

 This work is licensed under a
This work is licensed under a Abstract
Proteinuric renal disease is prevalent in congenital or acquired forms of generalized lipodystrophy. In contrast, an association between familial partial lipodystrophy (FPLD) and renal disease has been documented in very few cases. A 22-year-old female patient presented with impaired glucose tolerance, hyperinsulinemia, hirsutism and oligomenorrhea. On examination, there was partial loss of subcutaneous adipose tissue in the face, upper and lower limbs, bird-like facies with micrognathia and low set ears and mild acanthosis nigricans. Laboratory investigations revealed hyperandrogenism, hyperlipidemia, elevated serum creatine kinase and mild proteinuria. A clinical diagnosis of FPLD of the non-Dunnigan variety was made; genetic testing revealed a heterozygous c.1045C > T mutation in exon 6 of the LMNA gene, predicted to result in an abnormal LMNA protein (p.R349W). Electromyography and muscle biopsy were suggestive of non-specific myopathy. Treatment with metformin and later with pioglitazone was initiated. Due to worsening proteinuria, a renal biopsy was performed; histological findings were consistent with mild focal glomerular mesangioproliferative changes, and the patient was started on angiotensin-converting enzyme inhibitor therapy. This is the fourth report of FPLD associated with the c.1045C > T missense LMNA mutation and the second with co-existent proteinuric renal disease. Patients carrying this specific mutation may exhibit a phenotype that includes partial lipodystrophy, proteinuric nephropathy, cardiomyopathy and atypical myopathy.
Learning points:
Lipodystrophy is a rare disorder characterized by the complete or partial loss of subcutaneous adipose tissue, insulin resistance, diabetes mellitus and hyperlipidemia.
Proteinuric renal disease is a prevalent feature of generalized lipodystrophy but rare in familial partial lipodystrophy.
Patients carrying the c.1045C > T missense LMNA mutation (p.R349W) may present with familial partial lipodystrophy, proteinuric nephropathy, cardiomyopathy and atypical myopathy.
Background
Lipodystrophy is a rare group of clinically heterogeneous disorders characterized by the complete or partial loss of subcutaneous adipose tissue (1, 2). This condition is often accompanied by metabolic complications such as insulin resistance, impaired glucose tolerance or diabetes mellitus, dyslipidemia and hepatic steatosis or steatohepatitis. The severity of these metabolic derangements is dependent on the extent of fat loss (3, 4).
Patients with congenital or acquired generalized lipodystrophy have a high prevalence of proteinuric renal disease that may take several forms, especially focal segmental glomerulosclerosis (FSGS) and membranoproliferative glomerulonephritis (MPGN) (5). On the other hand, an association between familial partial lipodystrophy (FPLD) and renal disease has been documented in very few cases (6, 7, 8, 9).
We describe the case of a patient with FPLD, atypical myopathy and proteinuric renal disease.
Case presentation
A 22-year-old female patient was referred to our department due to impaired glucose tolerance (IGT) and hyperinsulinemia. She had previously been investigated at the age of 13 years in a pediatric hospital for myalgias, and she was found to have elevated liver transaminases, creatine kinase (CK) and fasting insulin levels. A 75 g oral glucose tolerance test (OGTT) revealed IGT and hyperinsulinemia. At that time, further investigation including a muscle biopsy was advised but the patient was lost to follow-up. There was no other past medical history of note apart from oligomenorrhea (8 menstrual cycles per year) with menarche at the age of 12 years.
The patient’s father had a history of cardiomyopathy, proteinuric renal disease and atypical myopathy and died suddenly at the age of 43 years. According to his medical records, he had features of partial lipodystrophy with loss of subcutaneous adipose tissue in the extremities and face but genetic analysis had never been performed. The patient’s mother was 55 years old, with no signs of lipodystrophy and had a history of arterial hypertension, hypothyroidism due to Hashimoto’s thyroiditis and hyperlipidemia.
