

Abstract
Exomiser is an application that prioritizes genes and variants in next-generation sequencing (NGS) projects for novel disease-gene discovery or differential diagnostics of Mendelian disease. Exomiser comprises a suite of algorithms for prioritizing exome sequences using random-walk analysis of protein interaction networks, clinical relevance and cross-species phenotype comparisons, as well as a wide range of other computational filters for variant frequency, predicted pathogenicity and pedigree analysis. In this protocol, we provide a detailed explanation of how to install Exomiser and use it to prioritize exome sequences in a number of scenarios. Exomiser requires ~3 GB of RAM and roughly 15–90 s of computing time on a standard desktop computer to analyze a variant call format (VCF) file. Exomiser is freely available for academic use from http://www.sanger.ac.uk/science/tools/exomiser.
INTRODUCTION
Whole-exome sequencing (WES) refers to massively parallel NGS of exonic sequences. WES has been enormously successful in the identification of novel Mendelian disease-associated genes1,2, and it has recently begun to be used for clinical diagnostics3–6. WES uses hybridization methods to enrich (‘capture’) sequences of interest, representing mainly the exon sequences of nearly all protein-coding genes, but these sequences may additionally include miRNA sequences or other genomic regions of interest. Common linkers or adapters are used as primers to amplify the target sequences in a single PCR, and to subject the enriched fragments to NGS7. The underlying assumption of this strategy is that the great majority of mutations in Mendelian disease will be located in or near coding exon sequences.
WES has substantially accelerated the pace of discovery of disease-associated genes8,9. Nonetheless, the analysis of WES remains challenging. There are at least 7,000 Mendelian (monogenic) diseases, but to date only about half of the genes mutated in these disorders have been identified7. An individual exome typi-cally harbors over 30,000 variants compared with the genomic ref-erence sequence, and up to roughly 10,000 of them are predicted to lead to nonsynonymous amino acid substitutions, alterations of conserved splice site residues, or small insertions or deletions9. Even after filtering out common variants, additional methods are needed to predict the variants that may have serious functional con-sequences and prioritize them for validation10–12. Many methods exist to predict which variants deleteriously affect the function of individual proteins on the basis of characteristics such as conservation, physicochemical properties of the wild-type and variant amino acids, and other protein features. However, each genome is thought to harbor ~100 genuine loss-of-function vari-ants with ~20 genes completely inactivated13. This implies that merely ranking candidate genes on the basis of the rarity and predicted pathogenicity of variants found in them will not result in good identifications of disease-associated genes in WES data. Therefore, a number of different methods for computational disease-gene prioritization have been developed that aim to integrate complex and heterogeneous data sets, including expression data, genetic sequences, functional annotations, protein-protein interaction networks and information from the medical literature in order to derive a ranked list of genes that provide investigators clues about those genes that warrant closer investigation14. WES data analysis can combine the assessment of individual variants with the prioritization of affected genes.
Traditional rare-disease workups including clinical evaluation, chromosomal analysis, fluorescence in situ hybridization (FISH), array comparative genomic hybridization (CGH), single-gene Sanger sequencing, biochemical studies and gene panel analysis identify the molecular diagnosis in less than half of the patients seen in typical medical genetics settings15. Although WES is expected to substantially improve on this diagnostic yield, it is currently difficult to provide an estimate of the magnitude of the improvement to be expected in a general clinical environment. However, initial studies of clinical WES on large cohorts have reported diagnostic yields of 16–25% (refs. 3,4,16). We have addressed the question of what additional yield can be expected by WES for patients who have already had extensive clinical genetic evaluation (physical examination by medical geneticists, array CGH, and often targeted Sanger gene sequencing) but remained without a diagnosis, and we have developed a phenotype-driven prioritization procedure that can be used with WES or with large gene panels comprising all currently known disease-associated genes. In this group of patients, the additional diagnostic yield was 28% (ref. 5).
Here we describe a protocol for the Exomiser suite that comprises several methods that use clinical data5, model organism phenotype data5,17–19, as well as random-walk analysis of protein interactome data20,21 to perform prioritization. The Exomiser has been used in a number of projects for disease-gene discovery and diagnostics22–24. The US National Institutes of Health (NIH) Undiagnosed Diseases Program (UDP) has incorporated Exomiser into its WES analyses and used it to aid in the identification of novel disease-gene associations, as well as in the diagnosis of known disorders25. The PhenomeCentral portal (https://phenomecentral.org/) uses Exomiser to help clinicians find similar rare disease patients from deep phenotype and WES data, and it integrates Exomiser’s gene prioritization in its matchmaking algorithms.
Overview of analysis with Exomiser
The inputs to Exomiser are the called variants resulting from exome sequencing of a rare disease patient and, optionally, other affected and unaffected family members. These variants are stored using VCF, a tab-separated file format with columns for the chromosome, position, Single Nucleotide Polymorphism Database (dbSNP) ID, reference allele, alternative allele, variant quality, a filter column containing information on whether the variant has passed or failed various filters, a column containing extra annotations, and finally columns that define the genotype of the variant26.
