

Abstract
The discovery of disease-causing mutations typically requires confirmation of the variant or gene in multiple unrelated individuals, and a large number of rare genetic diseases remain unsolved due to difficulty identifying second families. To enable the secure sharing of case records by clinicians and rare disease scientists, we have developed the PhenomeCentral portal (https://phenomecentral.org). Each record includes a phenotypic description and relevant genetic information (exome or candidate genes). PhenomeCentral identifies similar patients in the database based on semantic similarity between clinical features, automatically prioritized genes from whole-exome data, and candidate genes entered by the users, enabling both hypothesis-free and hypothesis-driven matchmaking. Users can then contact other submitters to follow up on promising matches. PhenomeCentral incorporates data for over 1,000 patients with rare genetic diseases, contributed by the FORGE and Care4Rare Canada projects, the US NIH Undiagnosed Diseases Program, the EU Neuromics and ANDDIrare projects, as well as numerous independent clinicians and scientists. Though the majority of these records have associated exome data, most lack a molecular diagnosis. PhenomeCentral has already been used to identify causative mutations for several patients, and its ability to find matching patients and diagnose these diseases will grow with each additional patient that is entered.
Keywords: deep phenotyping, HPO, patient matchmaking, semantic similarity, Matchmaker Exchange
Introduction
The availability of low-cost whole-exome sequencing (WES) has revolutionized the study and diagnosis of rare diseases. WES has enabled the discovery of the genetic cause for well over 180 Mendelian diseases [Boycott et al., 2013], opening up new avenues to diagnostics and treatment. While some disorders have been solved by identifying a cohort and discovering a commonly mutated gene, or by investigating the inheritance of a suspected genetic variant through a large family (linkage), many others are not amenable to such approaches due to the lack of family history, including extremely rare recessive disorders and dominant disorders caused by de novo mutations [Beaulieu et al., 2014]. Even when WES screening identifies a single candidate variant in a previously uncharacterized disease gene, this is typically insufficient evidence to implicate it in the disease.
A promising method for identifying the causes of such rare conditions is the comparison of the exomes or genomes of unrelated individuals with a specific disorder. However, identifying multiple unrelated patients with similar indications is a nontrivial task. Such patients will likely be seen by different clinicians at different hospitals, and the clinician working with one family will often be unaware of other cases. Historically, such cases have been shared by clinicians through case reports, while attending “unsolved” symposia, or through other interactions, where one clinician may present an unsolved case and another recalls a similar case. While the causes of some rare diseases have been identified in this manner, such interactions are serendipitous and may take years to occur, if ever. A centralized Web portal that enables clinicians to effectively share undiagnosed patient information could allow similar patients to be instantly found anywhere in the world and significantly shorten the time to gene discovery and diagnosis.
The effective sharing of phenotypic and genetic data between clinicians is fraught with social and technical challenges. On the social level, in addition to privacy constraints, it is necessary to build trust between the two individuals; information about rare disease patients is scientifically valuable, and many clinicians have a natural tendency not to share the data if it may be used without appropriate attribution. Thus, in any solution, one user should not be able to access data contributed by another user without that person’s knowledge or consent. Once trust is established, there are additional difficulties. Sharing of standard rare disease names, for example, based on OMIM [Amberger et al., 2015] or Orphanet (http://www.orpha.net/) is not practical when the very goal of data sharing is the diagnosis of a disorder. Further, standardized data such as billing codes (ICD-9) and even SNOMED CT terms typically have insufficient granularity and completeness to describe rare disorders. Finally, sharing free-form textual descriptions of patients makes comparison complicated, as different clinicians (depending on their specialization) may collect and record different granularity of patient data, and may use different terminology to describe the same set of clinical features.
The recording of detailed and standardized phenotypes for patients displaying a broad variety of indications requires a rich vocabulary with clear semantic relationships between the terms, to allow for the identification of similar (yet not identical) indications. Although the London Dysmorphology Database presented one of the first efforts to organize phenotypes typically seen by a clinical geneticist, the Human Phenotype Ontology (HPO) [Köhler et al., 2014] is currently the most complete vocabulary available for recording patient phenotypes for genetic diseases [Winnenburg and Bodenreider, 2014]. However, the broad use of the HPO is hindered by its size and complexity: the HPO has over 11,000 terms, and only a small fraction of these are relevant for a specific patient. Intuitive user interfaces, such as PhenoTips [Girdea et al., 2013], can help clinicians record precise descriptions of their patients and allow for the use of synonyms and variable granularity of presentations. Further, when clinical features are encoded using an ontology such as the HPO, cases can be compared not just based on the annotated features, but also the corresponding semantic annotations of these features. The HPO defines the terminology (and synonyms), as well as the relationship between terms (e.g., both “focal seizures” and “tonic/clonic seizure” are subtypes of “seizures,” which in turn is a subtype of “neurological abnormality”), allowing for computational reasoning about similarities between patient descriptions.
Many similarity measures exist for comparing terms, or sets of terms, from an ontology. Foundational methods, such as Resnik’s measure [Resnik, 1995] and Jiang’s and Conrath’s measure [Jiang and Conrath, 1997], were developed for lexical analysis and rose to prominence in the field of bioinformatics with their application to the gene ontology (GO) [Ashburner et al., 2000]. Additional measures, such as simGIC [Pesquita et al., 2007], were developed specifically for use with the GO. Pesquita et al. [2009] provides an excellent review of the most popular similarity measures and their performance on GO-related tasks. More recently, the HPO has enabled the use of semantic similarity measures to predict clinical diagnoses [Köhler et al., 2009; Bauer et al., 2012; Zemojtel et al., 2014], to find representative model organisms for gene prioritization [Smedley et al., 2013], and most recently, to identify similar patients [Gottlieb et al., 2015].
The HPO also maintains links between phenotypic terms and known rare diseases (including those in Orphanet and OMIM) and their associated genes [Köhler et al., 2014], which provides an effective bridge from phenotype to genotype within the rare disease domain. A number of recently published methods have focused on using HPO terms to improve the prioritization of candidate genes from exome sequence data, including PHIVE [Robinson et al., 2014], Phevor [Singleton et al., 2014], Phen-Gen [Javed et al., 2014], and PhenIX [Zemojtel et al., 2014]. However, to our knowledge, no existing methods attempt to address these two problems simultaneously: finding similar patients and identifying associated genes for those matches.
