

Abstract
Voltage-gated CaV1.2 channels (L-type calcium channel α1C subunits) are critical mediators of transcription-dependent neural plasticity. Whether these channels signal via the influx of calcium ion (Ca2+), voltage-dependent conformational change (VΔC), or a combination of the two has thus far been equivocal. We fused CaV1.2 to a ligand-gated Ca2+-permeable channel, enabling independent control of localized Ca2+ and VΔC signals. This revealed an unexpected dual requirement: Ca2+ must first mobilize actin-bound Ca2+/calmodulin-dependent protein kinase II, freeing it for subsequent VΔC-mediated accumulation. Neither signal alone sufficed to activate transcription. Signal order was crucial: Efficiency peaked when Ca2+ preceded VΔC by 10 to 20 seconds. CaV1.2 VΔC synergistically augmented signaling by N-methyl-D-aspartate receptors. Furthermore, VΔC mistuning correlated with autistic symptoms in Timothy syndrome. Thus, nonionic VΔC signaling is vital to the function of CaV1.2 in synaptic and neuropsychiatric processes.
Voltage-gated CaV1.2 channels (L-type calcium channel α1C subunits) play an important role in transcription-dependent forms of synaptic and homeostatic plasticity (1–6), and CaV1.2 alterations have been linked to severe neuropathologies (7, 8). The influx of Ca2+ is required in CaV1.2-mediated transcription (4), but it remains unclear whether voltage-dependent conformational change (VΔC) provides a necessary additional signal. There is precedence for VΔC signaling: CaV1.1 uses only VΔC to initiate skeletal-muscle contraction (9, 10). However, a signaling role for VΔC has been difficult to establish in excitation-transcription coupling, because eliminating voltage-dependent opening of the channel also prevents Ca2+ influx through the CaV [but see (11)].
We fused CaV1.2 to an adenosine triphosphate (ATP)–gated, Ca2+-permeable, tandem-trimeric P2X2 channel (ttP2X) (Fig. 1A). The ttP2X was used to provide Ca2+ influx independently of whether CaV1.2 was open or closed; the tethering between the channels (Fig. 1A) localized ttP2X influx to the CaV1.2 nanodomain (12, 13).
Fig. 1. Decoupling CaV1.2 Ca2+ and VΔC signals reveals dual requirement for CREB activation.
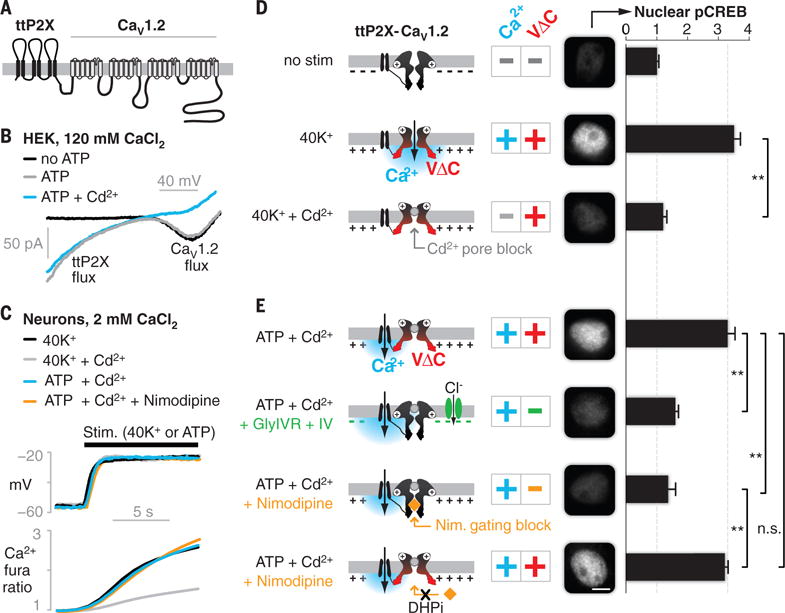
(A) The engineered chimeric construct, showing transmembrane domains and intra- and extracellular loops. (B) Ca2+ currents mediated by the chimeric channel in HEK cells. (C) Changes in voltage (top, current clamp recording) and Ca2+ influx (bottom, fura-2 imaging) under key stimulation conditions (N = 6 cells; fig. S2, A and B, shows SEM). (D and E) Nuclear pCREB in cultured cortical neurons assayed after 3 min of stimulation (conditions are indicated on the left; the cartoons represent the ttP2X-CaV1.2 chimera, with plus and minus signs indicating the presence and absence of the signals, respectively). pCREB was elevated only with dual Ca2+ and VΔC stimuli [second row in (D) and first and fourth rows in (E)]. Elimination of either Ca2+ [third row in (D)] or VΔC [second and third rows in (E)] abrogated signaling. Black bars show means ± SEM (normalized to no stimulation) of 50 cells from three independent cultures. Scale bar, 5 μm. **P < 0.01, determined by one-way analysis of variance (ANOVA) followed by Fisher’s least significant difference test.
