneously to ULVWF strings secreted by HUVECs in the presence of PP2B inhibitors under flowing conditions. HUVECs were preincubated with the control reagents or PP2B inhibitors before the perfusion of normal fresh washed human platelets using a flow chamber (wall shear stress, 2.5 dyne/cm2). The number of ULVWF-platelet strings that formed was counted over the course of 2 min in 20 different microscope fields. Data are presented as +/- SEM of 2-6 experiments. Compared to DMSO, ‡P=0.02 (for 1 nM CsA), *P=0.0024 (for 10 nM CsA), ∞P=0.05 (for 1 nM FK506), +P=0.002 (for 10 nM FK506) and #P=0.0069 (for histamine). Compared to Tyrode's buffer treated cells, †P=0.05 (for 1 μM AI peptide), ♣P<0.01 for 2 μM AI peptide).
In order to determine if CsA can enhance VWF secretion in vivo, we treated mice with CsA and analyzed plasma VWF antigen levels. Compared to the mice intraperitoneally injected with saline (NaCl) or DMSO vehicle control, CsA caused an ∼1.6-fold increase in plasma VWF antigen within 15 minutes, suggesting that CsA enhances VWF secretion in vivo (Figure 3). A functional effect of PP2B inhibition on HUVEC VWF release was further evaluated using short interference RNA (siRNA) to knockdown the expression of endogenous α and β isoforms of the PP2B-A subunit in HUVECs. Knockdown of PP2B-Aα and PP2B-Aβ was confirmed by RT-PCR studies (Figures 4A and 4C) and immunoblotting with isoform specific antibodies (Figures 4B and 4D). There was no evidence of any compensatory increase in PP2B-Aβ protein levels in PPP3CA knockdown and vice versa. Compared to control siRNA treated cells, the phosphatase activity in α and β knock down ECs were decreased by ∼48% (Figure 4E). Targeting the α isoform leaves active β isoform that may account for the remaining ∼50% phosphatase activity observed in PP2B-Aα knockdown cells and vice versa. More importantly, knockdown of α and β isoforms of PP2B-A subunit resulted in a ∼2.5 -2 fold increased VWF release from HUVECs, respectively (Figure 4F).
Figure 3. Increased VWF in the plasma of mice treated with CsA.

Data are mean +/-SE from 3 experiments. Compared to treatment with DMSO alone, *P=0.042 for CsA treatment.
Figure 4. Increased VWF in the supernatants of HUVECs in PP2B-Aα and PP2B-Aβ knockdown cells.
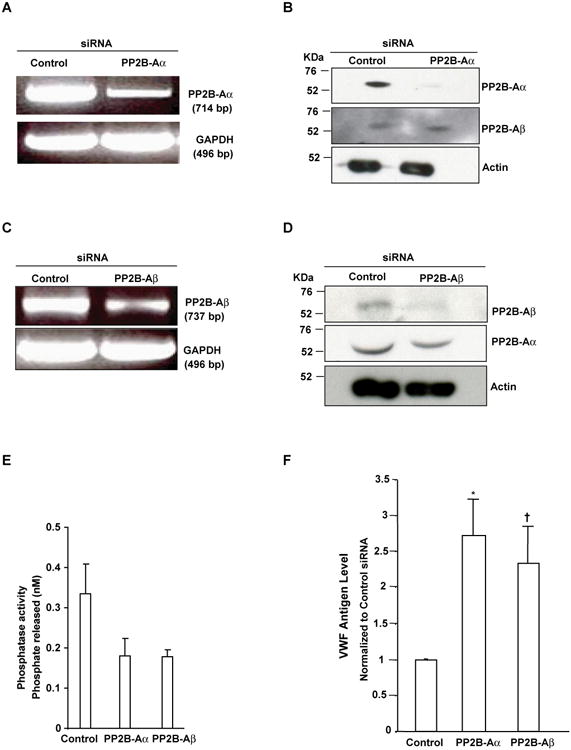
HUVECs were transfected with either control siRNAs or specific siRNAs targeting PPP3CA and PPP3CB using DharmaFect. (A and C) RT-PCR analysis. After 48 hours, total cellular RNA was extracted and RT-PCR performed using primers specific to α, β and GAPDH (loading control). (B and D) Imunoblotting. Lysates were immunoblotted with antibodies to PP2B-Aα, PP2B-Aβ and actin (loading control). (E) PP2B-Aa and PP2B-Aβ knockdown in HUVECs inhibited PP2B activity. Data are the mean +/- SEM of 3-4 experiments. Compared to control siRNA treated cells, knockdown of α and β isoform of PP2B-A subunit decreased PP2B activity by ∼48%. (F) PP2B-Aa and PP2B-Aβ knockdown in HUVECs promoted VWF secretion. PP2B-Aα and PP2B-Aβ protein was depleted by a siRNA, and HUVEC supernatants were collected after 48 hours, centrifuged, and VWF antigen levels were quantified by the Spectro VWF ELISA assay kit (Ramco Laboratories). Results are mean +/- SEM of 4-5 experiments performed in triplicates. Compared to control siRNA treated cells *P=0.001 for α knockdown and †P=0.05 for β knockdown.