On clinical examination, the patient’s blood pressure was 120/80 mmHg, pulse rate 76/min regular and her body mass index was 20 kg/m2 (height 158 cm and body weight 50 kg) with partial loss of subcutaneous adipose tissue in the face and both upper and lower limbs. Bird like facies with micrognathia and low set ears, mild acanthosis nigricans in the axillae, hirsutism (Ferriman–Gallwey score: 8) and scoliosis were also noted. Neurological examination showed no evidence of muscle weakness, atrophy or contractures.
Investigation
Laboratory investigations showed elevated liver transaminases, CK, aldolase, lactate dehydrogenase (LDH) and fasting insulin levels, low levels of serum adiponectin, hyperlipidemia, hyperandrogenism and autoimmune thyroiditis with normal serum thyroid hormone levels (Table 1). Additional testing included a 75 g OGTT that revealed IGT with marked hyperinsulinemia (Table 2) and a 24-h urine collection that showed mild proteinuria (urine protein (UPR): 180 mg/24 h, normal values: <150 mg/24 h). An abdominal ultrasound was consistent with fatty liver disease, whereas dual-energy X-ray absorptiometry revealed low bone mineral density (BMD) at the femoral neck (Z-score: −3).
Table 1.
Patient’s initial biochemical and hormonal characteristics.
| Parameters | Patient’s values | Normal values |
|---|---|---|
| Glucose (mg/dL) | 79 | 70–100 |
| Insulin (μIU/mL) | 50.6 | 1.9–23 |
| HbA1c (%) | 5.5 | 4.3–5.6 |
| HOMA-IR | 9.87 | |
| Total cholesterol (mg/dL) | 226 | 110–200 |
| HDL cholesterol (mg/dL) | 46 | 35–70 |
| LDL cholesterol (mg/dL) | 151 | 60–130 |
| Triglycerides (mg/dL) | 146 | 40–175 |
| Leptin (μg/L) | 26.1 | 2.4–24.4 |
| Adiponectin (mg/L) | 2.3 | 4.5–17.7 |
| Urea (mg/dL) | 34 | 11–54 |
| Creatinine (mg/dL) | 0.6 | 0.6–1.2 |
| Albumin (g/dL) | 4.2 | 3.5–5 |
| AST (IU/L) | 40 | 10–35 |
| ALT (IU/L) | 51 | 10–35 |
| Alp (IU/L) | 45 | 30–125 |
| γGT (IU/L) | 15 | 6–32 |
| Total bilirubin (mg/dL) | 0.4 | 0.1–1 |
| LDH (U/L) | 234 | 115–230 |
| CK (IU/L) | 894 | 25–160 |
| Aldolase (IU/L) | 10.4 | 0–7.6 |
| TSH (μIU/mL) | 1.17 | 0.34–5.6 |
| Free T4 (ng/dL) | 0.8 | 0.6–1.37 |
| T3 (ng/mL) | 1.24 | 0.56–1.56 |
| Anti-TPO (IU/mL) | 938 | <35 |
| Anti-TG (IU/mL) | 1181 | <40 |
| FSH (IU/L) | 5.7 | 3.85–8.8 |
| LH (IU/L) | 7.3 | 2.1–10.9 |
| Estradiol (pmol/L) | 190 | 88–418 |
| Testosterone (ng/mL) | 0.7 | <0.75 |
| Calculated free testosterone (pg/mL) | 18.6 | 0.8–7.4 |
| SHBG (nmol/L) | 15.7 | 18.2–135.5 |
| DHEAS (μg/dL) | 422 | 18–391 |
γGT, gamma-glutamyl transferase; Alp, Alkaline phosphatase; ALT, alanine aminotransferase; anti-TG, antibody against thyroglobulin; anti-TPO, antibody against thyroid peroxidase; AST, aspartate aminotransferase; CK, creatine kinase; DHEAS, dehydroepiandrosterone sulfate; HDL, high density lipoprotein; HOMA-IR, homeostatic model assessment for insulin resistance; LDH, lactate dehydrogenase; LDL, low density lipoprotein; SHBG, sex hormone binding globulin.