Here, we present the PhenomeCentral Web portal, a restricted-access network for clinicians, researchers, and scientific consortia to share patient phenotype and genotype data and discover similar patients across the world. PhenomeCentral allows clinicians and researchers to enter deidentified patient phenotype and genotype data through a user-friendly Web interface, discover the existence of other similar patients along with potential shared genetic mechanisms, and contact the submitters of these cases to share case reports and foster worldwide collaborations. To enable the discovery of genetic mechanisms for a disease in a hypothesis-free manner, PhenomeCentral simultaneously identifies patients likely to have the same disease and predicts genes that are potentially responsible. PhenomeCentral serves as the data repository for several rare disease consortia, including the FORGE Canada and Care4Rare Canada projects, US NIH Undiagnosed Diseases Project, Neuromics, and ANDDIrare, and at the time of writing had 1,027 case reports and 391 users across five continents. PhenomeCentral is part of the Matchmaker Exchange (MME) [Philippakis et al., 2015], and has implemented the MME API [Buske et al., 2015] to enable submitted cases to be matched with records at other MME sites.
Results
The PhenomeCentral Web Portal
Portal overview
The PhenomeCentral Web portal enables clinicians and researchers to quickly and easily find similar patients submitted by other contributors. Users can enter deidentified patient data directly into PhenomeCentral through a user-friendly Web interface based on the PhenoTips software [Girdea et al., 2013], by pushing existing patient records from private PhenoTips installations, or by uploading them in bulk (Fig. 1A–C).
Figure 1.

Finding similar patients in PhenomeCentral. Patient data can be contributed to PhenomeCentral through the PhenoTips user interface, including the phenotype quick search box that enables rapid entry of phenotype terms from the HPO (A), or selected records can be automatically deidentified and transferred from any PhenoTips instance automatically. The patient record can contain both present and absent phenotypic features (B) as well as genetic information, including candidate genes and VCF files (C). The patient’s features are then immediately compared with all other patients in PhenomeCentral (D), and the best matches are shown to the user. A detailed breakdown of the phenotypic (E) and genotypic (F) similarity is shown for each match, enabling the user to see the underlying reasons for the match and determine whether or not the match is worth following up. A customizable email template (G) facilitates contacting the (potentially undisclosed) submitter of another patient record.
Rather than a traditional database that users query using a sophisticated language, users “query” the PhenomeCentral repository simply by contributing a patient record. The critical fields for match-making are “Clinical symptoms and physical findings,” which allows for the selection (presence or absence) of relevant phenotypic terms from the HPO, and “Genotype information,” where genetic variants can be entered and uploaded. The phenotype terms can be selected either using quick search functionality, or through a set of (expandable) check boxes. PhenomeCentral supports both entering a curated set of candidate genes and uploading a VCF file (with patient consent). The VCF file is automatically processed using the Exomiser software (http://www.sanger.ac.uk/resources/databases/exomiser/) to identify an additional, computationally prioritized set of candidate genes. The set of selected phenotypic terms and candidate genes for each patient are compared with those in all other patient records in the repository, using algorithms described below. This “query by example” approach frees the users from the responsibility of composing the right interrogation, and gives them incentive to contribute data and participate in the growth of PhenomeCentral. The user can immediately see information about the most similar other patients in the database and contact those submitters, in a way that preserves the privacy of the patients (Fig. 1D–G).
The PhenomeCentral user interface incorporates many other popular components of the PhenoTips software on which it is based, including support for entering case notes, relevant medical records, and measurements; automatic plotting of growth charts; and drawing of pedigrees. Combined, this functionality enables the recording of all relevant study data within a single portal, and allows research consortia to use PhenomeCentral as their primary data repository. The Clinical Genetics journal has also adopted PhenomeCentral as the preferred repository for depositing structured phenotype data associated with case reports published in the journal.
PhenomeCentral facilitates collaboration between clinicians
Each patient record in PhenomeCentral can be set to one of three different visibility settings:
-
-
Private: the record is visible only to the submitter unless explicitly shared with other users or groups, and does not participate in any matchmaking activity.
-
-
Matchable: the record is not directly visible unless explicitly shared, but similar patients are shown to the submitter, and other contributors with similar patients can discover the existence of the record. The matched phenotypes and genomic variants are obfuscated (phenotypes are made more general and only gene-level information is provided).
-
-
Public: the record is visible to all registered users on PhenomeCentral and participates in matchmaking activity. Contributors of similar patients are shown the submitter’s contact details and the matched phenotypes and genomic variants. Whenever a whole exome is provided for a public case, only the top 10 potential causal variants (ranked by the Exomiser) are shown to other users.
Private records are most useful when a consortium enforces a period of direct data sharing among its scientists before broader sharing is allowed (e.g., in the Neuromics consortium there is a 6-month waiting period before any sharing). Matchable records allow patients that are not yet published or consented for full sharing to participate in matchmaking activities with enhanced patient and submitter privacy. Contacting the submitter of a matchable case is simplified with a customizable message template, allowing the user to quickly and easily choose what patient information to include in the message and add a personal message (Fig. 1G).
A rapidly growing repository of patients for matchmaking
Matches can also be found in other databases outside of PhenomeCentral through the MME API [Buske et al., 2015]. To further increase the number of potential matches, contributors can opt to include their patient records in matchmaking across the MME [Philippakis et al., 2015], a federated network of patient databases of which PhenomeCentral is a core founding member. The contributor is then shown the most similar patients across partner sites including GeneMatcher [Sobreira et al., 2015] and DECIPHER [Chatzimichali et al., 2015], and users on these partner sites can discover the existence of this record if they have contributed a similar patient (additional details in the Methods). This enables clinicians to instantly find and compare similar patients, even across different patient databases. Supp. Fig. S1 shows this user interface for a promising match that is currently being validated.
Submitting a patient record to PhenomeCentral enables high-quality matchmaking against a rapidly growing number of cases. PhenomeCentral has seen a consistent rise in the number of patient records and user accounts since the first collaborators started submitting data in June 2013, with the number of patients tripling since the official launch on Rare Disease Day, February 28, 2014 (Fig. 2A). As of April 19, 2015, PhenomeCentral contains data from 1,027 clinically phenotyped patients with rare genetic diseases (1,243 records in total, including unaffected relatives) collectively entered by 391 user accounts spanning five continents (Fig. 2B).
Figure 2.
