

Abstract
Patient: Female, 24
Final Diagnosis: Primary Sjögren syndrome
Symptoms: Kidney stones • nocturia • polyuria
Medication: —
Clinical Procedure: —
Specialty: Nephrology
Objective:
Rare co-existance of disease or pathology
Background:
Sjögren’s syndrome is an autoimmune disorder caused by the infiltration of monocytes in epithelial glandular and extra-glandular tissues. Hallmark presentations include mouth and eye dryness. Although renal involvement is uncommon in primary Sjögren’s syndrome (pSS), patients may experience renal tubular acidosis type I (RTA I), tubulointerstitial nephritis, diabetes insipidus (DI), nephrolithiasis, and Fanconi syndrome. However, it is atypical to see more than 1 of these manifestations in a single patient.
Case Report:
We present the case of a 24-year-old woman with polyuria and polydipsia, who was initially diagnosed with nephrogenic diabetes insipidus. She also had chronic hypokalemia and nephrolithiasis. Based on clinical presentation and work up, she was diagnosed with pSS and treated accordingly.
Conclusions:
This was a pSS patient with tubulointerstitial nephritis, diabetes insipidus, renal tubular acidosis, hypokalemia, and nephrolithiasis, who was receiving symptomatic treatment for diabetes insipidus. Diagnosis and treatment of pSS led to significant improvement in systemic and renal presentations of the patient. pSS should be considered as one of the differential diagnoses in patients with diabetes insipidus and renal tubular acidosis.
MeSH Keywords: Acidosis, Renal Tubular; Diabetes Insipidus; Sjögren’s Syndrome
Background
Primary Sjögren’s syndrome is an autoimmune connective tissue disorder characterized by infiltration of B-lymphocytes in epithelial tissues of exocrine glands (salivary and lacrimal glands). The B-lymphocytes are also found in extra-glandular epitheliums such as the lungs, gastrointestinal tract, hepatobiliary tract, and renal epithelium [1]. Classic clinical manifestations of pSS include sicca, defined as dry eyes and mouth, which occurs in more than 90% of the patients [2]. Mouth dryness can lead to pain swallowing and also dental caries, and eye dryness (keratoconjunctivitis sicca) can present with eye pain, irritation, and foreign body sensation in the eyes. Extra-glandular manifestations of pSS are variable and can involve most organ systems. Some of the known presentations include interstitial pneumonia and tracheobronchial sicca in the respiratory system, and pericarditis and pulmonary hypertension in the cardiovascular system. Other signs and symptoms can include dysphagia, arthralgia, myalgia, hypothyroidism, and skin manifestations, including non-blenching rashes. Since the condition is caused by a B-lymphocyte proliferation, development of lymphoproliferative disorders is a major concern [3]. Sjögren’s syndrome can also be secondary to other autoimmune conditions, including, but not limited to, systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and systemic sclerosis (scleroderma) [4].
The prevalence of renal involvement in pSS has been reported to range approximately from 10% [2,5] to 30% [6]. For example, a recent review by François et al. estimated the prevalence of renal disease in pSS to be less than 10% [7].
Renal disease in pSS is due to 2 distinct pathophysiological processes: (1) Epithelial disease with significant lymphocytic infiltration, resulting in different conditions like tubulointerstitial nephritis (TIN), electrolyte disturbances like hypokalemia, distal renal tubular acidosis, proximal renal tubular acidosis, Fanconi syndrome, diabetes insipidus, Gitelman syndrome, nephrolithiasis and nephrocalcinosis, and (2) Non-epithelial disease that can lead to glomerulopathy as a result of an immune complex-mediated process [7,8]. However, most of the patients present only 1 of the above-mentioned signs and symptoms. Furthermore, it is uncommon for 1 patient to present multiple features of renal disease associated with pSS.