The Exomiser analyzes these VCF files using a single command to first filter the variants and then to prioritize the remaining candidates to help researchers identify the causative variant or variants. The filtering step is critical in order to reduce the 30,000+ variants seen in a typical exome to a more manageable size. However, this still typically leaves more candidates (up to 1,000) than can be reasonably manually assessed, so some sort of ranking by a prioritization algorithm is necessary. Figure 1 summarizes the rationale behind Exomiser’s filtering and prioritization steps, and it illustrates the four prioritization algorithms that are incorporated in Exomiser.
Figure 1.
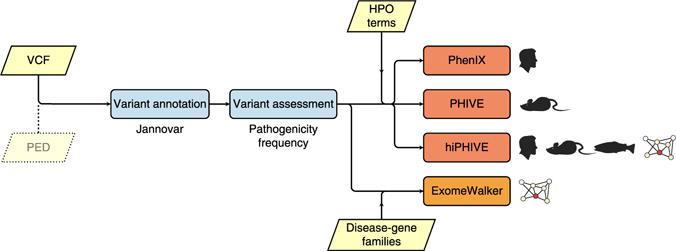
Overview of the processing steps of Exomiser. The Exomiser comprises a suite of four algorithms for the analysis of NGS data for diagnostics or novel disease-gene discovery in the field of rare disease. As input, Exomiser requires a VCF file containing the called variants. If the VCF file comprises samples from multiple family members, then a PED file is additionally required. The Exomiser initially annotates variants using Jannovar27, and then it removes variants that are off the exomic target or that are more common than a user-supplied threshold; it then ranks the remaining variants according to their predicted pathogenicity. Finally, the clinical relevance of the genes harboring these variants is assessed using one of three phenotype-driven algorithms (PHIVE, PhenIX or hiPHIVE) or by a random-walk algorithm that assesses the vicinity of the genes to members of disease-gene family on the protein-protein interactome. Users are required to supply HPO terms for the phenotype-driven algorithms or a list of seed genes representing the disease-gene family for ExomeWalker.
Filtering
The first step Exomiser performs is to annotate each variant relative to the University of California, Santa Cruz (UCSC) hg19 transcript set using the Jannovar software library27. This annotation describes the location within or between transcripts, the type of variant (missense, nonsense, intergenic and so on) and the predicted consequence of the variant on the protein-coding sequence. By default, Exomiser then removes any variants that are off target (intergenic, intronic, upstream, downstream or intronic) or synonymous, although this can be switched off (Box 1). As candidates for rare disease are being sought, users typically specify an upper threshold of minor allele frequency (MAF) for Exomiser’s frequency filter (under the assumption that a common variant cannot be the cause of a rare disease). Other optional filters allow the user to remove variants that are below a particular quality in the QUAL column of the VCF file or to ignore those that are not in predefined set of genes or within a genomic interval (Box 1).
Box 1. Modifying the behavior of Exomiser.
The behavior of the prioritizers described in Step 2A–D can be modified by adding the following arguments to the command.
(A) Inheritance model
If you know the suspected inheritance pattern for the patient being analyzed, this can be specified in Step 2A–D by supplying the extra option ‘--inheritance-mode <arg>’ or ‘-I <arg>’ where <arg> is one of AR, AD or X for autosomal recessive, autosomal dominant or X-linked inheritance.
(B) Family-based analysis
If your VCF file is a multisample exome file containing data from affected and unaffected related people, then these relationships should be specified in a pedigree file (Box 2) in Step 2A–D by supplying the extra option ‘--ped <file>’ or ‘-p <file>’ where <file> is the full path to your PED file. The mode of inheritance should additionally be specified. An example multisample VCF (Pfeiffer-quartet. vcf) and a PED file (Pfeiffer-quartet.ped) are provided in the installation directory, and they can be analyzed with the following command:
java -Xms2g -Xmx3g -jar exomiser-cli-6.0.0.jar --prioritiser=hiphive --max-freq 1 --hpo-ids HP:0001156, HP:0001363, HP:0011304, HP:0010055 --vcf data/Pfeiffer-quartet.vcf --ped data/Pfeiffer-quartet.ped -I AD --out-format=TSV-GENE,TSV-VARIANT,VCF,HTML --out- file=results/Pfeiffer-quartet
(C) Specifying a disease instead of a list of phenotype terms
If the specific phenotypes exhibited by the patient are not to hand, but the suspected or diagnosed disease is, then this can be specified by replacing the ‘--hpo-ids <arg>’ option in Step 2A,B with ‘--disease-id <arg>’ or ‘-D <arg>’ where <arg> is an OMIM, Orphanet or DECIPHER disease identifier, e.g., OMIM:101200. This enables Exomiser to use the generic HPO annotations for the disease (available at http://www.human-phenotype-ontology.org). Therefore, the disease must be present in that resource.
(D) Retaining off-target and synonymous variants
By default, off-target (intergenic, intronic, upstream, downstream or intronic noncoding RNA) and synonymous variants are removed by Exomiser before any other user-defined filtering and prioritization steps. To be more conservative and to detect possible noncoding or synonymous causative variants, the ‘–keep-off-target –keep-non-pathogenic’ options should be added to Step 2A–D.