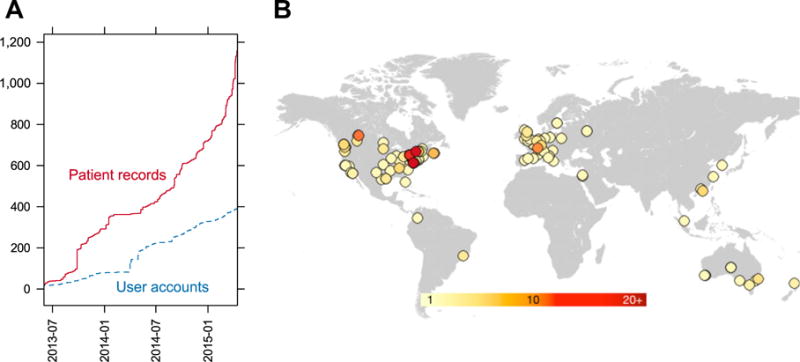
A: The number of patient records (red solid line) and user accounts (blue dashed line) on PhenomeCentral overtime. B: The locations of PhenomeCentral users, estimated from the domain name of institutional email addresses associated with user accounts. The approximate region was identified by querying freegeoip.net with the IP address associated with the domain name of each email address. One point is plotted per domain name, with the color corresponding to the number of users with that domain (the darker the color, the more users with email addresses on that domain).
Finding matches and identifying causal genes
For a given query patient, PhenomeCentral seeks to identify similar patients for which there is a plausible common genetic mechanism. PhenomeCentral first calculates the phenotypic similarity between the query and every other patient in the database. Because clinical features are encoded using terms in an ontology (the HPO), distinct terms can be compared using semantic similarity measures that take into account the structure of the ontology and the specificity of each term. Similar patients can therefore be identified even when the two patients have no annotated terms in common. For the most similar patients, we identify genes with predicted harmful variants in both patients, and rank these genes based on their relevance to the observed phenotypes. Patients with overlapping manually curated candidate genes are considered similar even in the absence of phenotypic similarity.
Benchmarking PhenomeCentral’s Ability to Find Matches and Identify Causal Genes
Test data sets
We validated our approach to matchmaking using two data sets (additional details in the Methods). First, we created a synthetic test set of 1,000 patients, each with an OMIM diagnosis, an exome spiked with a pathogenic variant in a disease-associated gene, and a set of HPO terms associated with the disease. Patients were sampled in pairs with the same OMIM diagnosis, so every synthetic patient had at least one match with the same disease. Second, we selected a subset of the real patient records in PhenomeCentral. As of April 20, 2015, PhenomeCentral contained 720 patient records with at least five observed clinical features. We considered two patients to match if they were submitted as part of the same cohort, diagnosed with the same disease, or annotated with the same gene as a likely candidate or confirmed cause. These criteria resulted in 225 real cases with at least one match in the database. To benchmark the gene prioritization methods, we selected the subset of deeply phenotyped records that also had WES data available and one or more known candidate or causal genes. This resulted in 112 real cases with detailed phenotype and genotype data with a human-curated strong candidate gene, which we used as our gold standard.
Phenotypic matching
Because PhenomeCentral uses the HPO to represent phenotypic features, we can measure the similarity between pairs of patients using well-established semantic similarity measures. We compared 13 measures from the literature (see Supp. Table S1). The two best-performing measures were the PhenoDigm score [Smedley et al., 2013] and the simGIC score [Pesquita et al., 2007]. On simulated data, the PhenoDigm score outperformed all other measures and ranked the true match first for 69% of the patients after introducing random phenotypic noise and imprecision, and within the top five patients 90% of the time. The performance was similar when the information content was computed using a smaller corpus and when it was computed using the topology of the HPO (Supp. Fig. S2). On real data, the PhenoDigm and simGIC scores performed comparably, ranking the true match first for 54%–60% of patients, and within the top five matches 70%–71% of the time (Fig. 3A). However, when phenotypic noise and imprecision were added to these real patients, the performance dropped considerably, with respective top one and top five recall rates of 11% and 29% for PhenoDigm, and only 3% and 9% for simGIC.
Figure 3.
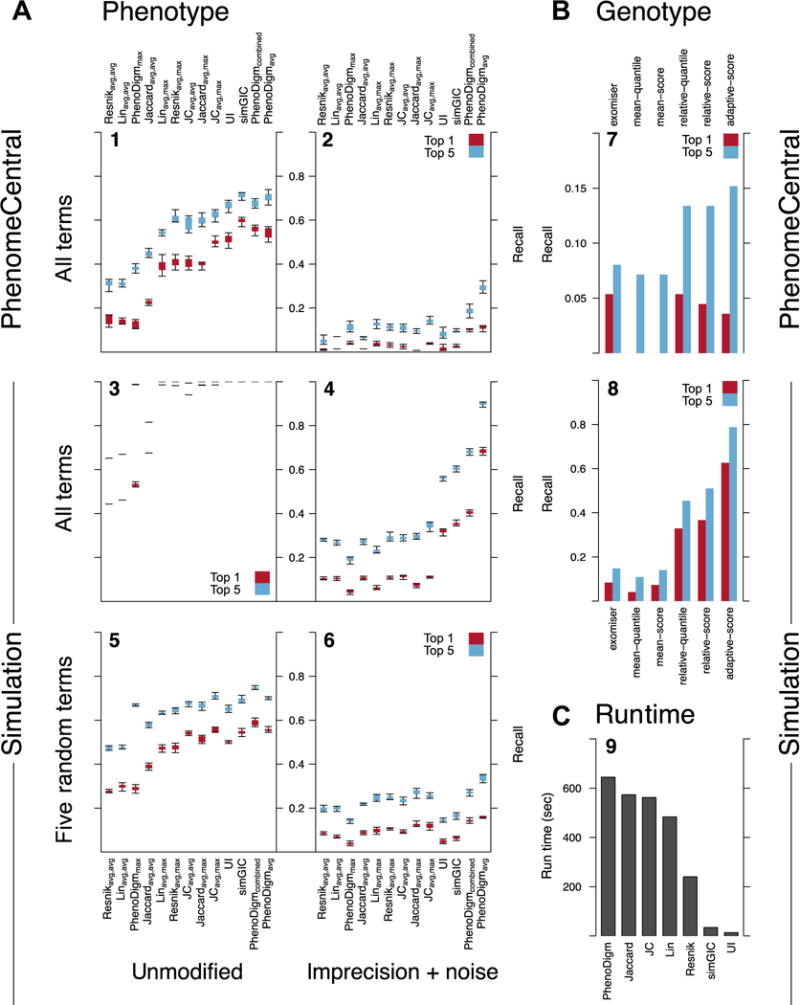
A: Comparison of the performance of 13 semantic similarity measures at finding similar patients in real PhenomeCentral cases (N = 720; panels 1 and 2) and simulated cases (N = 1,000; panels 3–6). For simulated patients, either all disease-associated phenotype terms were selected (panels 3 and 4), or five terms were randomly selected (panels 5 and 6). Noise (40% additional random phenotype terms) and imprecision (replacing terms with a random ancestor) were then introduced (panels 2, 4, 6). Cases were considered similar if they were sampled from the same OMIM disease in simulated cases, and if they shared a candidate gene, shared a diagnosis, or were submitted as part of the same cohort for real cases. To control for variable cohort size (2–12 for simulated cases, 2–14 for real cases), two cases were randomly selected from each cohort for each of 10 iterations. The performance of each measure is the fraction of cases for which the matching case was ranked within the top one (red/dark) or five (blue/light) most similar cases. The box extends from the first to third data quartile, with whiskers extending to the most extreme data point at most 1.5 times the interquartile range away from the box. Measures were ordered by mean top-five performance across all experiments. B: Comparison of the performance of six methods at prioritizing causal