We confirmed the integrity of the chimeric protein and the functionality of its components (fig. S1). In human embryonic kidney (HEK) 293 cells, the Ca2+ current appeared only at depolarized potentials in the absence of ATP (Fig. 1B, black u-shaped trace), as expected for a voltage-gated CaV1.2. On addition of ATP, the ttP2X portion of the chimera also supported Ca2+ influx, evident as an additional inward current at negative potentials (Fig. 1B, gray trace). Cd2+, which blocks the CaV1.2 pore (14) but not the opening of ttP2X, did not prevent the ttP2X-mediated entry of Ca2+ (Fig. 1B, blue trace). We further validated the function of the chimeric components in cultured neurons, rendering the CaV1.2 portion dihydropyridine-insensitive (DHPi) to enable its distinction from endogenous channels (6) and confirming that ATP had no effect on untransfected neurons (fig. S1). Whereas ttP2X on its own distributed uniformly over the somatodendritic surface, ttP2X fused to CaV1.2 formed puncta and signaled more potently (fig. S1), consistent with localization to signaling hotspots.
These control experiments framed critical tests of the roles of Ca2+ and VΔC in signaling to nuclear CREB (cyclic adenosine monophosphate response–element binding protein), a transcription factor that is critical in many forms of learning and memory (Fig. 1, D and E) (1, 2, 15). Providing Ca2+ and VΔC in combination via the depolarization of neurons (3 min of exposure to 40 mM K+ (40K+)] (Fig. 1C, black traces) increased the phosphorylation of nuclear CREB (pCREB) at Ser133 threefold (Fig. 1D, second row) (3–5). The pCREB response was abolished by Cd2+ (Fig. 1D, third row), which prevents CaV1.2 from conducting Ca2+ without affecting depolarization (Fig. 1C, gray traces) or voltage-dependent gating (14); this confirms the known requirement for Ca2+ influx in signaling to pCREB (4, 6, 13). Next, we rerouted Ca2+ through the neighboring ttP2X by blocking the CaV1.2 pore with Cd2+ and opening the ttP2X portion of the chimera with ATP. This generated depolarizations and increases in bulk Ca2+ that were nearly identical to those achieved with 40K+ (Fig. 1C, compare blue with black traces, and fig. S2, A and B) and increased pCREB to the same degree as 40K+ (compare Fig. 1E, top row, with Fig. 1D, second row), confirming the utility of the chimeric channel approach.
We thus could use the chimera to determine whether localized Ca2+ influx can drive CREB activation in the absence of VΔC. In a first test, we provided the localized influx of Ca2+ by means of ATP activation of the chimeric ttP2X, but we attenuated VΔC signals through hyperpolarization, which was achieved by coexpressing and activating an ivermectin-gated chloride channel (GlyIVR) (16). This manipulation prevented neuronal depolarization (fig. S2C) and thus increased Ca2+ flux through the chimeric ttP2X (Fig. 1B, blue). Nonetheless, this manipulation inhibited signaling to CREB (Fig. 1E, second row). The attenuation of CREB signaling required a combination of GlyIVR expression and ivermectin (fig. S2D) and could not be attributed to increased Ca2+ influx (fig. S2E).
In a second test, we inhibited CaV1.2 conformational opening with nimodipine. This CaV1-selective agent (17, 18) blocked ATP-mediated CREB signaling (Fig. 1E, third row) without affecting depolarization or Ca2+ influx (Fig. 1C, compare orange with blue). Nimodipine did not block the pCREB response when the chimeric CaV1.2 was rendered nimodipine-insensitive (Fig. 1E, bottom row), excluding potential off-target effects of the drug. Similar results were obtained with CREB-dependent expression of the immediate early gene c-fos (fig. S3). Thus, CaV1.2 signaling to pCREB and gene expression requires two distinct messages: The Ca2+ signal works in conjunction with an equally indispensable VΔC signal arising from CaV1.2.