(E) Restricting to a defined set of genes
If you are only interested in identifying causative variants in a defined set of the genes, then this can be specified by adding the ‘–genes-to-keep <arg>‘ option to Step 2A–D where <arg> is a comma-separated list of EntrezGene identifiers.
Another optional but frequently used filter is to remove variants that do not fit the expected inheritance pattern: autosomal dominant (AD), autosomal recessive (AR) or X-linked. For single sample VCF files, this simply restricts the output to genes containing the following: (i) one or more heterozygous variants for AD inheritance, (ii) X-chromosomal genes for X-linked variants or (iii) a homozygous or two heterozygous variants for AR inheritance. For multisample, family-based analysis, the filtering is more sophisticated and powerful in terms of reducing the number of candidates. For AD inheritance, the filter demands that all affected persons and no unaffected person carry a specific variant in a heterozygous state. We note that although this filter can be used to search for de novo variants, to avoid false-positive results, it is preferable to first analyze the alignment data using a tool (such as DeNovoGear28) that is designed to analyze de novo mutations from familial sequencing data, before ranking candidate genes with Exomiser. For AR diseases, the filter searches for a variant that is homozygous in all affected individuals, heterozygous in both parents of each affected person and heterozygous or not present in unaffected siblings. The filter additionally recognizes compound heterozygous mutations that are both present in all affected individuals and each present in only one parent, and of which at most one is present in unaffected siblings. The filters for X-chromosomal recessive and dominant inheritance function analogously. To analyze family data, users are required to input a PED file representing the family structure. There must be one entry for each family member being sequenced (Box 2). We note that the pedigree filtering capabilities of Exomiser are intended for use with the relatively small and simple family structures such as trios and nuclear families with multiple affected children that are typical of current WES experiments. For larger pedigrees, it may be useful to first examine the sequence data using formal linkage analysis in order to filter out regions that are incompatible with linkage before Exomiser analysis29.
Box 2. Generating a PED file.
PED files use a tab-separated file format and require the following columns:
Family ID
Individual ID
Paternal ID (0=unknown)
Maternal ID (0=unknown)
Sex (1=male; 2=female; other=unknown)
Phenotype (1=unaffected; 2=affected)
An example PED file (Pfeiffer-quartet.ped) is provided in the data directory of the installation:
| FAM1 | ISDBM322016 | 0 | 0 | 1 | 1 |
| FAM1 | ISDBM322018 | 0 | 0 | 2 | 1 |
| FAM1 | ISDBM322015 | ISDBM322016 | ISDBM322018 | 1 | 1 |
| FAM1 | ISDBM322017 | ISDBM322016 | ISDBM322018 | 2 | 2 |
This represents a family with unaffected father (ISDBM322016), mother (ISDBM322018) and son (ISDBM322015), and an affected daughter (ISDBM322017). These individual IDs refer to the sample columns used in the multisample VCF (Pfeiffer-quartet.vcf) that store the genotype for each patient per variant. Currently, Exomiser supports only PED files that represent a single family.
Prioritization
The Exomiser suite contains a number of different methods for variant prioritization based on protein-protein interactions and/or phenotype comparisons between a patient and existing human disease databases and model organisms. Each of these is detailed below, and the decision tree for choosing which prioritization method to use is described in Figure 2.
Figure 2.
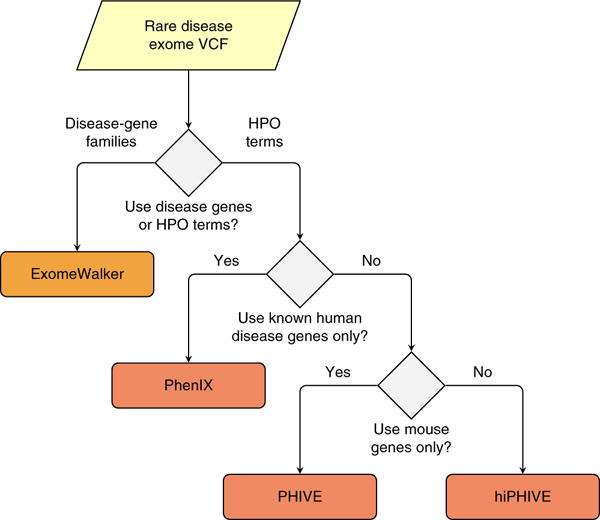
Choice of Exomiser prioritization method. Exomiser can be run using a set of seed genes if a disease-gene family can be identified. For instance, numerous genes mutated in retinitis pigmentosa (RP) have been identified. If it is suspected that a patient may have a mutation in a novel gene for RP, then Exomiser can be run with the ExomeWalker prioritization method together with a list of NCBI Entrez gene IDs for genes already known to be involved in RP (e.g., 6121 for RPE65, 130557 for ZNF513 and so on). Alternatively, Exomiser can be run using phenotypic similarity–based algorithms (PHIVE, PhenIX or hiPHIVE). Here the user needs to enter a list of HPO terms representing the clinical manifestations observed in the individual being investigated (Box 3). The choice of the prioritization method and parameters will depend on the clinical or research goal. PhenIX will interrogate only known Mendelian disease genes, and it will use only human phenotypic data to calculate phenotypic similarities. PHIVE will use mouse phenotypic data to identify candidates. Finally, hiPHIVE can use mouse, zebrafish and human clinical data, and it can additionally integrate further candidates on the basis of an analysis of the protein-protein interaction network. All of the analyses can be combined with additional filters (Box 1).