and candidate genes in 112 cases from PhenomeCentral (top; panel 7) and 1,000 simulated cases with noise and imprecision introduced (bottom; panel 8; same parameters as panel 4). As a baseline method, the Exomiser was run on each case individually and genes ordered by their PHIVE score. This was compared with five methods that first identify the most phenotypically similar patents (using the PhenoDigm score), and then score genes separately for each match. The performance of each method was measured in two ways: the fraction of cases where one of the causal or candidate genes was ranked as the top gene for the most similar patient (red/dark) or among the top five genes (blue/light; either the top gene for one of the four most similar patients or the top gene from the Exomiser directly). C: Execution time of each class of similarity measure on the 1,000 simulated cases (N = 499,500 pairwise comparisons). Measures were implemented in Python using memoization and executed on a single thread of a 32-core Intel Xeon 2.70GHz CPU.
This suggests that PhenoDigm is more robust than simGIC to the type of phenotypic noise we introduce, potentially due to the two very different ways the scores operate. The PhenoDigm score is a normalized average of the best pairwise match for each phenotype in the patient, whereas the simGIC score assesses the overall shared information present across the clinical manifestations of the two patients. This makes PhenoDigm less sensitive to spurious phenotypes and changes in the number of terms. However, the PhenoDigm measure is also more complex to implement and slower to compute (10 min to perform all pair-wise comparisons of 1,000 simulated cases vs. 34 sec for simGIC in our implementations, as shown in Fig. 3C), so the relative utility of each score depends on the size of the data set and the amount of noise in the data.
Gene prioritization
PhenomeCentral currently incorporates the Exomiser for annotating and filtering exome sequence data, and for prioritizing genes by phenotypic relevance. The Exomiser was selected because the source code was readily available and because the PHIVE score [Robinson et al., 2014] uses model organism data instead of human data, decreasing the likelihood that disease-gene associations (e.g., those found in OMIM) published using PhenomeCentral patients were included in the training data for the algorithm. This means that the success rate we achieve on the real data set should be a reasonable estimate of our ability to identify novel genes associated with undiagnosed rare diseases, rather than overfitting to existing knowledge.
By leveraging the genomic data across multiple patients in the database to improve gene prioritization, PhenomeCentral is able to outperform a baseline method that only uses the data for a single patient. Our adaptive scoring method (described in more detail in the Methods) first selects a variant harmfulness threshold, and then discounts the average score for every other case with a variant above the threshold, weighted by the phenotypic similarity of the other case. This reduces the score of genes that are ubiquitously prioritized (such as HLA-A and TTN), while preserving high scores if a large cohort of phenotypically similar patients all have deleterious variants in the same gene. We evaluate the performance of this approach by identifying, for each patient, the most phenotypically similar patients (using the PhenoDigm measure, discussed above) and then combining the top genes for each of the top matches using several approaches, described in the Methods. We compare these genes to the top genes found with Exomiser’s PHIVE score based on the single patient. We find that an adaptive scoring method outperforms all other methods, increasing sevenfold the number of correctly identified causal genes in simulations with phenotypic noise and imprecision (8% vs. 63% rank the causal gene first), and doubling the number of real patients with correctly identified genes (8% vs. 15% of patients having a causal or candidate gene ranked within the top five genes) over using the Exomiser separately on each patient (see Fig. 3B).
PhenomeCentral Identifies Unrelated Patients with Similar Phenotype and Genotype
Here, we describe two successful matches of undiagnosed patients with rare genetic diseases enabled by PhenomeCentral. The first match (Fig. 4A) involves an initially undiagnosed patient who was matched with a group of mandibulofacial dysostosis with microcephaly (MFDM) patients. A mutation identified in EFTUD2, the gene responsible for MFDM [Lines et al., 2012], confirmed the diagnosis. The patient record listed a few typical features of MFDM including microcephaly, micrognathia, and developmental delay; however, many other common features were absent, including most ear abnormalities: microtia/dysplastic pinna(e) (present in ~98% MFDM patients); malformations of the auditory canal, and/or middle ear with associated conductive hearing loss (~77% of MFDM patients) [Lines et al., 2014]. Furthermore, the patient had abnormalities of the heart and hand, but the specific features were atypical compared with those commonly reported in MFDM patients, as well as subglottic stenosis and vocal cord paralysis, which are not characteristic of MFDM. Despite this atypical presentation, PhenomeCentral matched this patient with a patient previously diagnosed with MFDM, and correctly identified EFTUD2 as the causal gene for the pair of patients.
Figure 4.