Previous work has implicated Ca2+/calmodulin-dependent protein kinase II (CaMKII) in mediating the signaling from CaV1.2 to nuclear CREB (3–5, 19). CaMKIIs are activated and recruited into puncta near CaV1.2 upon depolarization (4, 5), where they play a critical role in dispatching a signal to the nucleus (19). We confirmed the critical role of α- and βCaMKII by means of pharmacology (Fig. 2A and fig. S4A) and selective knockdowns of either isoform with small hairpin RNA (shRNA) (Fig. 2, B and C, and fig. S4, B and C). In contrast, signaling remained intact when protein kinase C, protein kinase A, or mitogen-activated protein kinase pathways (3, 6, 20) were blocked (fig. S4A). We further established that αCaMKII forms an activity-dependent complex with CaV1.2: Immunoprecipitation with a CaV1.2 antibody pulled down αCaMKII, and the degree of the coimmunoprecipitation was increased by stimulation with 40K+ (Fig. 2D).
Fig. 2. Redistribution of CaMKII to CaV1.2 puncta requires dual Ca2+ and VΔC signals.
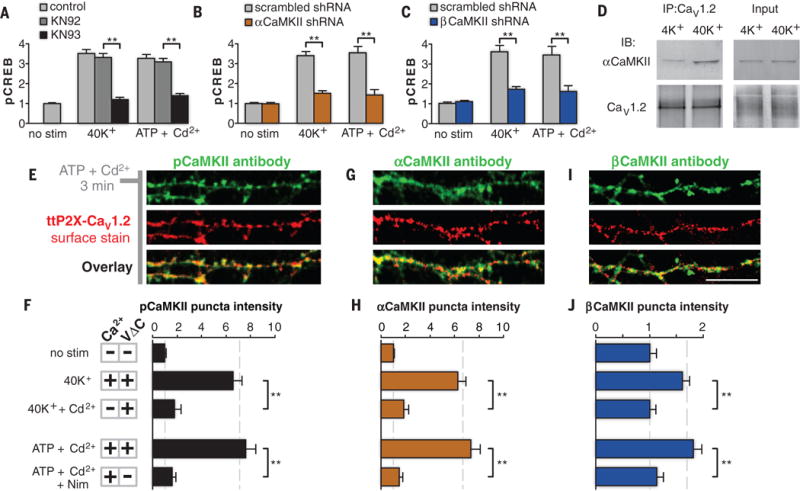
(A to C) pCREB signaling (3 min of 40K+ or ATP-plus-Cd2+ stimulation) was blocked by the CaMKII inhibitor KN93 (KN92 is an inactive analog) (A), or by shRNA against either αCaMKII (B) or βCaMKII (C). (D) Co-immunoprecipitation with antibodies against CaV1.2 indicated that the association between CaV1.2 and αCaMKII increased after stimulation (IB, immunoblotting; IP, immunoprecipitation). (E, G, and I) pCaMKII (E), αCaMKII (G), and βCaMKII (I) in dendrites of cultured cortical neurons after stimulation (ATP plus Cd2+ for 3 min). Their staining is punctate (green) and colocalizes with surface ttP2X-CaV1.2 channels (red). Scale bar, 10 mm. (F, H, and J) Quantification of puncta intensity for pCaMKII (F), αCaMKII (H), and βCaMKII (J) (Nim, nimodipine). Puncta intensity increases with dual Ca2+ and VΔC signals (second and fourth rows) but not with Ca2+ or VΔC signals in isolation (third and fifth rows). The bars show means ± SEM (normalized to no stimulation) of 50 cells from three independent cultures. **P < 0.01, determind by one-way ANOVA followed by Fisher’s least significant difference test.
We next assessed the contributions of Ca2+ and VΔC signals in mediating the spatial recruitment of CaMKII to the CaV1.2 channel. Staining with a phospho-specific CaMKII antibody revealed the formation of intense phospho-CaMKII (pCaMKII) puncta near surface ttP2X-CaV1.2 channels after dual Ca2+ and VΔC stimulation (Fig. 2, E and F), but not after stimulation by Ca2+ or VΔC alone (Fig. 2F). A similar pattern was observed for isoform-specific antibodies against αCaMKII (Fig. 2, G and H, and fig. S4, D and E) or βCaMKII (Fig. 2, I and J, and fig. S4E), which is consistent with CaMKII isomers relocating as heteromultimers (21, 22). Thus, CaV1.2 communication to multiple isoforms of CaMKII follows the signaling logic observed in CaV1.2-mediated CREB signaling; all demand a conjunction of Ca2+ and VΔC signals.