Exomiser calculates variant-based and method-specific, gene-based scores and combines them using a logistical regression model to generate a final combined score that is used for ranking. The variant scores are a combination of how rare the variant is as observed in the 1000 Genomes Project30 and Exome Server Project (ESP 6500) data sets, together with its predicted pathogenicity. How the gene score is calculated varies, depending on which prioritization method is chosen by the user.
Cross-species mouse-human phenotype comparisons: PHIVE
The original implementation of Exomiser used the PhenoDigm algorithm31 to calculate the phenotypic similarity between a patient’s clinical signs and symptoms and observed phenotypes in mouse mutants associated with each gene candidate in the exome. The rationale behind this approach is that if a mouse model exists for the gene containing the disease-associated mutation, then it is likely to exhibit phenotypic similarity to the clinical phenotypes. These mouse data come from the Mouse Genome Database32 (MGD) and the International Mouse Phenotyping Consortium33 (IMPC). The current coverage of human protein-coding genes with mouse phenotype data is only ~33%, but the IMPC plans to achieve near-complete coverage by 2021.
Clinical diagnostics: PhenIX
In clinical diagnostics, it is not always possible or appropriate to follow up on interesting candidate genes for which today no known disease association has been proven. Therefore, we have developed a phenotypic prioritization procedure that analyzes only those genes that have been associated with a Mendelian disease5. This strategy may be desirable in certain clinical settings in which the search for novel disease genes in a research context is not possible or not desired by the affected families. The algorithm uses the semantic similarity approach to differential diagnostics that we previously implemented as the Phenomizer34. The PhenIX algorithm evaluates and ranks variants on the basis of pathogenicity and semantic similarity of patients’ phenotypes as described by Human Phenotype Ontology (HPO) terms to those of Mendelian diseases whose molecular etiology has been clarified (corresponding to 3,101 genes in the current HPO version). The approach achieved a high diagnostic yield of 97% as the top hit in simulations5. Users may want to consider first using PhenIX to search for known diseases that might explain the clinical manifestations of the patient being investigated, and then using the other prioritization methods in the Exomiser suite to search for novel disease-gene candidates if no diagnosis can be made with PhenIX.
Protein-protein interactions: ExomeWalker
In genetically heterogeneous diseases such as Bardet-Biedl syndrome, mutations in different genes lead to a single disease with an identical or nearly identical spectrum of clinical manifestations. In addition, genetic diseases with lesser degrees of similarity to one another, such as type I congenital disorders of glycosylation, are often grouped into so-called disease-gene families35. In both cases, the genes involved are often part of the same pathway or interact closely, such that a mutation in any of them results in similar phenotypic manifestations. The ExomeWalker prioritization algorithm is designed to identify new causative genes by identifying which of the mutated genes in the exome interacts closely with previously implicated genes for the disease21. The user supplies the list of implicated or suspected seed genes, and a random walk with restart algorithm is used to score how close each candidate gene is to these in a protein-protein association network20. An overall score for how close each candidate is to each of the seeded genes is then used in the Exomiser ranking algorithm. Thus, Exome-Walker is suitable for cases in which the user can define a set of seed genes. Although we have tested the algorithm using disease-gene families based on phenotypic similarity, Exomiser can be run with any set of seed genes that the user deems to be relevant (encoded as a list of National Center for Biotechnology Information (NCBI) gene36 identifiers).
Integrated phenotypic and interactome analysis: hiPHIVE
Exomiser has been extended to use data sources from the previous three algorithms into an algorithm called human/interactome-PHIVE, or hiPHIVE. The phenotypic similarity is calculated not only with mouse data as in the original implementation of PHIVE, but also with zebrafish37 and human phenotype data38. The human data come from the disease-gene associations maintained by Online Mendelian Inheritance in Man (OMIM)39 and Orphanet40 and the phenotype annotations maintained by us and encoded as HPO terms38,41. The zebrafish data originate from the Zebrafish Model Organism database (ZFIN) using the Zebrafish Anatomical Ontology37, Gene Ontology42 and the Phenotype and Trait Ontology (PATO) ontology of qualities43, and they are subsequently converted to a combined zebrafish phenotype term. In hiPHIVE, the human comparisons allow known disease-gene associations to be detected with high specificity and sensitivity, and the mouse and zebrafish data allow novel candidate genes to be flagged as in the original PHIVE algorithm. Finally, for genes that have no phenotype data from any of these sources, we use a random walk with restart algorithm20 to score how close the candidate is in a protein-protein association network to genes with strong phenotypic similarity to the patient. The interaction network consists of high-confidence (>0.7) interactions from STRING44 version 9.05 and contains direct (physical) and indirect (func-tional) protein-protein interactions, as well as associations transferred by orthology from other species or obtained through text mining.