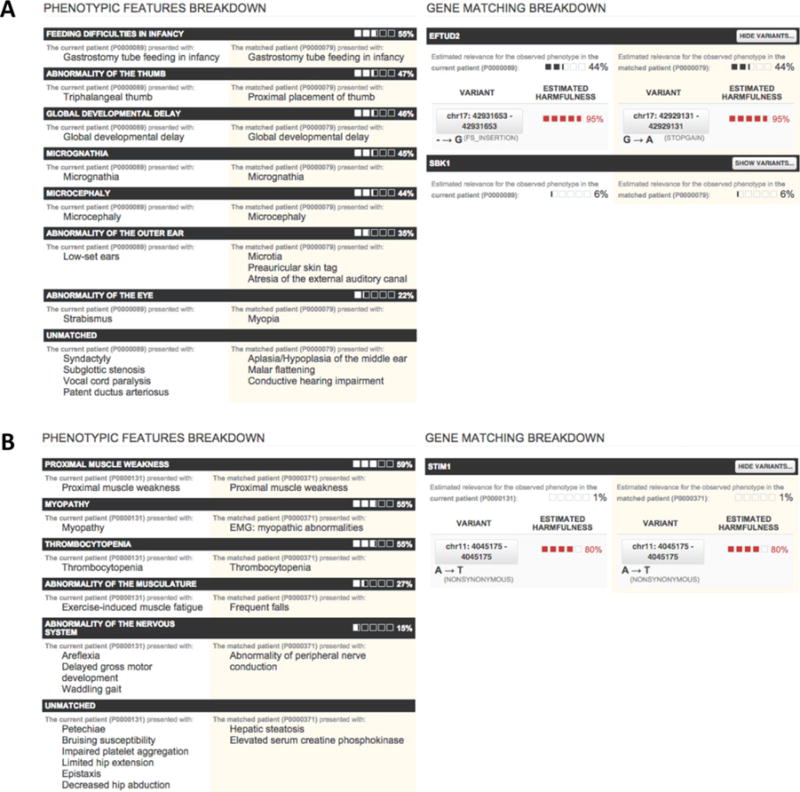
Two validated PhenomeCentral matches, each showing a breakdown of the phenotypic similarity between the two patients on the left, and the genotypic similarity between the two patients on the right. The phenotypes are grouped via a greedy iterative process. In each iteration, the most informative common ancestor is found and all descendants of that term in each patient are removed and displayed as a group. A: The match between two patients with EFTUD2 mutations, where only one was classified as having mandibulofacial MFDM at the outset (the other was described as “CHARGE-like”). B: The match between two patients with STIM1 mutations, subsequently diagnosed with York Platelet syndrome.
In the second match (Fig. 4B), a pair of patients were matched together based on various overlapping abnormal phenotypes in multiple organ systems including myopathy, thrombocytopenia, and peripheral nerve conduction abnormality. Although the patients had overlapping phenotypes, the phenotypes were not specific enough to confirm a diagnosis in either patient. The differential diagnoses included: thrombocytopenia, X-linked, with or without dysthropoietic anemia (MIM #300367), Quebec Platelet Disorder (MIM #601709), and platelet disorder, familial, with associated myeloid malignancy (MIM #601399). However, in addition to these over-lapping phenotypic findings, both patients had the same mutation in the STIM 1 gene, ranked as the top candidate for the pair of patients by PhenomeCentral. Follow-up studies were completed by the clinicians who contributed these patients into PhenomeCentral, and these patients were diagnosed with York platelet syndrome (MIM #185070), which is characterized by thrombocytopenia, striking ultrastructural platelet abnormalities, and deficiency of platelet Ca(2+) storage in delta granules [Markello et al., 2015; Bone et al., in preparation].
Discussion
PhenomeCentral addresses the increasing need for computational approaches to identify individuals affected by the same or overlapping phenotypes and mutations in the same gene, thereby enabling novel gene discovery. We compared the performance of a number of popular semantic similarity measures, and found that the PhenoDigm score is best able to accurately and robustly match patients based on their phenotypes in a diagnosis-free manner. However, for situations where speed and simplicity are important, the simGIC score is an easy-to-implement alternative that provides comparable performance on real data. We also show that combining genetic information across phenotypically similar patients dramatically improves the prioritization of candidate genes. Although the performance of the best method is only 15%, this is an intentionally conservative estimate and reflects our performance on novel diseases using clinical exomes without using any human data for gene prioritization. Because in practice many of the cases submitted may be known disorders with a novel mutation or where the causative mutation was missed by the submitter, PhenomeCentral incorporates an improved scoring measure within the Exomiser [hiPHIVE; Bone et al., in preparation; Smedley et al., in preparation] that combines model organism information in the original PHIVE score with known human disease–gene associations and protein–protein interaction data. This version was not used for benchmarking in this manuscript as it would lead to overly optimistic results, but this functionality is included in PhenomeCentral and clinicians are able to immediately see the top genes associated with each matching patient.
We have tested PhenomeCentral’s ability to identify phenotypically related cases as well as prioritize known disease-causing and candidate genes using both real data available at PhenomeCentral and simulations. These results highlight our ability to identify similar patients and causal genes using only HPO terms and whole-exome VCF files, demonstrating the feasibility of hypothesis-free matchmaking of rare disease cases using computational tools. Simultaneously, hypothesis-driven queries, where a gene of interest is already known, remain a prominent use case, and are also enabled by PhenomeCentral by boosting the similarity score in cases where patients share manually curated candidate genes or diagnoses.
PhenomeCentral is based on the popular PhenoTips software, which makes its user interface familiar to many clinical geneticists, and also allows for the direct transfer of patient records from any other PhenoTips instance to PhenomeCentral. This enables institutional workflows (such as at the NIH Undiagnosed Diseases Program) in which clinicians use the PhenoTips software clinically, storing full patient records within the institutional firewall and then exporting the deidentified phenotypic records (HPO terms and additional needed demographics) to PhenomeCentral to enable matchmaking.
One important functionality, not available in PhenomeCentral but requested by several users and recently implemented in the GeneYenta system [Gottlieb et al., 2015], is the ability to indicate the importance of specific phenotypes for matching. In clinical practice, a specific phenotype may be extremely prominent or severe, and other patients with this phenotype are of especial interest. While PhenomeCentral takes the frequency of a phenotype (in the OMIM corpus) into account, allowing the user to additionally specify such constraints is likely to improve performance as the links between the HPO and OMIM, used in our computation of information content, are incomplete. We find our results to be similar across several different methods and corpora for computing information content, but this incompleteness in mappings can also affect the accuracy of our simulations, as well as simulations by previous authors who utilize these links to “sample” realistic patients. We are also currently exploring adjustments to the similarity score based on additional phenotypic metadata (such as age of onset and severity).
Since its release, PhenomeCentral has been rapidly growing and now contains more than 1,100 deeply phenotyped patients with rare genetic disorders, with accounts for nearly 400 scientists and clinicians. Most of these patients are undiagnosed, and most have exome sequence data. The coordination with the MME also increases the number of potential matches, with the MME API enabling automated querying of GeneMatcher and DECIPHER for records with same candidate genes. With additional sites planning to implement the MME API in the near future, storing deep phenotype and genotype data for all patients in PhenomeCentral will help ensure the maximum potential for matchmaking for these rare disease patients.