We looked for dynamic changes of CaMKII produced by Ca2+ or VΔC signals in isolation. Whereas VΔC-only stimuli had no effect, Ca2+-only stimuli (10 s) mobilized βCaMKII [Fig. 3, A (green trace) and B (top two images)], which is consistent with its known dissociation from F-actin upon Ca2+ stimulation (21, 22). The mobilization of βCaMKII (Fig. 3A, green trace) outlasted bulk Ca2+ elevation (Fig. 3A, blue trace) but returned to the initial distribution within 60 s, which is consistent with the kinetics of calmodulin trapping (23). Evidently, mobilized βCaMKII reverted to its cytoskeleton-bound state in the absence of VΔC signaling (Fig. 3B). To find out when VΔC signaling was most effective, we varied the timing of brief (10-s) pulses of Ca2+ and VΔC inputs (Fig. 3C), quantifying efficacy by downstream CREB activation (Fig. 3D). Synchronous stimuli (difference in time, Δt = 0) were suboptimal. Instead, VΔC potency peaked at Δt = 10 to 20 s after Ca2+ influx and persisted at Δt = 40 s (Fig. 3D, black bars). Reversing the order of stimuli (VΔC first) resulted in little signaling to CREB (Fig. 3D; Δt = −10 s), a pronounced temporal asymmetry. The dynamics of VΔC potency (Fig. 3D, black bars) and of βCaMKII mobilization (Fig. 3D, green trace from Fig. 3A) were similar. This suggests that Ca2+ mobilization of βCaMKII [or α/βCaMKII heteromultimers (21)] is a prerequisite for their subsequent VΔC-mediated accumulation at CaV1.2 channels and for downstream signaling to pCREB (fig. S5) (19).
Fig. 3. Temporal dissection of Ca2+ and VΔC signals reveals a slow coincidence detection scheme.
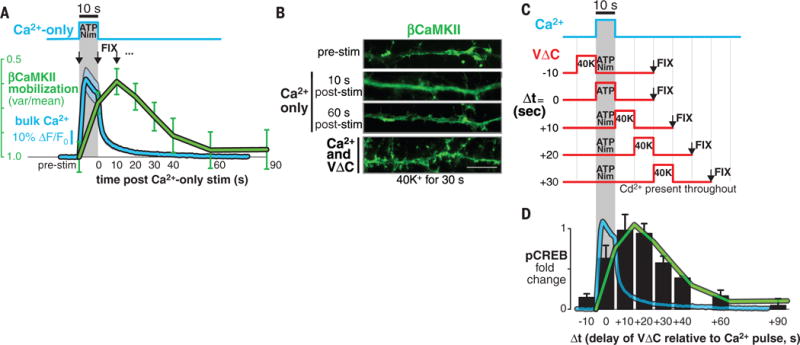
(A) The protocol for the 10-s Ca2+-only pulse produced with ATP plus Nim (top) and the resulting bulk Ca2+ elevation (bottom, blue waveform; ΔF/F0, ratio of fluorescence difference to basal value). Cells were fixed at various time points (black arrows) and βCaMKII mobilization (green waveform) was determined by a variance-over-mean (var/mean) analysis. Blue shading and green bars show means ± SEM of 25 cells from three cortical cultures. (B) Sample images of βCaMKII distribution in response to different stimuli. Ca2+ alone transiently mobilizes βCaMKII (top three images) but does not form βCaMKII puncta (compare with the bottom image). Scale bar, 10 μm. (C) The protocol for temporally shifted 10-s pulses of Ca2+ and VΔC. (D) Nuclear pCREB signaling in response to stimuli in (C) (black bars), overlaid with waveforms from (A). The bars show means ± SEM of 120 cells from four independent cortical cultures.
To test whether VΔC signals interact with other Ca2+ sources, such as N-methyl-D-aspartate receptors (NMDARs) (21, 22), we examined CaMKII puncta formation in response to NMDAR activity in isolation (Fig. 4A, right; nimodipine present) or in combination with CaV1.2 VΔC (Fig. 4A, left; nimodipine absent and CaV1.2 pore blocked with Cd2+). VΔC inclusion enhanced the amplitude of the NMDAR-induced signal (CaMKII puncta formation) (Fig. 4, B and C). Thus, VΔC signals not only interact with Ca2+ entry from the home CaV1.2 (Fig. 3) but also synergistically modulate the signaling potency of other Ca2+ sources (Fig. 4).