Simulations, run as in the original publication of Exomiser5 and based on spiking known disease mutations into unaffected exomes from the 1000 Genomes Project, demonstrated that the correct variant(s) can be detected as the top hit in 97% of samples. Further experiments in which the known disease-gene associations were masked from the database to represent discovery of a novel association revealed that the causative variant(s) could be detected as the top hit 87% of the time. Hence, the hiPHIVE prioritization method is the tool of choice for patients in whom the causative variant could be a known or novel association45.
Use of Exomiser as a stand-alone application or in larger analysis pipelines
The Exomiser can be run as a stand-alone application that will output an HTML page that summarizes the results of the analysis (Fig. 3). For low-volume or demonstration use of the hiPHIVE method, the online version at http://www.sanger.ac.uk/science/tools/exomiser can be used instead of the protocol detailed in this paper. If desired, Exomiser can also be used within larger analysis pipelines. For this purpose, Exomiser outputs tab-separated value (TSV) and VCF files containing details on the analysis results in a form that can be easily used as input for other software designed for visualization, for specialized analysis or for storage of the results in a database.
Figure 3.

Screenshot of Exomiser output. The Exomiser outputs the results of its analysis as an HTML page that can be opened in any web browser. Equivalent but more detailed results can be output as a text file if desired. The results are shown for a single gene, and include a list of diseases that are known to be caused by mutations in that gene, phenotypic similarity matches to human and model organisms, and indications of proximity in the protein interaction network to further phenotypically similar genes. A list of affected transcripts and the predicted pathogenicity of the variant are shown. The full output has a ranked list of genes, each of which are provided with this information.
For example, The NIH UDP46 has incorporated Exomiser into its standard analysis pipeline. Before running Exomiser, family VCF files are annotated and filtered on the basis of allele frequencies in the UDP cohort and other available databases, basic transcript requirements and Mendelian modes of inheritance. The variants that pass these filters and the HPO terms of the proband are input into Exomiser for each mode of inheritance. The Exomiser VCF output is used to record the variant rankings and scores, and the Exomiser HTML output is used to evaluate the underlying reason for the rankings provided by Exomiser. Both of these output files are taken into account by the clinicians and researchers to prioritize candidate variants.
PhenomeCentral (https://phenomecentral.org/) also uses Exomiser to help clinicians find additional patients with the same rare genetic disease. In addition to providing deep phenotype data encoded as HPO terms, clinicians can upload VCF files for patients, which are then automatically processed with Exomiser. The Exomiser is used to filter the variants by allele frequency and predicted pathogenicity, to annotate them with their effect and then to score the genes on the basis of their phenotypic relevance. PhenomeCentral displays the top results from the VCF output to the clinician, and it incorporates the gene and variant scores into its matchmaking algorithms.
Limitations of the protocol and the software
The Exomiser is designed only for the analysis of Mendelian disease—i.e., it searches for single genes with predicted pathogenic mutations that can best explain the clinical symptoms. The prioritization currently encompasses only those variants that affect the coding region of protein-coding genes, as well as the highly conserved splice consensus sequences at the exon-intron boundaries. The Exomiser integrates numerous data sources including dbSNP47; the 1000 Genomes Project30; the Exome Variant Server (NHLBI GO Exome Sequencing Project 2013, http://evs.gs.washington.edu/EVS/); OMIM39; Orphanet40; the HPO38; mouse phenotype data from the Mouse Genome Database32 at the Mouse Genome Informatics resource and from the IMPC33; zebrafish data from ZFIN using the Zebrafish Anatomical Ontology37, Gene Ontology42 and the PATO ontology of qualities43 (and subsequently converted to a combined zebrafish phenotype term); pathogenicity predictions by MutationTaster10,48, Polyphen2 (ref. 49) and SIFT50, as provided by dbNSFP51; variant annotations by Jannovar27; and gene and transcript definitions by UCSC Genome Browser database52. The data are collected as a consortial effort within the context of the Monarch Initiative (http://monarchinitiative.org/), where each source is versioned and integrated; however, the data sources may still contain inaccuracies or omissions that can change from release to release, and these would affect the performance of Exomiser. Finally, Exomiser does not yet support the analysis of copy-number variants or other large structural variants. The Exomiser will endeavor to include new relevant data sources in the future—for instance, the beta version of Exomiser has incorporated data from the Exome Aggregation Consortium (ExAC; http://exac.broadinstitute.org/), which has aggregated WES data from over 60,000 unrelated individuals from a variety of large-scale sequencing projects, thereby providing a useful reference set of allele frequencies. In addition, support for the new gVCF format will be added to future versions of Exomiser.