Table 2.
75 g oral glucose tolerance test at diagnosis.
| Time (min) | |||||
|---|---|---|---|---|---|
| 0 | 30 | 60 | 90 | 120 | |
| Glucose (mg/dL) | 79 | 150 | 157 | 188 | 172 |
| Insulin (μIU/mL) | 50.6 | >300 | >300 | >300 | >300 |
In view of the consistently raised serum CK, aldolase and LDH levels, the patient underwent electromyography, which revealed non-specific myopathic patterns. Antibody testing for autoimmune disorders (anti-nuclear, anti-mitochondrial, anti-smooth muscle and anti-neutrophil cytoplasmic antibodies as well as antibodies against extractable nuclear antigens) was negative and a muscle biopsy was suggestive of non-specific myopathy.
On the basis of the clinical and laboratory findings outlined above as well as the patient’s family history, a working diagnosis of FPLD of the non-Dunnigan variety was made and genetic testing was performed. Patient’s DNA was extracted from peripheral blood leukocytes, and fluorescent sequencing analysis (Mutation Surveyor, SoftGenetics, LLC) revealed the presence of a heterozygous c.1045C > T mutation in exon 6 of the LMNA gene, which is predicted to result in an abnormal LMNA protein, (p.R349W). The alleles were present in equal proportions and gave no indication of being mosaic.
In view of the fact that this specific LMNA mutation has been associated with dilated cardiomyopathy (10), the patient underwent a transthoracic echocardiography and electrocardiography, which did not reveal any pathological findings.
The patient was initially treated with metformin and later with pioglitazone and atorvastatin and follow-up was arranged. One year later, plasma glucose, serum cholesterol, free testosterone and dehydroepiandrosterone sulfate levels returned to normal, and the patient reported no menstrual irregularities. In addition, abdominal ultrasound revealed improvement of her previously noted hepatic steatosis and BMD of femoral neck was slightly improved (Z-score: −2.4). Echocardiography was normal. Hyperinsulinemia persisted during OGTT (Table 3) but improved compared to basal levels and serum CK was 515 IU/L. She was clinically well and again, no signs of muscle weakness, atrophy or contractures were noted.
Table 3.
75 g oral glucose tolerance test one year after pioglitazone initiation.
| Time (min) | |||||
|---|---|---|---|---|---|
| 0 | 30 | 60 | 90 | 120 | |
| Glucose (mg/dL) | 80 | 104 | 119 | 120 | 103 |
| Insulin (μIU/mL) | 11.8 | 64.6 | 94.4 | 103.1 | 92.5 |
The patient’s condition remained stable over a five-year period; gradual deterioration of the proteinuria was however observed over a 12-month period (UPR: 2.2 g/24 h). Renal function and serum albumin levels remained normal (urea: 27 mg/dL, serum creatinine: 0.67 mg/dL, eGFR: 118 mL/min/1.73 m2, albumin: 4.2 g/dL). Ultrasonography revealed normal sized kidneys with no abnormal findings. Autoimmune screen (anti-nuclear, anti-neutrophil cytoplasmic antibodies, serum immunoglobulins, complement levels) was once again negative and serum and urine protein electrophoresis were normal.
A renal biopsy was performed, which was consistent with mild focal glomerular mesangioproliferative changes (Fig. 1), although the interpretation of the histopathologic findings was limited by the small tissue sample and the absence of tissue for ultrastructural examination by electron microscopy. Examination by immunofluorescence (scale intensity 0–4+) in 3 glomeruli showed no significant granular or linear stain for immunoglobulins (IgG, IgA and IgM), κ and λ light chains and complement component C3 in glomeruli. A granular pattern for C1q (1–2+) was observed in the mesangium.
Figure 1.