Fig. 4. CaV1.2 VΔC synergistically augments NMDAR signaling and is mistuned in TS mutations.
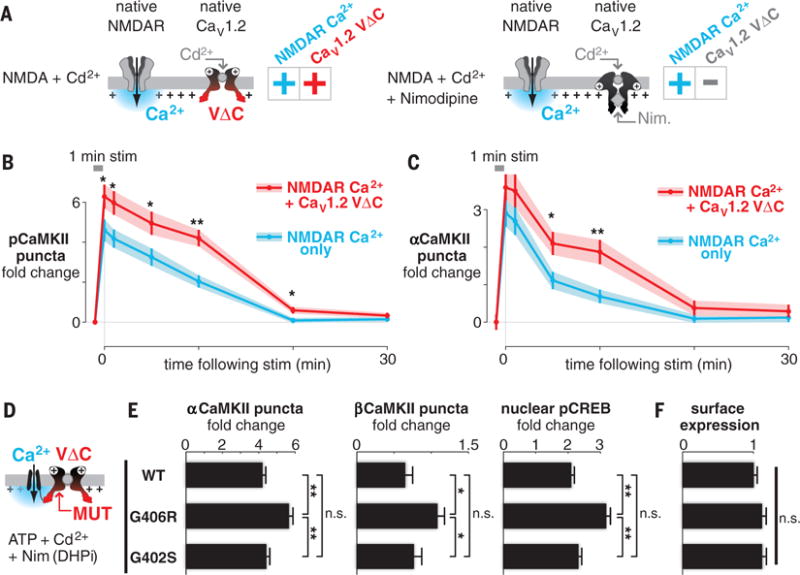
(A) Scheme for examining the functional crosstalk of NMDAR and CaV1.2 VΔC. (B and C) pCaMKII (B) and αCaMKII (C) puncta intensity analysis in neurons after 1 min of stimulation. Shown are means (points) ± SEM (bars) of 50 cells for each condition. *P < 0.05 and **P < 0.01, determined by a two-tailed independent Student’s t test. (D) Approach to examining the VΔC potency of CaV1.2 variants by means of chimeras (MUT, mutation). (E) Signaling to both CaMKII and CREB is substantially elevated (~30 to 70%) in G406R relative to either WT or G402S CaV1.2. Bars show means ± SEM of 50 cells from three cultures. *P < 0.05 and **P < 0.01, determined by one-way ANOVA followed by Fisher’s least significant difference test. (F) Surface expression of TS mutants is indistinguishable from WT CaV1.2 (live-cell surface stain).
With regard to disease, Timothy syndrome (TS) (7) arises from either of two point mutations in helix IS6 of CaV1.2 (fig. S6A). Both the G406R and G402S TS variants cause a prolonged Ca2+ current (arising from the slowing of CaV1.2 inactivation), and both variants produce a long-QT cardiac arrhythmia (fig. S6B) (7, 8). Whereas G406R produces autism spectrum disorder with >60% penetrance, G402S patients are neurologically intact, suggesting that Ca2+ influx is not what determines the neurological phenotype (fig. S6B, black symbols) (8, 24, 25). To test the role of VΔC in TS, we made TS variants in the CaV1.2 portion of the chimera while holding the Ca2+ flux fixed via the fused ttP2X (Fig. 4D). Mutant-specific effects on CaV1.2 Ca2+ flux (7, 8, 24–27) were suppressed with Cd2+, and nimodipine eliminated endogenous CaV1 VΔC contributions, whereas chimeric channels were nimodipine-insensitive (Fig. 4D). We found that G406R CaV1.2 exhibited gain-of-function VΔC signaling: α/βCaMKII and CREB signaling were substantially elevated (~30 to 70%). However, chimera surface expression was no different than in wild-type (WT) CaV1.2 (Fig. 4, E and F). In contrast, G402S CaV1.2 did not differ at all from WT CaV1.2 (Fig. 4E). Thus, mistuning of VΔC is associated with the G406R (28) but not with the G402S variant of TS (fig. S6B, colored symbols), in correlation with the autistic symptoms in TS.