Alternative analysis packages
The analysis of whole-exome data involves a pipeline of steps, including quality control53, the reference-guided alignment of NGS reads by programs such as BWA54, variant calling with tools such as GATK55, and the interpretation of the called variants. Exomiser is designed to aid in the interpretation of called variants, and we will not discuss any of the previous steps in the pipeline here, although practitioners need to be aware of the fact that different pipelines will call divergent sets of variants, with obvious consequences for downstream analysis56, and that the provenance of steps needs to be carefully considered and tracked. Analysis packages for VCF26 files can be divided into three categories: those that annotate and filter the sequence variants, packages that apply statistical algorithms for rare disease association to identify candidate genes and packages that use one of a number of algorithms to prioritize genes and variants in order to place the most likely disease gene near the top of the list of candidates. Annotation packages, such as ANNOVAR57, Ensembl’s Variant Effect Predictor58 and Jannovar27 transform the chromosomal coordinates that result from aligning WES reads to the reference genome (e.g., chr14:g.88401213T>C) to the corresponding coordinates for affected transcripts, as well as the category of variant (e.g., c.1843A>G; p.T615A, a missense variant in exon 17 of the GALC gene (encoding galactosylceramidase)). Such applications are not intended to be used as stand-alone programs for the interpretation of WES data, as their output, a list of tens of thousands of annotated variants, does not attempt to pinpoint likely candidate genes or mutations. A number of applications combine annotation, filtering by allelic frequency and predicted pathogenicity, and segregation analysis or intersection of multiple unrelated affected individuals59–63.
A second category of tools comprise those that have been developed to rank genes and variants in rare disease studies on the basis of different probabilistic frameworks that analyze the background variation in genes, as well as the nature and frequency of variants in affected individuals64–66. These tools are especially useful for cohort studies with multiple affected families or individuals. A third category of tools, to which Exomiser belongs, makes use of phenotypic data to prioritize candidate genes using one of a number of algorithms6,67–70. All of these tools use the HPO and deep phenotyping71,72 (Box 3) to power the phenotypic analysis.
Box 3. Selecting HPO terms.
The HPO (http://www.human-phenotype-ontology.org) provides a structured, comprehensive and well-defined set of more than 11,000 terms describing human phenotypic abnormalities, and it provides annotations of nearly 7,300 human hereditary syndromes that yield computable representations of the diseases, associated disease genes, as well as the signs, symptoms, laboratory findings and other phenotypic abnormalities that characterize the diseases38. The HPO is widely used in the rare disease community, having been adopted by the Sanger Institute’s DECIPHER and DDD projects74, the rare disease section of the UK 100,000 Genomes Project, the NIH Undiagnosed Diseases Program and Network, the Matchmaker Exchange and many others. HPO is designed to be interoperable with model organism vocabularies and the gene ontology, in support of cross-species bioinformatics analyses.
Users should enter clinical data into the Exomiser software in the form of a list of HPO terms that describe the clinical features of the patient being analyzed. Terms of the HPO describe individual phenotypic abnormalities, such as ‘Hypoglycemia’ or ‘Macrocephaly’. The HPO website offers a browser (http://www.human-phenotype-ontology.org) with which users can explore the HPO to find appropriate terms17,41. Alternatively, the HPO can be explored in a number of other websites including the Ontology Lookup Service7,75 of the European Bioinformatics Institute (EBI) and the BioPortal of the National Center for Bio-Ontologies76. Terms can also be entered in the clinic using PhenoTips77 and exported.
For each phenotypic feature found in the individual being examined, choose the most specific term possible. Features should be entered to cover all of the important phenotypic abnormalities seen in the patient. However, some medical judgment may be required to decide whether a particular term should be used or not. For instance, in a patient with a disease that is otherwise not related to the eyes, it may not be appropriate to enter the HPO term for ‘Myopia’ (HP:0000545) if the patient has mild short-sightedness of −0.75 diopters with no other eye problems, as this is commonly found in the general population. However, severe myopia of −8 diopters might be related to an underlying genetic defect. In addition, some diseases are associated with large numbers of phenotypic abnormalities (currently, 942 diseases have over 30 HPO annotations in the current version of the HPO). We have found that it often suffices to enter up to 5–10 abnormalities to obtain high-quality search results, but it is difficult to provide a general rule. If no plausible candidate genes are revealed using a particular set of HPO terms, it may be useful to restrict the terms to a smaller set of terms with balanced coverage of all affected organ systems. We have posted guidelines for best practices for HPO annotation, and we also supply a metric to assist the sufficiency of the phenotype profile against the corpus of all known disease-gene and model-gene associations78.
A main advantage of the version of Exomiser, as presented here, is that it can be downloaded and run within hospital firewalls, thereby avoiding data security issues involved in sending patient data to a web server. Exomiser runs quickly (~15–90 s on a typical desktop computer depending on the algorithm chosen), and it can produce output files in HTML format for human consumption or as VCF or tab-separated files that can be incorporated into larger bioinformatics pipelines.
MATERIALS
EQUIPMENT
Data: A VCF file and clinical data (phenotype, pedigree), as described in the text
Exomiser software (the FTP site is accessible through the main Exomiser website http://www.sanger.ac.uk/science/tools/exomiser or directly at ftp://ftp.sanger.ac.uk/pub/resources/software/exomiser/downloads/exomiser/)
Java runtime environment. The Exomiser is written in Java and requires at least version 7 (available at www.oracle.com/java for a wide variety of operating systems)
Free disk space of 25 GB (to store the Exomiser program code and the integrated database)
Hardware (64-bit computer with at least 3 GB free RAM (8 GB preferred); see Equipment Setup
EQUIPMENT SETUP
▲ CRITICAL Most of the commands described in this protocol are meant to be run from the shell prompt (‘command line’) and have been tested under Linux, Mac OS X (10.7 Lion or later) and Windows. Commands to be executed from the shell prompt are prefixed with a dollar sign (‘$’).