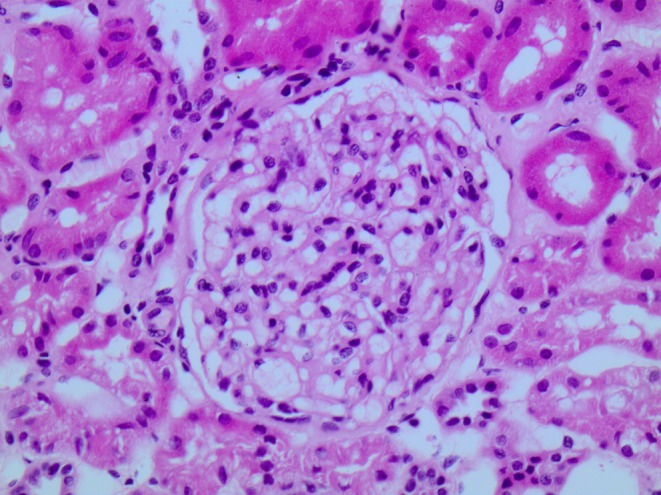
Ten glomeruli were identified in serial sections, one globally sclerosed. The rest of the glomeruli showed focal mild mesangial matrix expansion in association with mild mesangial cell proliferation (Η&Ε ×400).
Outcome and follow-up
In view of the progressive proteinuria, low-dose angiotensin-converting-enzyme inhibitor treatment was initiated. Six months after initiation of treatment, the patient had preserved renal function (serum creatinine: 0.68 mg/dL, eGFR: 117 mL/min/1.73 m2), while proteinuria persisted but improved (UPR: 1.1 g/24 h).
Discussion
We describe the case of a patient with a c.1045C > T missense LMNA mutation (p.R349W) presenting with FPLD of the non-Dunnigan variety associated with atypical myopathy and proteinuric nephropathy.
Lipodystrophies are grossly categorized based on etiology (genetic or acquired) and distribution of adipose tissue loss (generalized or partial) (11). However, genetic lipodystrophies can be further subdivided on the basis of their clinical phenotype, complications and genetic etiology. The most prevalent form of FPLD is the autosomal dominant type 2, also called as Dunnigan variety, caused by mutation in the LMNA gene (12). These patients are characterized by gradual loss of adipose tissue from the extremities and trunk, although phenotypic heterogeneity in the pattern of body fat loss can be observed (13, 14).
LMNA is mapped to chromosome 1q21–22 and contains 12 exons (15). This gene produces two major protein isoforms, lamin A and lamin C, through alternative splicing, that polymerize with type B lamins to form the nuclear lamina, a complex meshwork of proteins underlying the inner nuclear membrane (6). The physiological role of these proteins involves several cellular processes. LMNA mutations are responsible for a group of genetic disorders described under the term ‘laminopathies’, including lipodystrophies, diseases affecting muscle tissue and axonal neurons, as well as progeroid syndromes (16).
Laminopathies are most frequently characterized by skeletal muscle involvement with three main phenotypes being reported so far: limb-girdle muscular dystrophy type 1B, autosomal dominant Emery-Dreifuss muscular dystrophy and a form of congenital muscular dystrophy (16). Atypical cases, sharing only parts of the above phenotypes, have also been described (17, 18).
On the other hand, cardiac involvement, especially dilated cardiomyopathy and cardiac conduction defects, is another common feature of laminopathies and can be life threatening. Patients may die suddenly due to malignant ventricular tachyarrhythmias, even in the absence of dilated cardiomyopathy (19). The prevalence of cardiac involvement in LMNA mutation carriers, increases with age, so that almost all patients with laminopathy after the age of 60 years, have cardiac disease (14).