In order to investigate a potential mechanism by which CsA promotes endothelial cell secretion, we investigated the effect of PP2B inhibition by CsA on the serine phosphorylation of Munc18c. Munc18c phosphorylation is an essential post-translational modification of this vesicle trafficking protein that is associated with increased vesicle exocytosis in several other cell systems [14]. Treatment of HUVECs with 1 and 10 nM of CsA resulted in the increased serine phosphorylation of Munc18c by about ∼1.5 or 2-fold, respectively, compared to control DMSO vehicle-treated cells (Figures 5A and 5B). When the catalytic subunit of PP2B (PP2B-Aα subunit) was expressed as a glutathione S-transferase (GST) fusion protein in E.coli, GST “pull-down” assays revealed that GST-PP2B-Aα but not GST alone, interacted with Munc18c (Figure 5C). Taken together, these data indicate that the catalytic subunit of PP2B can associate with Munc18c at least in vitro, and that increased serine phosphorylation of Munc18c is associated with both inhibition of PP2B activity by CsA and augmented secretion of ULVWF.
Figure 5. Serine phosphorylation of the vesicle trafficking protein, Munc18c, was enhanced in CsA-treated HUVECs. (A) CsA triggered serine phosphorylation of Munc18c.
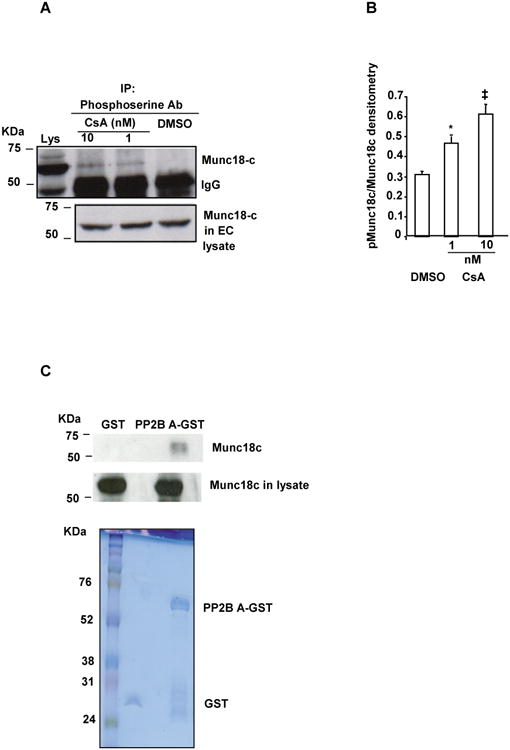
Lysates from HUVECs treated with either vehicle control DMSO or varying concentrations of CsA were immunoprecipitated (IP) with anti-phosphoserine antibody, separated by reducing SDS-PAGE, and immunoblotted using anti-Munc18c antibody (Upper panel). The position of the 50 KDa IgG γ heavy chain is indicated as IgG. EC lysate is shown to indicate the position of Munc18c as a positive control reference for the immunoprecipitated phospho-serine samples (Lys; upper panel). Lysate used for immunoprecipitation experiments contain Munc18c (lower panel). The upper panel is obtained at a relatively longer exposure compared to the lower panel and is responsible for the difference in the intensity of Munc18c in the lysate of the upper panel compared to the lower panel. (B) Densitometric quantification of serine phosphorylated Munc18c. Phosphorylated Munc18c (pMunc18c) is the ratio of phosphorylated Munc18c/total Munc18c (in arbitrary units) formed in response to CsA (N = 3). Compared to control DMSO vehicle treatment, *P=0.03 and ‡P=0.01 for 1 and 10 nM CsA, respectively. (C) Munc18c interacted with PP2B-Aa. PP2B-Aα-GST coupled to glutathione beads were used to pull down Munc18c from the HUVEC lysate, as described in Methods. Some Munc18c became bound to PP2B-Aα-GST beads, but not to beads coated with GST alone. The PP2B-bound Munc18c was then released by boiling in SDS-buffer, separated by reduced SDS-PAGE, and detected by immunoblotting with anti-Munc18c. The GST and PP2B-Aα-GST proteins in the pull down assays were detected by Coomassie blue staining, while the presence of Munc18c in the lysates used for pull down was detected by immunoblotting with anti-Munc18c.