Case Report
Our patient was a 24-year-old woman who had polyuria and polydipsia since her teens. Her symptoms began more than 10 years ago, when she started drinking 7–10 liters of water daily, had an abnormal increase in production of urine, and would wake up multiple times a night to urinate. Her other symptoms included chronic asymptomatic hypokalemia, kidney stones, and rashes. Kidney stones were diagnosed on an ultrasound 2 years prior to the referral, located in the upper and middle pole of the right and left kidney, respectively, each measuring 7 mm in diameter. The rash first started 3 years ago and initially consisted of scattered non-blanching, purpuric, and hyperpigmented macules on the calves, shins, and ankles. She also had 2 outbreaks of more generalized rash 2 years and 1 year ago, involving the entire lower extremities, each lasting for a few weeks and resolving spontaneously. There was no history of any photosensitivity, and the rash did not show any changes with sun exposure. She was seen by an outside nephrologist and was diagnosed with nephrogenic diabetes insipidus and chronic metabolic acidosis. These conditions were treated with amiloride and sodium bicarbonate. However, during the course of the disease, she experienced an increase in serum creatinine and chronic kidney disease stage III, with a baseline creatinine of 1.2–1.4, and subsequently was referred for a second opinion as her polyuria did not resolve. Even though she had a normal baseline urinalysis 5 years prior to the referral, 1 of her recent UAs demonstrated 500 mg of proteinuria. Multiple repeat urinary protein/creatinine ratios were within normal range after that 1 test. She had been worked up for connective tissue diseases and was found to have increased ANA, RF, and ESR on multiple occasions, without a definite diagnosis. The physical examination was positive for several acne forms erythematous macules behind her right ear with no exudate, slight erythema on the inner lip, and rare white plaques on the lateral edge of the tongue. Family history was negative for any autoimmune condition, and social history was non-contributory.
Upon further questioning, the patient mentioned long-standing severe dry mouth without any skin or eye dryness. Based on these symptoms, pSS was highly suspected. The anti-Sjögren’s antibody A and B were checked and were both found to be positive.
The patient was evaluated for diabetes insipidus. At presentation, she had elevated serum osmolality of 301, with a very low urine osmolality of 61, and serum sodium of 143. She also produced more than 9 liters of urine per day. A trial of desmopressin was administered, without a significant change in urine osmolality, compatible with a diagnosis of nephrogenic diabetes insipidus. She was then started on Chlorthalidone, and urine output decreased to 3 liters.
A basic metabolic panel, which is shown in Table 1, revealed metabolic acidosis with hypokalemia and elevated urine pH, consistent with distal RTA. She had a history of a non-obstructive kidney stone and a 24-hour urine collection was obtained, which is presented in Tables 2 and 3.
Table 1.
Basic metabolic panel.
| Na | K | HCO3 | Chloride | Osmolality | Creatinine | GFR | BUN | Uric acid |
|---|---|---|---|---|---|---|---|---|
| 143 mmol/L | 3.3 mmol/L | 16 mmol/L | 112 mmol/L | 301 mosm/kg | 1.5 mg/dl | 43 mL/min/1.73 m2 | 14 mg/dl | 3.5 mg/dl |
Table 2.
Urinalysis.
| Urine PH | Na | K | Creatinine | Ca | Osmolality |
|---|---|---|---|---|---|
| 7 | <20 mmol/L | 5 mmol/L | 7.0 mg/dl | 129 mmol/L | 61 mosm/kg |
Table 3.
24-hour urine collection.
| Volume | Citric acid | Oxalate | Phosphorus | Uric acid |
|---|---|---|---|---|
| 9893 ml | <108 mg/day (Nl: 320–1240) | 971 mg/day (Nl=4–31) | <14 mg/dl | <5.7 mg/dl |
The patient was extensively worked up to exclude secondary causes of SS, including, but not limited to, systemic lupus erythematosus and infections. Her thyroid function tests were normal and she was not receiving any long-term NSAIDs or cytotoxic medications. Moreover, in multiple urinalyses done over the course of the disease, she never had any sign of urinary tract infection, such as white blood cells in the urine. The results of these tests are summarized in Table 4.
Table 4.