Generalized forms of lipodystrophy (congenital or acquired) are associated with FSGS and MPGN. In addition, a connection between acquired partial lipodystrophy and MPGN is recognized (20). The underlying mechanisms are unclear, but hypocomplementemia and elevated C3 nephritic factor, an IgG autoantibody that causes C3 consumption and possibly adipocyte lysis, have been hypothesized as contributing factors (21, 22, 23). However, the presence of proteinuric nephropathy in patients with congenital lipodystrophy due to LMNA mutations has very rarely been reported (6, 7, 8, 9). All patients suffered from FPLD (of the Dunnigan and non-Dunnigan varieties) due to LMNA missense mutations c.1930C > T (p.R644C), c.1444C > T (p.R482W), c.1445G > A (p.R482Q) and c.1045C > T (p.R349W). A possible connection between LMNA mutations and FSGS through transforming growth factor-beta1 (TGF-β1) has been proposed (13). Lamin A/C controls the gene activity downstream of TGF-β1, whereas the overexpression of this profibrotic cytokine in transgenic mice leads to glomerulosclerosis and a lipodystrophy-like syndrome (24, 25).
To our knowledge, this is the fourth report of FPLD associated with the c.1045C > T missense LMNA mutation (p.R349W) and the second with co-existent proteinuric renal disease (Table 4). FPLD in the affected patients is mainly of the non-Dunnigan variety and proteinuric nephropathy as well as cardiac involvement are common features. Micrognathia has been reported in one patient, apart from ours. Interestingly, elevated serum CK levels were found in three out of seven patients (including ours), although no signs of muscular dystrophy were noted. CK levels were not determined in the other four patients.
Table 4.
Published cases of laminopathies due to an heterozygous c.1045C > T missense LMNA mutation (p.R349W).
| Reference | Sex | FPLD type | Cardiac disease | Renal disease | Myopathy |
|---|---|---|---|---|---|
| Thong et al. (9) | Female | Non-Dunnigan variety | No | Yes | ND |
| Thong et al. (9) | Female | Non-Dunnigan variety | No | Yes | ND |
| Thong et al. (9) | Male | Non-Dunnigan variety | Yes | Yes | ND |
| Thong et al. (9) | Female | Non-Dunnigan variety | Yes | Yes | ND |
| Mory et al. (14) | Male | Atypical (loss of subcutaneous fat in limbs including dorsal and palmar aspects of hand and feet) | No | ND | Yes |
| van Tintelen et al. (26) | Female | Dunnigan variety | Yes | ND | Yes |
| Present case | Female | Non-Dunnigan variety | No | Yes | Yes |
FPLD, familiar partial lipodystrophy; ND, not determined.
In conclusion, this case adds to the accumulating evidence of a possible genetic link between the c.1045C > T missense LMNA mutation and FPLD with proteinuric nephropathy.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Patient consent
Written informed consent has been obtained from the patient for publication of the submitted article.
Author contribution statement
A Fountas wrote the manuscript and was involved in the care of the patient. Z Giotaki, E Dounousi and A Tsatsoulis were involved in patient’s care. G Liapis conducted the pathology review. A Bargiota provided critical review of the manuscript. S Tigas is the patient’s physician and was responsible for the study supervision and manuscript writing. All listed authors contributed to the editing process.
Acknowledgment
We are very grateful to Dr David Savage, Department of Clinical Biochemistry, Metabolic Research Laboratories, University of Cambridge, UK, for his valuable help with the genetic analyses.