Discussion
Protein kinases and phosphatases regulate signaling pathways initiated by the binding of agonists to endothelial cell receptors, and the phosphorylation status of vesicular trafficking proteins can regulate the secretion of granule contents [14]. Any specific role for serine/threonine phosphatases in the secretion of VWF from the WPBs of endothelial cells is, however, currently unknown. In this study, we demonstrate that inhibition of HUVEC PP2B activity by CsA activates the serine phosphorylation of a PP2B-associated vesicle trafficking protein, Munc18c. Concurrently, CsA inhibition of HUVEC PP2B promotes the secretion of hyper-adhesive ULVWF multimeric strings.
CsA has been reported previously to potentiate agonist [thrombin, histamine and phorbol 12-myristate 13-acetate (PMA)]-induced VWF release from HUVECs [21]. Other investigators have found increased quantities of VWF antigen in the supernatants of HUVECs exposed to CsA for 5-6 days [22]. It is not known if this relatively long-term exposure of HUVECS to CsA caused cell damage and leakage of VWF, rather than CsA-triggered alteration in cell signaling and associated VWF secretion.
We observed increased ULVWF/VWF secretion from HUVECs within minutes of cell exposure to CsA, FK506 or a PP2B autoinhibitory peptide (AI). These data suggest that inhibition of PP2B promotes ULVWF/VWF secretion from HUVECs, in the absence of any other endothelial cell-stimulating agonist. We also obtained compatible results from in vivo studies of mice treated with CsA. It is not yet known if our findings are associated with the development of thrombotic microangiopathy in a subset of organ transplantation patients who are immunosuppressed with CsA or tacrolimus [23-24]. In addition to inhibition of PP2B, CsA also inhibits: the mitochondrial permeability transition pore, a voltage sensitive and Ca+2-activated channel in the inner mitochondrial membrane; c-Jun N-terminal kinase (JNK); and P38 signaling in T cells [25-26]. In order to determine if inhibition of PP2B by CsA contributes to the promotion of ULVWF/VWF secretion, we utilized siRNAs to knock down the α and β isoforms of PP2B-A subunits in HUVECs. Knockdown of PP2B-Aα or PP2B-Aβ resulted in a significant increase in ULVWF/VWF secretion by HUVECs compared to control siRNA-treated cells (Figure 4F). We conclude from these studies that CsA inhibition of PP2 activity helps promote the secretion of ULVWF/VWF from endothelial cells.