Methods
Patient Entry Using the PhenoTips User Interface
PhenomeCentral records all phenotypic information as HPO terms, using the PhenoTips software for patient phenotyping. Two resources are available to help ensure users properly and completely enter the patient’s phenotype into PhenomeCentral. First, the Monarch Initiative and PhenomeCentral jointly developed a set of annotation guidelines and best practices for clinical phenotyping using PhenoTips and the HPO (https://phenomecentral.org/annotation-guidelines). Second, a widget is included in PhenomeCentral that displays the Monarch Initiative’s annotation sufficiency metric [Washington et al., 2014] as a rating from one to five stars. This provides the user with real-time feedback on the specificity of the patient description and encourages the user to enter more terms and more specific terms to phenotype the patient.
In addition to facilitating the accurate recording of detailed phenotypic descriptions in a standardized terminology, the PhenoTips interface used by PhenomeCentral has been configured to record nonidentifiable demographic information such as month and year of birth, sex, ethnicity, family history, as well as any suspected genetic causes of the patient’s disease, expressed either as a list of genes or captured in detail in a VCF file. Currently, only whole-exome data are supported, as whole-genome VCF files are too large to reliably upload through a Web browser. When a VCF file is uploaded for a patient record, the Exomiser is automatically run to identify candidate genes. The Exomiser uses the VCF file, the HPO terms annotated in the patient record, and the mode of inheritance of the patient’s disease if specified, to score genes according to their phenotypic relevance and the estimated harmfulness of variants identified in exome sequencing. The highest-ranked genes are then shown to the user and incorporated in patient matchmaking, both internally and when matching in other databases through the MME.
PhenomeCentral as a Portal to the Matchmaker Exchange
After entering a patient record into PhenomeCentral, the user can opt to have their patient record participate in the MME, a federated network of rare disease patient repositories. This allows the case to be matched to and matched by similar cases in other repositories in the Exchange, currently GeneMatcher and DECIPHER. PhenomeCentral uses the MME API to identify similar patients within other patient repositories. The MME API uses a “query by example” philosophy, where a match request consists primarily of a set of HPO terms and several genetic features (candidate genes or variants). The match request is securely sent to other sites in the MME, and each site instantly responds with a description of the most similar patients in their database and contact details to connect with the submitter of each match. After inspecting the phenotypic and genotypic evidence for each match, the user can follow up on promising matches directly.
At the same time, patient records in PhenomeCentral that participate in the MME can be discovered by users that submit cases into other sites in the MME when those sites send match requests to PhenomeCentral. In these situations, a summary of the phenotypic and genotypic profile of the similar record will be returned, allowing the user of the other site to evaluate the match and follow up if it is promising. To reduce the identifiability of the case, patient details are obfuscated before responding to the match request: phenotypes are replaced by their ancestors and only gene-level genetic data are returned (variant-level details are left out).
Creation of a Test Data Set from Synthetic Patients
To benchmark the performance of phenotypic matching and gene prioritization algorithms, we generated 500 pairs of synthetic patients with the same genetic disease, following the standard protocols used in the literature [Javed etal., 2014; Robinson et al., 2014; Zemojtel et al., 2014]. Each pair of patients was randomly assigned a disease gene and associated OMIM diagnosis, with 250 pairs assigned an autosomal-dominant disease and 250 pairs an autosomal-recessive disease.
The phenotype of each patient was sampled from the set of HPO terms associated with the disease in several ways (HPO version 2014-06-09, disease–phenotype mappings and inheritance mode from the “phenotype_annotation.tab” file released with the HPO). Initially, all phenotype terms were included. We then introduced noise to model the sources of variability and error in real patient records using three methods from Zemojtel et al. (2014). Clinicians frequently use less precise terms, so we artificially introduced imprecision by randomly replacing every term with a term drawn uniformly from the set of ancestors of that term (including the term, excluding the root: HP:0000118 [Phenotypic abnormality]). We modeled variability in clinical presentation and unrelated clinical features both by sampling a random subset of five terms, and by adding two random terms for every five terms in the patient description (sampled uniformly from the set of disease–phenotype associations).
For each patient, we synthesized a corresponding whole-exome VCF file by taking the exome of a healthy control from the 1000 Genomes Project [phase 1 integrated calls; The 1000 Genomes Project Consortium, 2010] and spiking in random pathogenic variants from HGMD (v1.0.3) in the disease-associated gene (one heterozygous variant if the disease was dominant, one homozygous or two heterozygous variants if recessive). HGMD variants were filtered to only nonsynonymous variants overlapping RefSeq coding sequences, and variants explicitly labeled with “associated” or “susceptibility” were ignored. Disease–gene associations were taken from OMIM.org (accessed July 8, 2015). We only considered genes associated with a single OMIM disease, a single inheritance mode, and at least five HPO terms, resulting in 156 autosomal-dominant and 605 autosomal-recessive diseases.
Creation of a Test Data Set from Real PhenomeCentral Cases
The phenotypic similarity benchmark data set was composed of the 720 patients from PhenomeCentral annotated with five or more observed HPO terms. Of these, 225 cases had at least one matching case in the database, where we considered two patients to match if they were submitted as part of the same cohort, diagnosed with the same disease, or annotated with the same gene as a likely candidate or confirmed cause (candidate genes were only used in this case if at most two were specified).
The gene prioritization benchmark data set was composed of the 112 patients from PhenomeCentral annotated with five or more observed HPO terms, for which exome sequence data were available, and annotated with between one and five candidate or causal genes. Whole-exome sequence data were present for 692 of the 1,027 cases in PhenomeCentral. Of these, 20 were filtered out in quality control: 15 exomes were removed because only SNP calls (and not indel calls) were available; five samples were removed due to abnormally high numbers of exonic variants (four of these were the only samples in the data set sequenced using the AB SOLiD platform, and the other sample was processed using a deprecated pipeline on the NCBI36 assembly).
Semantic Similarity for Finding Phenotypically Similar Patients
We compared the performance of 13 different semantic similarity measures (two topological measures and 11 information content measures) by their ability to match patients with the same disease based on their annotated HPO terms. See Supp. Table S1 for the definitions of these terms and Figure 3A for additional description of the benchmarking method and a summary of the results. The Resnik, Lin, JC, and Jaccard measures score the semantic similarity between pairs of terms. To extend these measures to compare two sets of terms, P and Q, we employed two methods commonly found in the literature [Pesquita et al., 2009]: (1) simavg,avg: averaging the score across all pairs of terms; and (2) simavg,best: averaging the score of the best match for each term in P (it is worth noting that the latter produces an asymmetrical similarity measure). In contrast, the PhenoDigm, UI, and simGIC measures directly score the similarity between two sets of terms.