References
- 1.Chan JL, Oral EA. 2010. Clinical classification and treatment of congenital and acquired lipodystrophy. Endocrine Practice 16 310–323. ( 10.4158/EP09154.RA) [DOI] [PubMed] [Google Scholar]
- 2.Garg A. 2011. Lipodystrophies: genetic and acquired body fat disorders. Journal of Clinical Endocrinology and Metabolism 96 3313–3325. ( 10.1210/jc.2011-1159) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garg A. 2004. Acquired and inherited lipodystrophies. New England Journal of Medicine 350 1220–1234. ( 10.1056/NEJMra025261) [DOI] [PubMed] [Google Scholar]
- 4.Agarwal AK, Garg A. 2006. Genetic basis of lipodystrophies and management of metabolic complications. Annual Review of Medicine 57 297–311. ( 10.1146/annurev.med.57.022605.114424) [DOI] [PubMed] [Google Scholar]
- 5.Javor ED, Moran SA, Young JR, Cochran EK, DePaoli AM, Oral EA, Turman MA, Blackett PR, Savage DB, O’Rahilly S, et al. 2004. Proteinuric nephropathy in acquired and congenital generalized lipodystrophy: baseline characteristics and course during recombinant leptin therapy. Journal of Clinical Endocrinology and Metabolism 89 3199–3207. ( 10.1210/jc.2003-032140) [DOI] [PubMed] [Google Scholar]
- 6.Owen KR, Donohoe M, Ellard S, Clarke TJ, Nicholls AJ, Hattersley AT, Bingham C. 2004. Mesangiocapillary glomerulonephritis type 2 associated with familial partial lipodystrophy (Dunnigan-Kobberling syndrome). Nephron Clinical Practice 96 c35–c38. ( 10.1159/000076396) [DOI] [PubMed] [Google Scholar]
- 7.Rankin J, Auer-Grumbach M, Bagg W, Colclough K, Nguyen TD, Fenton-May J, Hattersley A, Hudson J, Jardine P, Josifova D, et al. 2008. Extreme phenotypic diversity and nonpenetrance in families with the LMNA gene mutation R644C. American Journal of Medical Genetics Part A 146 1530–1542. ( 10.1002/ajmg.a.32331) [DOI] [PubMed] [Google Scholar]
- 8.Imachi H, Murao K, Ohtsuka S, Fujiwara M, Muraoka T, Hosokawa H, Ishida T. 2009. A case of Dunnigan-type familial partial lipodystrophy (FPLD) due to lamin A/C (LMNA) mutations complicated by end-stage renal disease. Endocrine 35 18–21. ( 10.1007/s12020-008-9127-1) [DOI] [PubMed] [Google Scholar]
- 9.Thong KM, Xu Y, Cook J, Takou A, Wagner B, Kawar B, Ong AC. 2013. Cosegregation of focal segmental glomerulosclerosis in a family with familial partial lipodystrophy due to a mutation in LMNA. Nephron Clinical Practice 124 31–37. ( 10.1159/000354716) [DOI] [PubMed] [Google Scholar]
- 10.Tesson F, Saj M, Uvaize MM, Nicolas H, Płoski R, Bilińska Z. 2014. Lamin A/C mutations in dilated cardiomyopathy. Cardiology Journal 21 331–342. ( 10.5603/CJ.a2014.0037) [DOI] [PubMed] [Google Scholar]
- 11.Brown RJ, Araujo-Vilar D, Cheung PT, Dunger D, Garg A, Jack M, Mungai L, Oral EA, Patni N, Rother KI, et al. 2016. The diagnosis and management of lipodystrophy syndromes: a multi-society practice guideline. Journal of Clinical Endocrinology and Metabolism 101 4500–4511. ( 10.1210/jc.2016-2466) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nolis T. 2014. Exploring the pathophysiology behind the more common genetic and acquired lipodystrophies. Journal of Human Genetics 59 16–23. ( 10.1038/jhg.2013.107) [DOI] [PubMed] [Google Scholar]
- 13.Jacob KN, Garg A. 2006. Laminopathies: multisystem dystrophy syndromes. Molecular Genetics and Metabolism 87 289–302. ( 10.1016/j.ymgme.2005.10.018) [DOI] [PubMed] [Google Scholar]