Autoimmune work up.
| ANA | Sm Ab | RNP | Anti dsDNA (NL <200 IU/mL) | Anti- SSA | Anti-SSB | C3 (NL: 14–46 mg/dl) | C4 (NL: 76–165 mg/dl) | Rheumatoid Factor (NL: <25 IU/mL) | Cyclic Citrulline Ab IgG (NL: <20 Unit) | Beta 2 Macroglobulin |
|---|---|---|---|---|---|---|---|---|---|---|
| 1: 1280 | Negative | Negative | <200 | 138 | 94 | 33 | 80 | 211 | 16 | 177143 mcg/mg |
A skin biopsy of the lesion on the lower extremities was performed, which led to a diagnosis of leukocytoclastic vasculitis and ruled out SLE.
A kidney biopsy was obtained, which revealed tubular interstitial fibrosis with infiltration of lymphocytes and plasmocytes. This infiltration focally extended into intact cortical parenchyma and was consistent with active chronic interstitial nephritis. Even though the overall features were non-specific, they were compatible with pSS. Out of 19 glomeruli studied in the specimen, 7 were completely or near-completely sclerotic, with no endocapillary hypercellularity, crescent formation, or evidence of thrombosis. Immunofluorescence microscopy was also performed on frozen sections stained with hematoxylin and eosin, periodic Acid-Schiff and fluoresceinated antisera to human IgG, IgA, IgM, C1q, C3, albumin, fibrinogen, and kappa and lambda immunoglobulin light chains. There was no significant glomerular staining for any of the tested immunoreactants (Figures 1, 2).
Figure 1.

(A, B) Inflammation in both scarred and non-scarred areas of the tubulointerstitium accompanied by foci of tubulitis in non-atrophic tubules (A: 10×, B: 20×).
Figure 2.
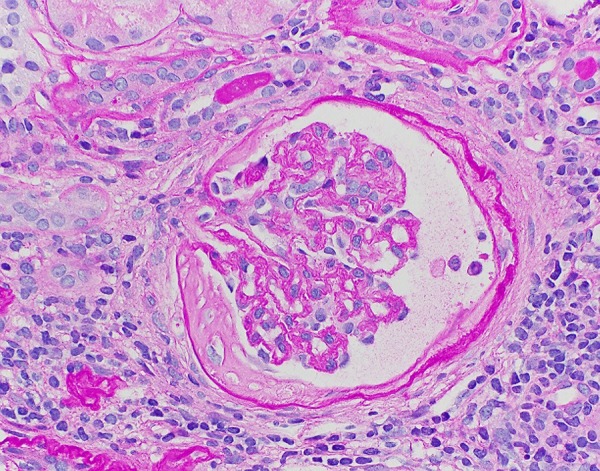
Glomerulus (20×).
For pSS treatment, she was started on azathioprine, hydroxychloroquine, and steroids. She had severe adverse reactions to the steroids, including weight gain, moon face, buffalo hump, and acne striae; therefore, steroids were discontinued. Before the initiation of hydroxychloroquine, she was referred to an ophthalmologist and was diagnosed with dry eyes, even though she was completely asymptomatic. However, as a result of the treatment, acidosis improved and her bicarbonate level normalized. Nocturia and polyuria, which were her chief concerns, improved. The patient also had improvements in energy level and experienced less fatigue.
In summary, this patient presented renal manifestations of pSS, including diabetes insipidus, renal tubular acidosis type I, tubulointerstitial nephritis, and nephrolithiasis. None of these findings are common presentations of pSS. The presence of all of these symptoms in one individual makes this patient an interesting and unique case.
Discussion
We presented the case of a female patient who presented polyuria and polydipsia. She was primarily diagnosed with nephrogenic DI, but her underlying condition was determined to be pSS.
Primary Sjögren syndrome is a disorder of lymphocyte proliferation in the exocrine glands, which usually presents with dryness of eyes and mouth, as a result of impaired saliva and tear production. Our patient had a long-standing history of dry mouth, which was compatible with pSS. Even though she never complained of dry eyes, periodic ophthalmologic examinations revealed that she has been also suffering from dry eyes, both of which are compatible with pSS.