To assess the sensitivity of the information-content-based measures to the particular corpus used to compute it, we evaluated each measure using three different methods for calculating information content (two different corpora and a topological method). The primary method used the disease–phenotype associations from the HPO, the same corpus used to simulate patients. We compared this with information content computed from the corpus of disease–phenotype associations provided by OMIM, as well as directly from the topology of the graph using the same method as GeneYenta.
Prioritization of Candidate Genes for Patient Matches
We used the PHIVE algorithm within the Exomiser to score genes within whole-exome sequence data using Exomiser version 7.0.0beta (built from commit a25c26c3) and the following options:
We then compared this baseline method, which scores genes for a single patient individually, to five other methods that combine these gene scores across patients.
In the first method, mean-score, wetookthemean of the PHIVE score for each gene appearing in the filtered Exomiser output of both cases. For mean-quantile, we instead averaged the quantile score of each gene, where the lowest-scoring gene has a quantile score of 0.0, and the highest-scoring gene has a quantile score of 1.0. For each mean measure, we also implemented a corresponding relative measure that scales the mean score by the mean value across all other cases in the database. For example, the relative-quantile score for a gene g and a pair of cases P and Q is defined as:
If the gene is not in the filtered Exomiser output for the other patient, R, a score/quantile of 0.0 is used. An adaptive measure, adaptive-score, was also compared, in which a per-gene variant harmfulness threshold is calculated, and the score is adjusted by the phenotypic similarity of any other patients with as or more harmful variants in the same gene. Formally:
where sim(P, Q) is the phenotypic similarity between cases P and Q, var(P, g) is the variant harmfulness score of gene g for patient P, and I (P, Q, R, g) is an indicator variable that is 1 if R’s variant harmfulness in gene g is at least that of P or Q and 0 otherwise.
These methods were compared on 1,000 simulated cases, and 112 real cases from PhenomeCentral in which exome data were available, causal or candidate genes were known, and at least five HPO terms were annotated as present. Performance was measured as the fraction of these cases in which a target gene was listed in the top one or top five exome wide. For the Exomiser method, genes were prioritized for each case in isolation. For the five pair-wise measures described above, the most phenotypically similar patients were first found using the PhenoDigm measure. Gene prioritization was then performed by selecting either the top gene from the top match or the combination of the top gene from the top four matches and the top gene directly from the Exomiser (see Fig. 3B for further details).
Supplementary Material
Acknowledgments
We would like to thank Jonathan Zung and Jialin Song for their work on phenotypic similarity measures, and Jules Jacobsen and Damian Smedley for their invaluable assistance with the Exomiser.
Contract grant sponsors: Care4Rare Canada Consortium funded by Genome Canada; Canadian Institutes of Health Research; Ontario Genomics Institute; Ontario Research Fund; Genome Quebec; Children’s Hospital of Eastern Ontario Foundation; Hospital for Sick Children; NSERC/CIHR Collaborative Health Research Project (CHRP); Garron Family Cancer Centre and Hospital for Sick Children Foundation Student Scholarship Program; NSERC Undergraduate Student Research Award.
Footnotes
Additional Supporting Information may be found in the online version of this article.
O.B., M.G., S.D., and M.B. wrote the manuscript. M.G., S.D., O.B., and A.M. developed the software. M.G., S.D. performed PhenoTips integration. B.G., T.H., H.T., C.B., and M.H. entered data and curated patient records. T.H. helped evaluate matches and test the software. K.B. and W.G. contributed data and helped evaluate matches. A.M. implemented the MME API. T.F. and O.B. simulated patients. T.F. mapped user locations. O.B. developed and evaluated matching methods. W.B. and A.L. provided data and assistance. B.G. and M.H. developed annotation guidelines. P.R. provided immense support for use of the HPO.
Disclosure statement: The authors declare no conflict of interest.
Patient data in PhenomeCentral have been obtained in a manner conforming with IRB and/or granting agency ethical guidelines. All presented patient data have been approved by submitting institutions’ respective Research Ethics Boards.
References
- Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015;43:D789–D798. doi: 10.1093/nar/gku1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer S, Köhler S, Schulz MH, Robinson PN. Bayesian ontology querying for accurate and noise-tolerant semantic searches. Bioinformatics. 2012;28(19):2502–2508. doi: 10.1093/bioinformatics/bts471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu CL, Majewski J, Schwartzentruber J, Samuels ME, Fernandez BA, Bernier FP, Brudno M, Knoppers B, Marcadier J, Dyment D, Adam S, Dennis E, et al. Forge Canada Consortium: outcomes of a 2-year national rare-disease gene-discovery project. Am J Hum Genet. 2014;94:809–817. doi: 10.1016/j.ajhg.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bone WP, Washington NL, Buske OJ, Adams DR, Davis J, Draper D, Flynn ED, Girdea M, Godfrey R, Golas G, Groden C, Jacobsen J, et al. Computational evaluation of exome sequence data using human and model organism phenotypes improves diagnostic efficiency. 2015 doi: 10.1038/gim.2015.137. In preparation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boycott KM, Vanstone MR, Bulman DE, MacKenzie AE. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat Rev Genet. 2013;14:681–691. doi: 10.1038/nrg3555. [DOI] [PubMed] [Google Scholar]