We investigated the molecular basis for the relationship between inhibition of PP2B and the release of ULVWF multimers from endothelial cells. The rapid secretion of ULVWF/VWF multimers from HUVECs upon CsA or AI peptide treatment precludes as the explanation increased VWF synthesis via transcriptional up-regulation of the VWF gene. The absence of overt morphological alterations or decreased metabolism in PP2B inhibitor-treated-HUVECs indicates that increased release of ULVWF/VWF multimers also does not occur as a consequence of PP2B inhibitor-induced endothelial damage. On the other hand, a possible explanation for our observations is that PP2B inhibition by CsA or tacrolimus initiates (or participates in) ULVWF/VWF multimer secretion from endothelial cells via regulation of the phosphorylation status of a vesicle trafficking protein. We found that CsA increased the serine phosphorylation of Munc18c, a SNARE-associated protein involved in the phosphorylation related release of granule contents from endothelial and other secretory cells [13], [27]. Consistent with the capacity of PP2B to regulate the serine phosphorylation of Munc18c, we also observed that PP2B-Aα interacts with Munc18c in vitro. We suggest that increased serine phosphorylation of Munc18c in the presence of CsA-induced (or tacrolimus-induced) PP2B inhibition may lead to decreased association of Munc18c with syntaxin-4. This may enable syntaxin-4 to associate with the vesicle-associated membrane protein 3 (VAMP-3) on WPBs to form a SNARE complex that facilitates the secretion of WPB contents, including ULVWF/VWF multimers (Figure 6).
Figure 6. Proposed mechanism for the regulation of VWF secretion by cyclosporine A (CsA).

The mechanism described is an adaption of a model described in other secretory system [35]. In ECs, granule exocytosis is likely to occur through a core complex formed between the target (t) SNAREs called syntaxin-4 (Syx-4) and soluble N-ethylmaleimide-sensitive factor attachment protein 23 (SNAP-23) and a vesicle (v) SNARE called vesicle associated membrane protein 3 (VAMP-3). Ser/Thr phosphatases may interact with SNARE- associated proteins like Munc18c and possibly other vesicle trafficking proteins directly or indirectly to maintain a dephosphorylated state. In the absence of CsA, Munc18c interacts with syntaxin-4 (Syx-4), and prevents VAMP-3 binding. CsA treatment results in the CsA-cyclophilin complex formation that inhibits PP2B and triggers serine phosphorylation of Munc18c. This induces a conformational change in Munc18c such that Munc18c has a decreased affinity for Syx-4. Munc18c dissociates from this complex thereby enabling VAMP-3 on the WPB to interact with the Syn-4 and SNAP-23 complex to form a core complex that promotes the exocytosis of VWF from the ECs.
It is unlikely that endothelial cell vesicle trafficking proteins are the only targets of PP2B. Cytoskeletal contractile forces facilitate the secretion of granule contents by centralizing, or clustering, organelles. The phosphorylation status of cytoskeletal proteins may be involved in the regulation of granule content secretion. This latter possibility is compatible with observations that generic inhibitors of protein phosphatase type 1, 2A and 4 (as by okadaic acid) enhance WPB clustering, which precedes the secretion of WPB contents by cAMP stimulating secretagogues [11]. Studies using CsA and tacrolimus indicate that PP2B may also participate in the regulation of granular exocytosis in other cells, including neurons [28-31] pancreatic islet cells [32], mast cells [33] and T-lymphocytes [34].
In summary, our studies demonstrate that inhibition of endothelial cell PP2B by CsA or tacrolimus promotes the secretion of ULVWF/VWF multimers. These observations are compatible with the clinically observed association of CsA treatment and increased plasma VWF levels in humans, as well as the association between prolonged exposure to CsA and thrombotic microangiopathy in a subset of exposed patients. This result implies that uninhibited PP2B is normally involved in suppressing ULVWF/VWF secretion. Chemical or pharmacologic manipulation of PP2B activity designed to control ULVWF/VWF secretion by endothelial cells may have potential clinical utility in the treatment of thrombotic disorders.