Rash is not a common finding in pSS, and is reported in 10– 30% of patients. The rash is most commonly a nonpalpable purpura in the lower extremities, but can have other presentations as well. The most important pathological findings in the skin biopsies of pSS include benign hypergammaglobulinemic purpura of Waldenstrom, leukocytoclastic vasculitis, and urticarial vasculitis [3]. Our patient had a history of rash in the lower extremities, with no photosensitivity. Biopsy of the lesion revealed the diagnosis to be leukocytoclastic vasculitis, which is compatible with pSS. The skin biopsy also ruled out SLE, which can be the underlying cause of SS in some cases.
Nephrogenic DI is one of the complications of renal involvement with pSS that has been reported in a few cases. For example, only 5 out of 48 pSS patients with renal involvement were diagnosed with DI [9], and this number was 7 out of 149 patients in another prospective cohort [10]. Nephrogenic DI is usually reported to be mild [6,11].
Epithelial involvement is the most common type of kidney disease, and is found in 75%–80% of the patients, while the rest of the patients present glomerular disease [12]. This epithelial involvement can present as RTA (73.1%), hypokalemic paralysis (7%), Fanconi syndrome (3%) and DI (2.3%) versus 20% glomerulonephritis [11]. However, not all studies report the same conclusion about the predominance of TIN. In a cohort study of 33 biopsies, 52% had glomerulonephritis, 35% had TIN, and 12% had both at the same time [5].
In another review, proximal lesions were reported in 10–42%, RTA I in 5–24%, along with a concentrating defect in 17–28% [13].
Our patient was suffering from tubular damage, which is in line with the more prevalent pathology of pSS renal involvement in the literature, leading to metabolic acidosis and subsequent compensatory respiratory alkalosis. Even though the epithelial presentation is seen in pSS, only 1 case report in the literature has found the presence of both RTA and nephrogenic DI in one patient [14]. Immunosuppressive therapy of pSS was effective in improving the acid/base level; her CO2 levels increased to baseline after therapy.
Nephrocalcinosis and nephrolithiasis in pSS have been known to occur as a result of RTA and hypercalciuric state [14,15]. Treatment of pSS depends on the severity of the disease: For mild disease, treatment is focused on protection of the eyes and mouth against dryness, to avoid the complications mentioned above. Currently available options include pilocarpine and cevemeline [16]. For moderate to severe disease, systemic immunosuppressive treatment might be necessary. Hydroxychloroquine is the current first-line treatment for systemic therapy, and methotrexate and steroids can be used in patients non-responsive to treatment [3,16,17]. The treatment in our patient started with corticosteroids, which were very effective in decreasing systemic symptoms and rash. However, due to severe adverse effects, most importantly weight gain, the patient was unable to tolerate the steroids and was switched to hydroxychloroquine, which was successful in decreasing the symptoms.
Conclusions
This patient was first diagnosed with nephrogenic DI and was started on treatment without addressing the underlying condition. We report this case to raise the clinical suspicion of pSS in patients with nephrogenic DI and other tubular disturbances. Treatment of pSS can result in improved tubular function and associated metabolic disturbances, as well as slowing the progression of kidney disease and other systemic manifestations, which highlights the importance of prompt diagnosis of the disease in treatment.