Since the classic discovery of flux-independent CaV1.1 signaling (9, 10), other nonionic modes of channel signaling have been reported to operate independently from ionic signaling (11, 28–30). In this study, we found that conformational and ionic signals from the same channel can work in concert to regulate transcription. Such dual-mode signaling offers enhanced specificity: Maximal CaV1.2-mediated CREB activity requires a sequence of Ca2+ influx followed ~10 to 20 s later by VΔC signaling, a form of coincidence detection that is reminiscent of spike timing–dependent plasticity (31) but more than a thousand times slower (fig. S5). A second benefit is an increase in the voltage dependence of signaling. Voltage-dependent contributions from Ca2+ and VΔC signals render CaV1.2-mediated transcription more sensitive to small changes in depolarization (4). Taken together, these two advantages—heightened temporal specificity and voltage sensitivity—would make suboptimal patterns of activity less effective, while allowing temporally optimal stimuli of even small magnitudes to signal strongly. This may explain why synaptic plasticity that is dependent on CaV1-mediated transcription typically requires prolonged bouts of activity or multiple bursts spread over tens of seconds (2, 32).
Yet another advantage of dual-mode signaling is the possibility of interaction between VΔC signals and recent Ca2+ entry from other sources. We found that CaV1.2 VΔC synergizes with NMDAR signaling (Fig. 4), raising the possibility of interactions with Ca2+ signals derived from other sources, such as internal Ca2+ stores or Ca2+-permeable AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptors. Looking beyond CaV1.2, our chimeric-channel approach may provide a generalizable strategy to investigate nonconducting roles of NMDARs (29) and of other Ca2+-permeant channels such as CaV1.3. CaV1.3 variants that produce autism (33) consistently display negative shifts in voltage dependence that would be expected to enhance VΔC signaling, much as we found for CaV1.2 G406R (Fig. 4E).
Acknowledgments
We thank B. Mensh, A. Lee, N. Spruston, and H. Bito for critical feedback on the manuscript; R. Groth, S. Sanchez, S. Cohen, J. Emery, and B. Shields for assistance with primary neuronal cultures; and H. Ma and other Tsien laboratory members for discussions throughout the execution of this project. The chimera construct is available from M.R.T. upon request. This work was supported by research grants to R.W.T. from the National Institute of General Medical Sciences; the National Institute of Mental Health; the National Institute of Neurological Disorders and Stroke; and the Simons, Mathers, and Burnett Family Foundations. M.R.T. was supported by the Howard Hughes Medical Institute, the Jane Coffin Childs Fellowship, and the Stanford Dean’s Fellowship.
Footnotes
The authors declare no conflicts.
Author contributions were as follows: R.W.T. framed the question and provided overall guidance. M.R.T. conceived of the approach, engineered the chimera, discovered the coincidence of Ca2+ and VΔC in activating CREB in WT and TS CaV1.2, and designed the Δt and NMDAR experiments. B.L. showed that coincidence detection was mediated by CaMKII signaling, implemented the Δt and NMDAR experiments, and performed biochemistry analyses. M.R.T. led the writing, B.L. shortened the manuscript to its published form, and R.W.T. provided editorial oversight.
The data are included in the main manuscript and the supplementary materials.