Acknowledgments
This work was supported in part by grants HL081613 (K.V.V), HL071895 (J.F.D) from the National Institute of Health (NIH), and the Mary Gibson Foundation (J.L.M). K.V.V and F.C.G are supported by National Scientist Development grants 0435017N and 0630180N respectively from the American Heart Association. S.P is supported in part by a NIH grant T-32HL072754. We acknowledge Vimal Patel for his excellent technical help in experiments involving the phosphorylation of Munc18c and measuring phosphatase activity in HUVECs.
References
- 1.Liu J, Farmer JD, Jr, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66:807–15. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- 2.Zakarija A, Bennett C. Drug-induced thrombotic microangiopathy. Semin Thromb Hemost. 2005;31:681–690. doi: 10.1055/s-2005-925474. [DOI] [PubMed] [Google Scholar]
- 3.Schrama YC, van Dam T, Fijnheer R, Hene RJ, de Groot P, Rabelink TJ. Cyclosporine is associated with endothelial dysfunction but not with platelet activation in renal transplantation. Neth J Med. 2001;59:6–15. doi: 10.1016/s0300-2977(01)00127-9. [DOI] [PubMed] [Google Scholar]
- 4.Dong JF, Moake JL, Nolasco L, Bernardo A, Arceneaux W, Shrimpton CN, Schade AJ, McIntire LV, Fujikawa K, Lopez JA. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood. 2002;100:4033–39. doi: 10.1182/blood-2002-05-1401. [DOI] [PubMed] [Google Scholar]
- 5.Nolasco LH, Turner NA, Bernardo A, Tao Z, Cleary TG, Dong JF, Moake JL. Hemolytic uremic syndrome-associated Shiga toxins promote endothelial-cell secretion and impair ADAMTS13 cleavage of unusually large von Willebrand factor multimers. Blood. 2005;106:4199–4209. doi: 10.1182/blood-2005-05-2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rondaij MG, Sellink E, Gijzen KA, ten Klooster JP, Hordijk PL, van Mourik JA, Voorberg J. Small GTP-binding protein Ral is involved in cAMP-mediated release of von Willebrand factor from endothelial cells. Arterioscler Thromb Vasc Biol. 2004;24:1315–20. doi: 10.1161/01.ATV.0000131267.13425.45. [DOI] [PubMed] [Google Scholar]
- 7.de Leeuw HP, Fernandez-Borja M, Reits EA, Romani dW, Wijers-Koster PM, Hordijk PL, Neefjes J, van Mourik JA, Voorberg J. Small GTP-binding protein Ral modulates regulated exocytosis of von Willebrand factor by endothelial cells. Arterioscler Thromb Vasc Biol. 2001;21:899–904. doi: 10.1161/01.atv.21.6.899. [DOI] [PubMed] [Google Scholar]
- 8.Rondaij MG, Bierings R, van Agtmaal EL, Gijzen KA, Sellink E, Kragt A, Ferguson SS, Mertens K, Hannah MJ, van Mourik JA, Fernandez-Borja M, Voorberg J. Guanine exchange factor RalGDS mediates exocytosis of Weibel-Palade bodies from endothelial cells. Blood. 2008;112:56–63. doi: 10.1182/blood-2007-07-099309. [DOI] [PubMed] [Google Scholar]
- 9.Knop M, Aareskjold E, Bode G, Gerke V. Rab3D and annexin A2 play a role in regulated secretion of vWF, but not tPA, from endothelial cells. EMBO J. 2004;23:2982–92. doi: 10.1038/sj.emboj.7600319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Disse J, Vitale N, Bader MF, Gerke V. Phospholipase D1 is specifically required for regulated secretion of von-Willebrand factor from endothelial cells. Blood. 2008 doi: 10.1182/blood-2008-06-165282. [DOI] [PubMed] [Google Scholar]