- 14.Mory PB, Crispim F, Freire MB, Salles JE, Valério CM, Godoy-Matos AF, Dib SA, Moisés RS. 2012. Phenotypic diversity in patients with lipodystrophy associated with LMNA mutations. European Journal of Endocrinology 167 423–431. ( 10.1530/EJE-12-0268) [DOI] [PubMed] [Google Scholar]
- 15.Lin F, Worman HJ. 1993. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. Journal of Biological Chemistry 268 16321–16326. [PubMed] [Google Scholar]
- 16.Maggi L, Carboni N, Bernasconi P. 2016. Skeletal muscle laminopathies: a review of clinical and molecular features. Cells 5 E33 ( 10.3390/cells5030033) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carboni N, Mura M, Marrosu G, Cocco E, Marini S, Solla E, Mateddu A, Maioli MA, Piras R, Mallarini G, et al. 2010. Muscle imaging analogies in a cohort of patients with different clinical phenotypes caused by LMNA gene mutations. Muscle and Nerve 41 458–463. ( 10.1002/mus.21514) [DOI] [PubMed] [Google Scholar]
- 18.Maioli MA, Marrosu G, Mateddu A, Solla E, Carboni N, Tacconi P, Lai C, Marrosu MG. 2007. A novel mutation in the central rod domain of lamin A/C producing a phenotype resembling the Emery-Dreifuss muscular dystrophy phenotype. Muscle and Nerve 36 828–832. ( 10.1002/mus.20879) [DOI] [PubMed] [Google Scholar]
- 19.Pasotti M, Klersy C, Pilotto A, Marziliano N, Rapezzi C, Serio A, Mannarino S, Gambarin F, Favalli V, Grasso M, et al. 2008. Long-term outcome and risk stratification in dilated cardiolaminopathies. Journal of the American College of Cardiology 52 1250–1260. ( 10.1016/j.jacc.2008.06.044) [DOI] [PubMed] [Google Scholar]
- 20.Musso C, Javor E, Cochran E, Balow JE, Gorden P. 2006. Spectrum of renal diseases associated with extreme forms of insulin resistance. Clinical Journal of American Society of Nephrology 1 616–622. ( 10.2215/CJN.01271005) [DOI] [PubMed] [Google Scholar]
- 21.Williams DG. 1997. C3 nephritic factor and mesangiocapillary glomerulonephritis. Pediatric Nephrology 11 96–98. ( 10.1007/s004670050241) [DOI] [PubMed] [Google Scholar]
- 22.Misra A, Peethambaram A, Garg A. 2004. Clinical features and metabolic and autoimmune derangements in acquired partial lipodystrophy: report of 35 cases and review of the literature Medicine 83 18–34. ( 10.1097/01.md.0000111061.69212.59) [DOI] [PubMed] [Google Scholar]
- 23.Mathieson PW, Peters DK. 1997. Lipodystrophy in MCGN type II: the clue to links between the adipocyte and the complement system. Nephrology Dialysis Transplantation 12 1804–1806. ( 10.1093/ndt/12.9.1804) [DOI] [PubMed] [Google Scholar]
- 24.Clouthier DE, Comerford SA, Hammer RE. 1997. Hepatic fibrosis, glomerulosclerosis, and a lipodystrophy-like syndrome in PEPCK-TGF beta1 transgenic mice. Journal of Clinical Investigation 100 2697–2713. ( 10.1172/JCI119815) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Berlo JH, Voncken JW, Kubben N, Broers JL, Duisters R, van Leeuwen RE, Crijns HJ, Ramaekers FC, Hutchison CJ, Pinto YM. 2005. A type lamins are essential for TGF-β1 induced PP2A to dephosphorylate transcription factors. Human Molecular Genetics 14 2839–2849. ( 10.1093/hmg/ddi316) [DOI] [PubMed] [Google Scholar]
- 26.Van Tintelen JP, Hofstra RM, Katerberg H, Rossenbacker T, Wiesfeld AC, du Marchie Sarvaas GJ, Wilde AA, van Langen IM, Nannenberg EA, van der Kooi AJ, et al. 2007. High yield of LMNA mutations in patients with dilated cardiomyopathy and/or conduction disease referred to cardiogenetics outpatient clinics. American Heart Journal 154 1130–1139. ( 10.1016/j.ahj.2007.07.038) [DOI] [PubMed] [Google Scholar]