SUPPLEMENTARY MATERIALS
www.sciencemag.org/content/351/6275/863/suppl/DC1)
Materials and Methods
Figs. S1 to S6
References (34–52)
DNA Construct Sequence
REFERENCES AND NOTES
- 1.Kandel ER. Science. 2001;294:1030–1038. doi: 10.1126/science.1067020. [DOI] [PubMed] [Google Scholar]
- 2.Raymond CR. Trends Neurosci. 2007;30:167–175. doi: 10.1016/j.tins.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 3.Wu GY, Deisseroth K, Tsien RW. Proc Natl Acad Sci USA. 2001;98:2808–2813. doi: 10.1073/pnas.051634198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wheeler DG, Barrett CF, Groth RD, Safa P, Tsien RW. J Cell Biol. 2008;183:849–863. doi: 10.1083/jcb.200805048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wheeler DG, et al. Cell. 2012;149:1112–1124. doi: 10.1016/j.cell.2012.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME. Science. 2001;294:333–339. doi: 10.1126/science.1063395. [DOI] [PubMed] [Google Scholar]
- 7.Splawski I, et al. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 8.Splawski I, et al. Proc Natl Acad Sci USA. 2005;102:8089–8096. doi: 10.1073/pnas.0502506102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Armstrong CM, Bezanilla FM, Horowicz P. Biochim Biophys Acta. 1972;267:605–608. doi: 10.1016/0005-2728(72)90194-6. [DOI] [PubMed] [Google Scholar]
- 10.Adams BA, Tanabe T, Mikami A, Numa S, Beam KG. Nature. 1990;346:569–572. doi: 10.1038/346569a0. [DOI] [PubMed] [Google Scholar]
- 11.Kobrinsky E, Schwartz E, Abernethy DR, Soldatov NM. J Biol Chem. 2003;278:5021–5028. doi: 10.1074/jbc.M211254200. [DOI] [PubMed] [Google Scholar]
- 12.Tadross MR, Tsien RW, Yue DT. Proc Natl Acad Sci USA. 2013;110:15794–15799. doi: 10.1073/pnas.1313898110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deisseroth K, Bito H, Tsien RW. Neuron. 1996;16:89–101. doi: 10.1016/s0896-6273(00)80026-4. [DOI] [PubMed] [Google Scholar]
- 14.Lansman JB, Hess P, Tsien RW. J Gen Physiol. 1986;88:321–347. doi: 10.1085/jgp.88.3.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bourtchuladze R, et al. Cell. 1994;79:59–68. doi: 10.1016/0092-8674(94)90400-6. [DOI] [PubMed] [Google Scholar]
- 16.Lynagh T, Lynch JW. J Biol Chem. 2010;285:14890–14897. doi: 10.1074/jbc.M110.107789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hadley RW, Lederer WJ. Am J Physiol. 1995;269:H1784–H1790. doi: 10.1152/ajpheart.1995.269.5.H1784. [DOI] [PubMed] [Google Scholar]
- 18.Tadross MR, Johny MB, Yue DT. J Gen Physiol. 2010;135:197–215. doi: 10.1085/jgp.200910308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma H, et al. Cell. 2014;159:281–294. doi: 10.1016/j.cell.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosen LB, Ginty DD, Weber MJ, Greenberg ME. Neuron. 1994;12:1207–1221. doi: 10.1016/0896-6273(94)90438-3. [DOI] [PubMed] [Google Scholar]
- 21.Shen K, Meyer T. Science. 1999;284:162–167. doi: 10.1126/science.284.5411.162. [DOI] [PubMed] [Google Scholar]
- 22.Shen K, Teruel MN, Subramanian K, Meyer T. Neuron. 1998;21:593–606. doi: 10.1016/s0896-6273(00)80569-3. [DOI] [PubMed] [Google Scholar]
- 23.Meyer T, Hanson PI, Stryer L, Schulman H. Science. 1992;256:1199–1202. doi: 10.1126/science.256.5060.1199. [DOI] [PubMed] [Google Scholar]
- 24.Fröhler S, et al. BMC Med Genet. 2014;15:48. doi: 10.1186/1471-2350-15-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tallila J, Hiippala A, Myllykangas S, Alastalo TP, Koskenvuo JW. Heart Lung Circ. 2014;23:e4–e5. [Google Scholar]
- 26.Xiao RP, Cheng H, Lederer WJ, Suzuki T, Lakatta EG. Proc Natl Acad Sci USA. 1994;91:9659–9663. doi: 10.1073/pnas.91.20.9659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Erxleben C, et al. Proc Natl Acad Sci USA. 2006;103:3932–3937. doi: 10.1073/pnas.0511322103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krey JF, et al. Nat Neurosci. 2013;16:201–209. doi: 10.1038/nn.3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nabavi S, et al. Proc Natl Acad Sci USA. 2013;110:4027–4032. doi: 10.1073/pnas.1219454110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Atlas D. Annu Rev Biochem. 2013;82:607–635. doi: 10.1146/annurev-biochem-080411-121438. [DOI] [PubMed] [Google Scholar]
- 31.Bi GQ, Poo MM. J Neurosci. 1998;18:10464–10472. doi: 10.1523/JNEUROSCI.18-24-10464.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grover LM, Teyler TJ. Nature. 1990;347:477–479. doi: 10.1038/347477a0. [DOI] [PubMed] [Google Scholar]
- 33.Pinggera A, et al. Biol Psychiatry. 2015;77:816–822. doi: 10.1016/j.biopsych.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]