- 11.Rondaij MG, Bierings R, Kragt A, Gijzen KA, Sellink E, van Mourik JA, Fernandez-Borja M, Voorberg J. Dynein-dynactin complex mediates protein kinase A-dependent clustering of Weibel-Palade bodies in endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26:49–55. doi: 10.1161/01.ATV.0000191639.08082.04. [DOI] [PubMed] [Google Scholar]
- 12.Matsushita K, Morrell CN, Cambien B, Yang SX, Yamakuchi M, Bao C, Hara MR, Quick RA, Cao W, O'Rourke B, Lowenstein JM, Pevsner J, Wagner DD, Lowenstein CJ. Nitric oxide regulates exocytosis by S-nitrosylation of N-ethylmaleimide-sensitive factor. Cell. 2003;115:139–50. doi: 10.1016/s0092-8674(03)00803-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu J, Naren AP, Gao X, Ahmmed GU, Malik AB. Protease-activated receptor-1 activation of endothelial cells induces protein kinase Ca-dependent phosphorylation of syntaxin 4 and Munc18c: role in signaling p-selectin expression. J Biol Chem. 2005;280:3178–84. doi: 10.1074/jbc.M410044200. [DOI] [PubMed] [Google Scholar]
- 14.Snyder DA, Kelly ML, Woodbury DJ. SNARE complex regulation by phosphorylation. Cell Biochem Biophys. 2006;45:111–23. doi: 10.1385/CBB:45:1:111. [DOI] [PubMed] [Google Scholar]
- 15.Lorenzi O, Frieden M, Villemin P, Fournier M, Foti M, Vische UM. Protein kinase C-delta mediates von Willebrand factor secretion from endothelial cells in response to vascular endothelial growth factor (VEGF) but not histamine. J Thromb Haemost. 2008;6:1962–69. doi: 10.1111/j.1538-7836.2008.03138.x. [DOI] [PubMed] [Google Scholar]
- 16.Shibasaki F, Hallin U, Uchino H. Calcineurin as a multifunctional regulator. J Biochem (Tokyo) 2002;131:1–15. doi: 10.1093/oxfordjournals.jbchem.a003063. [DOI] [PubMed] [Google Scholar]
- 17.Kissinger CR, Parge HE, Knighton DR, Lewis CT, Pelletier LA, Tempczyk A, Kalish VJ, Tucker KD, Showalter RE, Moomaw EW, Louis NG, Noriyuki H, Xinghai C, Fausto M, John EB, Russell B&J, Ernest V. Crystal structures of human calcineurin and the human FKBP12-FK506-calcineurin complex. Nature. 1995;378:641–4. doi: 10.1038/378641a0. [DOI] [PubMed] [Google Scholar]
- 18.Hashimoto Y, Perrino BA, Soderling TR. Identification of an autoinhibitory domain in calcineurin. J Biol Chem. 1990;265:1924–27. [PubMed] [Google Scholar]
- 19.Nichols WC, Cooney KA, Mohlke KL, Ballew JD, Yang A, Bruck ME, Reddington M, Novak EK, Swank RT, Ginsburg D. von Willebrand disease in the RIIIS/J mouse is caused by a defect outside of the von Willebrand factor gene. Blood. 1994;83:3225–31. [PubMed] [Google Scholar]
- 20.Terada H, Matsushita M, Lu YF, Shirai T, Li ST, Tomizawa K, Moriwaki A, Nishio S, Date I, Ohmoto T, Matsui H. Inhibition of excitatory neuronal cell death by cell-permeable calcineurin autoinhibitory peptide. J Neurochem. 2003;87:1145–51. doi: 10.1046/j.1471-4159.2003.02098.x. [DOI] [PubMed] [Google Scholar]
- 21.Collins P, Wilkie M, Razak K, Abbot S, Harley S, Bax C, Zaidi M, Blake D, Cunningham J, Newland A. Cyclosporine and cremaphor modulate von Willebrand factor release from cultured human endothelial cells. Transplantation. 1993;56:1218–23. doi: 10.1097/00007890-199311000-00032. [DOI] [PubMed] [Google Scholar]