Required data
The Exomiser protocol is illustrated using several different example exome VCF files that are provided as part of the installation. For this protocol, we use VCF files obtained from sequencing of an unaffected individual and family73 into which we have inserted published disease-causing mutations.
Hardware setup
The Exomiser does not require unusual hardware resources, and it should run on any computer with a 64-bit architecture.
Downloading and installing software
Download Exomiser from the FTP site (see Equipment). At the time of this writing, the version is 6.0.0 and the file is called ‘exomiser-cli-6.0.0-distribution.zip’. This file is ~1.2 GB in size. Once the download is completed, extract the files using the unzip command
$ unzip exomiser-cli-6.0.0-distribution.zip
Alternatively, use any decompression program supplied by your operating system. This command will cause a directory to be created with most of the files needed to run Exomiser.
Exomiser makes use of a relational database to store information about variant frequency and predicted pathogenicity, gene-to-disease associations and model organism phenotype data. The download version of Exomiser uses an H2 database (http://www.h2database.com/), which supports disk-based tables so that users do not need to install a relational database management system such as MySQL or postgreSQL. To download the database, go to the Exomiser FTP site to the directory called ‘h2_db_dumps’ and download the ‘exomiser-6.0.0.h2.db.gz’ file (the version number should match or be higher than the version number of the Exomiser software you have downloaded). The database file is currently 5.5 GB in size. Uncompress the file in the exomiser-cli-6.0.0/data directory as follows:
$ gunzip exomiser-6.0.0.h2.db.gz $ mv exomiser-6.0.0.h2.db exomiser.h2.db
Ensure that the file is called ‘exomiser.h2.db’ and that it is located in the ‘data/’ directory in which ‘exomiser-cli-6.0.0-distribution.zip’ was unpacked (also see the TROUBLESHOOTING section).
To test whether the installation was successful, run the command below; if the installation was successful, you will see a help message.
$ java -jar exomiser-cli-6.0.0.jar
Alternative procedure to use Exomiser with a local PostgreSQL database
The Exomiser can be run with a file-based H2 database as described above, or alternatively with a local postgreSQL database version. In this case, download the ‘exomiser_dump.pg.gz’ file from the FTP server and load into your database.
$ gunzip -c exomiser-6.0.0.pg.gz | pg_restore -d <database> -U <user> -W
This will generate a schema Exomiser and all the tables that are stored under it. Edit the ‘application properties’ file (Box 4) if the data directory is on a different location or if you want to use postgreSQL for Exomiser.
Box 4. Running Exomiser from a settings file and in batch mode.
Instead of typing all the options specified in Step 2A–D each time, they can be defined in a settings file to save time. For example, a file called step1.settings can be created using example. settings from the Exomiser installation directory as a template and changing the following fields:
vcf=data/Pfeiffer.vcf prioritiser=hiphive max-freq=1.0 hpo-ids=HP:0001156, HP:0001363, HP:0011304, HP:0010055 out-file=results/Pfeiffer-phive out-format=TSV-GENE,TSV-VARIANT,VCF,HTML
and Step 2A(ii) can be run instead as:
java -Xms2g -Xmx3g -jar exomiser-cli-6.0.0.jar --settings-file step1.settings
Command-line parameters override the settings file, so to analyze another VCF file the following command can be used:
java -Xms2g -Xmx3g -jar exomiser-cli-6.0.0.jar --settings-file step1.settings --vcf=AnotherVCFFile.vcf
Exomiser can also be run in batch mode. This can give much faster performance as the software caches large objects such as the protein-protein interaction matrix in memory, so subsequent runs take a fraction of the time of the first run. To run in batch mode, generate a settings file for every analysis you wish to perform, and then create a file such as exomiser_batch.txt that contains the paths to all these settings files. Exomiser is then run in batch mode by the following command:
java -Xms2g -Xmx3g -jar exomiser-cli-6.0.0.jar --batch-file exomiser_batch.txt
The memory caching behavior can be configured by editing the cache option in the application.properties file in the installation directory. Setting it to ‘mem’ will cache variants already analyzed; alternatively, the ‘ehcache’ parameter can be used for finer tuning via the ehcache.xml file.
PROCEDURE
Data preparation ● TIMING ~5–10 min
1| Prepare a VCF file with the variants called from an exome-sequencing experiment.
Analysis of the exome data and ranking of genes
-
2| Select and run an Exomiser program using one of the following options (options A–D). See Figure 2 for an overview of how to choose the method(s) best suited to the analysis goals.
Option Method Description
A PHIVE/hiPHIVE Performs phenotype comparisons with human, mouse and fish genes and their neighbors in the interactome (hiPHIVE) or only to mouse (PHIVE) B PhenIX Performs phenotype comparisons with existing clinical data C ExomeWalker Allows network analysis against previously implicated genes D No phenotype prioritization Runs Exomiser without phenotype prioritization
▲ CRITICAL STEP Box 1 gives advice on how to modify the behavior of each Exomiser program. If applicable, collect additional information regarding a linkage interval, the family structure (in form of a PED file, Box 2), an inheritance model or other optional parameters. The –out-format and –out-file options can be adjusted to rename the output files and only create some of the formats. Alternatively, they can be removed and a single HTML file will be generated with a filename consisting of the original VCF file name appended with -exomiser-6.0.0-results.html.