References:
- 1.Sjögren H. Zur kenntnis der keratoconjunctivitis sicca II. Acta Ophthalmol. 1935;13(1–2):1–39. doi: 10.1111/j.1755-3768.1971.tb08678.x. [in German] [DOI] [PubMed] [Google Scholar]
- 2.Ramos-Casals M, Solans R, Rosas J, et al. Primary Sjögren syndrome in Spain: Clinical and immunologic expression in 1010 patients. Medicine (Baltimore) 2008;87(4):210–19. doi: 10.1097/MD.0b013e318181e6af. [DOI] [PubMed] [Google Scholar]
- 3.Fox RI, Liu AY. Sjögren’s syndrome in dermatology. Clin Dermatol. 2006;24(5):393–413. doi: 10.1016/j.clindermatol.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 4.Theander E, Jacobsson LTH. Relationship of Sjögren’s syndrome to other connective tissue and autoimmune disorders. Rheum Dis Clin North Am. 2008;34(4):935–47. doi: 10.1016/j.rdc.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 5.Goules A V, Tatouli IP, Moutsopoulos HM, Tzioufas AG. Clinically significant renal involvement in primary Sjögren’s syndrome: Clinical presentation and outcome. Arthritis Rheum. 2013;65(11):2945–53. doi: 10.1002/art.38100. [DOI] [PubMed] [Google Scholar]
- 6.Lim AKH, Choi MJ. Distal renal tubular acidosis associated with Sjögren syndrome. Intern Med J. 2013;43(12):1330–34. doi: 10.1111/imj.12300. [DOI] [PubMed] [Google Scholar]
- 7.François H, Mariette X. Renal involvement in primary Sjögren syndrome. Nat Rev Nephrol. 2016;12(2):82–93. doi: 10.1038/nrneph.2015.174. [DOI] [PubMed] [Google Scholar]
- 8.Evans RDR, Laing CM, Ciurtin C, Walsh SB. Tubulointerstitial nephritis in primary Sjögren syndrome: Clinical manifestations and response to treatment. BMC Musculoskelet Disord. 2016;17(1):2. doi: 10.1186/s12891-015-0858-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bossini N, Savoldi S, Franceschini F, et al. Clinical and morphological features of kidney involvement in primary Sjögren’s syndrome. Nephrol Dial Transplant. 2001;16(12):2328–36. doi: 10.1093/ndt/16.12.2328. [DOI] [PubMed] [Google Scholar]
- 10.Ram R, Swarnalatha G, Dakshinamurty KV. Renal tubular acidosis in Sjögren’s syndrome: A case series. Am J Nephrol. 2014;40(2):123–30. doi: 10.1159/000365199. [DOI] [PubMed] [Google Scholar]
- 11.Ren H, Wang W-M, Chen X-N, et al. Renal involvement and followup of 130 patients with primary Sjögren’s syndrome. J Rheumatol. 2008;35(2):278–84. [PubMed] [Google Scholar]
- 12.Maripuri S, Grande JP, Osborn TG, et al. Renal involvement in primary Sjögren’s syndrome: A clinicopathologic study. Clin J Am Soc Nephrol. 2009;4(9):1423–31. doi: 10.2215/CJN.00980209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Evans R, Zdebik A, Ciurtin C, Walsh SB. Renal involvement in primary Sjögren’s syndrome. Rheumatology (Oxford) 2015;54(9):1541–48. doi: 10.1093/rheumatology/kev223. [DOI] [PubMed] [Google Scholar]
- 14.Hirose W, Kawagoe M. A case of Sjögren’s syndrome complicated by nephrogenic diabetes insipidus and renal tubular acidosis. Mod Rheumatol. 2000;10(3):176–79. doi: 10.3109/s101650070028. [DOI] [PubMed] [Google Scholar]
- 15.Moutsopoulos HM. Sjögren’s syndrome: autoimmune epithelitis. Clin Immunol Immunopathol. 1994;72(2):162–65. doi: 10.1006/clin.1994.1123. [DOI] [PubMed] [Google Scholar]
- 16.Thanou-Stavraki A, James JA. Primary Sjögren’s syndrome: Current and prospective therapies. Semin Arthritis Rheum. 2008;37(5):273–92. doi: 10.1016/j.semarthrit.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 17.Tishler M, Yaron I, Shirazi I, Yaron M. Hydroxychloroquine treatment for primary Sjögren’s syndrome: Its eVect on salivary and serum inflammatory markers. Ann Rheum Dis. 1999;58:253–56. doi: 10.1136/ard.58.4.253. [DOI] [PMC free article] [PubMed] [Google Scholar]