- 22.Evans SM, Giddings JC, Muraki T, Yamamoto J. Expression of von Willebrand factor, P-selectin (CD62P) and thrombomodulin in human endothelial cells in culture modulated by cyclosporin A. Clin Lab Haematol. 1997;19:115–22. doi: 10.1046/j.1365-2257.1997.00058.x. [DOI] [PubMed] [Google Scholar]
- 23.Medina PJ, Sipols JM, George JN. Drug-associated thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Curr Opin Hematol. 2001;8:286–93. doi: 10.1097/00062752-200109000-00004. [DOI] [PubMed] [Google Scholar]
- 24.Moake JL, Byrnes JJ. Thrombotic microangiopathies associated with drugs and bone marrow transplantation. Hematol Oncol Clin North Am. 1996;10:485–97. doi: 10.1016/s0889-8588(05)70348-8. [DOI] [PubMed] [Google Scholar]
- 25.Hansson MJ, Persson T, Friberg H, Keep MF, Rees A, Wieloch T, Elmer E. Powerful cyclosporin inhibition of calcium-induced permeability transition in brain mitochondria. Brain Res. 2003;960:99–111. doi: 10.1016/s0006-8993(02)03798-8. [DOI] [PubMed] [Google Scholar]
- 26.Matsuda S, Koyasu S. Mechanisms of action of cyclosporine. Immunopharmacology. 2000;47:119–25. doi: 10.1016/s0162-3109(00)00192-2. [DOI] [PubMed] [Google Scholar]
- 27.Imai A, Nashida T, Shimomura H. Roles of Munc18-3 in amylase release from rat parotid acinar cells. Arch Biochem Biophys. 2004;422:175–82. doi: 10.1016/j.abb.2003.12.021. [DOI] [PubMed] [Google Scholar]
- 28.Victor RG, Thomas GD, Marban E, O'Rourke B. Presynaptic modulation of cortical synaptic activity by calcineurin. Proc Natl Acad Sci U S A. 1995;92:6269–73. doi: 10.1073/pnas.92.14.6269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nichols RA, Suplick GR, Brown JM. Calcineurin-mediated protein dephosphorylation in brain nerve terminals regulates the release of glutamate. J Biol Chem. 1994;269:23817–23. [PubMed] [Google Scholar]
- 30.Sihra TS, Nairn AC, Kloppenburg P, Lin Z, Pouzat C. A role for calcineurin (protein phosphatase-2B) in the regulation of glutamate release. Biochem Biophys Res Commun. 1995;212:609–16. doi: 10.1006/bbrc.1995.2013. [DOI] [PubMed] [Google Scholar]
- 31.Lin MJ, Lin-Shiau SY. Enhanced spontaneous transmitter release at murine motor nerve terminals with cyclosporine. Neuropharmacology. 1999;38:195–98. doi: 10.1016/s0028-3908(98)00178-6. [DOI] [PubMed] [Google Scholar]
- 32.Ebihara K, Fukunaga K, Matsumoto K, Shichiri M, Miyamoto E. Cyclosporin A stimulation of glucose-induced insulin secretion in MIN6 cells. Endocrinology. 1996;137:5255–63. doi: 10.1210/endo.137.12.8940343. [DOI] [PubMed] [Google Scholar]
- 33.Hultsch T, Brand P, Lohmann S, Saloga J, Kincaid RL, Knop J. Direct evidence that FK506 inhibition of FcepsilonRI-mediated exocytosis from RBL mast cells involves calcineurin. Arch Dermatol Res. 1998;290:258–63. doi: 10.1007/s004030050301. [DOI] [PubMed] [Google Scholar]
- 34.Grybko MJ, Bartnik JP, Wurth GA, Pores-Fernando AT, Zweifach A. Calcineurin activation is only one calcium-dependent step in cytotoxic T lymphocyte granule exocytosis. J Biol Chem. 2007;282:18009–17. doi: 10.1074/jbc.M702222200. [DOI] [PubMed] [Google Scholar]
- 35.Hodgkinson CP, Mander A, Sale GJ. Protein kinase-zeta interacts with munc18c: role in GLUT4 trafficking. Diabetologia. 2005;48:1627–36. doi: 10.1007/s00125-005-1819-y. [DOI] [PubMed] [Google Scholar]