- Buske OJ, Schiettecatte F, Hutton B, Dumitriu S, Misyura A, Huang L, Hartley T, Girdea M, Sobreira N, Mungall C, Brudno M. The matchmaker exchange API: Automating patient matching through the exchange of structured phenotypic and genotypic profiles. Hum Mutat. 2015;36:922–927. doi: 10.1002/humu.22850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CK, Mungall CJ, Gkoutos GV, Doelken SC, Köhler S, Ruef BJ, Smith C, Westerfield M, Robinson PN, Lewis SE, Schofield PN, Smedley D. MouseFinder: candidate disease genes from mouse phenotype data. Hum Mutat. 2012;33:858–866. doi: 10.1002/humu.22051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatzimichali E, Brent S, Hutton B, Perrett D, Wright CF, Bevan AP, Hurles ME, Firth HV, Swaminathan GJ. Facilitating collaboration in rare genetic disorders through effective matchmaking in DECIPHER. Hum Mutat. 2015;36:941–949. doi: 10.1002/humu.22842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girdea M, Dumitriu S, Fiume M, Bowdin S, Boycott KM, Chénier S, Chitayat D, Faghfoury H, Meyn MS, Ray PN, So J, Stavropoulos DJ, Brudno M. PhenoTips: patient phenotyping software for clinical and research use. Hum Mutat. 2013;34:1057–1065. doi: 10.1002/humu.22347. [DOI] [PubMed] [Google Scholar]
- Gottlieb MM, Arenillas DJ, Maithripala S, Maurer ZD, TarailoGraovac M, Armstrong L, Patel M, Karnebeek C, Wasserman WW. GeneYenta: a phenotypeBased rare disease case matching tool based on online dating algorithms for the acceleration of exome interpretation. Hum Mutat. 2015;36:432–438. doi: 10.1002/humu.22772. [DOI] [PubMed] [Google Scholar]
- Javed A, Agrawal S, Ng PC. Phen-Gen: combining phenotype and genotype to analyze rare disorders. Nat Meth. 2014;11:935–937. doi: 10.1038/nmeth.3046. [DOI] [PubMed] [Google Scholar]
- Jiang JJ, Conrath DW. Semantic similarity based on corpus statistics and lexical taxonomy. Proceedings of International Conference on Research in Computational Linguistics. 1997:19–33. [Google Scholar]
- Köhler S, Schulz MH, Krawitz P, Bauer S, Dolken S, Ott CE, Mundlos C, Horn D, Mundlos S, Robinson PN. Clinical diagnostics in human genetics with semantic similarity searches in ontologies. Am J Hum Genet. 2009;85:457–464. doi: 10.1016/j.ajhg.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler S, Doelken SC, Mungall CJ, Bauer S, Firth HV, Bailleul-Forestier I, Black GCM, Brown DL, Brudno M, Campbell J, FitzPatrick DR, Eppig JT, et al. The Human Phenotype Ontology project: linking molecular biology and disease through phenotype data. Nucleic Acids Res. 2014;42:D966–D974. doi: 10.1093/nar/gkt1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lines M, Hartley T, Boycott K. Mandibulofacial dysostosis with microcephaly. GeneReviews. 2014 Available from: http://www.ncbi.nlm.nih.gov/books/NBK214367.
- Lines MA, Huang L, Schwartzentruber J, Douglas SL, Lynch DC, Beaulieu C, Guion-Almeida ML, Zechi-Ceide RM, Gener B, Gillessen-Kaesbach G, Nava C, Baujat G, et al. Haploinsufficiency of a spliceosomal GTPase encoded by EFTUD2 causes mandibulofacial dysostosis with microcephaly. Am J Hum Genet. 2012;90:369–377. doi: 10.1016/j.ajhg.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markello T, Chen D, Kwan JY, Horkayne-Szakaly I, Morrison A, Simakova O, Maric I, Lozier J, Cullinane AR, Kilo T, Meister L, Pakzad K, et al. York platelet syndrome is a CRAC channelopathy due to gain-of-function mutations in STIM1. Mol Genet Metab. 2015;114:474–482. doi: 10.1016/j.ymgme.2014.12.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesquita C, Faria D, Bastos H, Falcão A, Couto F. Evaluating go-based semantic similarity measures. Proceedings of 10th Annual Bio-Ontologies Meeting. 2007;37:38. [Google Scholar]
- Pesquita C, Faria D, Falcão AO, Lord P, Couto FM. Semantic similarity in biomedical ontologies. PLoS Comput Biol. 2009;5:e1000443. doi: 10.1371/journal.pcbi.1000443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippakis AA, Azzariti DR, Beltran S, Brookes AJ, Brownstein CA, Brudno M, Brunner HG, Buske OJ, Carey WK, Doll C, Dumitriu S, Dyke SOM, et al. The Matchmaker Exchange: a platform for rare disease gene discovery. Hum Mutat. 2015;36:915–921. doi: 10.1002/humu.22858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnik P. Using information content to evaluate semantic similarity in a taxonomy. Proceedings of the 14th International Joint Conference on Artificial Intelligence. 1995:448–453. [Google Scholar]
- Robinson PN, Köhler S, Oellrich A, Wang K, Mungall CJ, Lewis SE, Washington N, Bauer S, Seelow D, Krawitz P, Gilissen C, Haendel M, Smedley D. Improved exome prioritization of disease genes through cross-species phenotype comparison. Gene Res. 2014;24:340–348. doi: 10.1101/gr.160325.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton MV, Guthery SL, Voelkerding KV, Chen K, Kennedy B, Margraf RL, Durtschi J, Eilbeck K, Reese MG, Jorde LB, Huff CD, Yandell M. Phevor combines multiple biomedical ontologies for accurate identification of disease-causing alleles in single individuals and small nuclear families. Am J Hum Gen. 2014;94:599–610. doi: 10.1016/j.ajhg.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedley D, Jacobsen J, Jäger M, Köhler S, Holtgrewe M, Schubach M, Siragusa E, Zemojtel T, Buske O, Bone W, Haendel M, Robinson PN. Next-generation diagnostics and disease gene discovery with the Exomiser. 2015 doi: 10.1038/nprot.2015.124. In preparation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedley D, Oellrich A, Köhler S, Ruef B, Sanger Mouse Genetics Project. Westerfield M, Robinson P, Lewis S, Mungall C. PhenoDigm: analyzing curated annotations to associate animal models with human diseases. Database. 2013;2013:bat025. doi: 10.1093/database/bat025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: A matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015;36:928–930. doi: 10.1002/humu.22844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington NL, Haendel MA, Köhler S, Lewis SE, Robinson P, Smedley D, Mungall CJ. How good is your phenotyping? Methods for quality assessment. Proceedings of Phenotype Day 2014. 2014 Available from: http://phenoday2014.bio-lark.org/pdf/6.pdf.
- Winnenburg R, Bodenreider O. Coverage of phenotypes in standard terminologies. Proceedings of Phenotype Day 2014. 2014 Available from: http://phenoday2014.bio-lark.org/pdf/5.pdf.
- Zemojtel T, Köhler S, Mackenroth L, Jäger M, Hecht J, Krawitz P, Graul-Neumann L, Doelken S, Ehmke N, Spielmann M, Øien NC, Schweiger MR, et al. Effective diagnosis of genetic disease by computational phenotype analysis of the disease-associated genome. Sci Trans Med. 2014;6:252ra123. doi: 10.1126/scitranslmed.3009262. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.