- PHIVE/hiPHIVE: phenotype comparisons with human, mouse and fish genes and their neighbors in the interactome ● TIMING ~90 s
- Prepare a list of HPO terms representing the phenotypic abnormalities observed in the patient (see Box 3 for more information). The following terms have been selected here as an example: HP:0001156 (brachydactyly syndrome), HP:0001363 (craniosynostosis) and HP:0011304 (broad thumb).
-
Run Exomiser using the hiPHIVE prioritizer with the following command:
$ java -Xms2g -Xmx3g -jar exomiser-cli-6.0.0.jar --prioritiser=hiphive --max-freq 1 --hpo-ids HP:0001156, HP:0001363, HP:0011304, HP:0010055 --vcf data/Pfeiffer.vcf --out-format=TSV-GENE, TSV-VARIANT,VCF,HTML --out- file=results/Pfeiffer-hiphive
▲ CRITICAL STEP This query analyzes Pfeiffer.vcf containing a causative FGFR2 (encoding fibroblast growth factor receptor 2) variant for Pfeiffer syndrome. The file ‘Pfeiffer.vcf’ is provided with the Exomiser distribution, and it is located in the data subdirectory, as shown. To perform the analysis using the original PHIVE algorithm, merely substitute ‘--prioritiser=hiphive’ with ‘ --prioritiser=phive’ in the command shown above.? TROUBLESHOOTING
- PhenIX: phenotype comparisons with existing clinical data ● TIMING ~15 s
- As with Step 2A(i), select a number of HPO terms to represent the phenotypic abnormalities observed in the patient (see Box 3).
-
Run Exomiser using the PhenIX prioritization algorithm.
$ java -Xms2g -Xmx3g -jar exomiser-cli-6.0.0.jar --prioritiser=phenix --max-freq 1 --hpo-ids HP:0001156, HP:0001363, HP:0011304, HP:0010055 --vcf data/ Pfeiffer.vcf --out-format=TSV-GENE, TSV-VARIANT, VCF, HTML --out- file=results/Pfeiffer-phenix
▲ CRITICAL STEP This query is equivalent to the one in Step 2A(ii), except that PhenIX is used instead of hiPHIVE.? TROUBLESHOOTING
- ExomeWalker: network analysis against previously implicated genes ● TIMING ~90 s
- Select a list of seed genes for analysis with the ExomeWalker. EntrezGene IDs for seed genes are entered here as an example: FGFR1 (2260), FGF1 (2246) and FGF8 (2253). This simulates a scenario in which FGFR1, FGF1 and FGF8 have previously been associated with Pfeiffer syndrome, and a patient with a novel FGFR2 causative variant is being analyzed.
-
Run Exomiser using the following ExomeWalker prioritization algorithm:
$ java -Xms2g -Xmx3g -jar exomiser-cli-6.0.0.jar --prioritiser exomewalker --seed-genes 2260, 2246, 2253 --max-freq 1 --vcf data/Pfeiffer.vcf --out- format=TSV-GENE, TSV-VARIANT, VCF, HTML --out-file=results/Pfeiffer-walker
▲ CRITICAL STEP This query is equivalent the one in Step 2A(ii), except that ExomeWalker is used instead of hiPHIVE, and phenotype terms are not entered.? TROUBLESHOOTING
- Prioritization using allele frequency and pathogenicity only ● TIMING ~20 s
-
Run Exomiser without phenotype prioritization.
$ java -Xms2g -Xmx3g -jar exomiser-cli-6.0.0.jar --prioritiser=hiphive --max-freq 1 --vcf data/Pfeiffer.vcf --out-format=TSV-GENE, TSV -VARIANT, VCF, HTML --out-file=results/Pfeiffer-no-patient-data
▲ CRITICAL STEP This query is equivalent to the one in Step 2A(ii), except that no HPO terms are entered for phenotypic ranking.? TROUBLESHOOTING
-
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 1.
TABLE 1.
Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| Equipment Setup | The H2 database is not found (error message states ‘h2Path variable from application.properties not found’) | The H2 database was not placed in the correct directory | Unpack the file in exomiser-cli-6.0.0/data directory and rename it as ‘exomiser.h2.db’ |
| 2A–D | Java Out Of Memory Error | The VCF file is too large | Increase the memory from the 3GB specified by the ‘-Xmx3g’ flag on the command line |
| 2D | The expected candidate gene was not found, although the variant is known to be present | Inappropriate parameter settings | Reconsider the parameters. For instance, a frequency threshold of 0.1% may be too low for autosomal recessive diseases with a higher prevalence of heterozygote carriers |
● TIMING
Step 1, data preparation: ~5–10 min
Step 2A, PHIVE/hiPHIVE: ~90 s
Step 2B, PhenIX: ~15 s
Step 2C, ExomeWalker: ~90 s
Step 2D, prioritization using allele frequency and pathogenicity only: ~20 s
ANTICIPATED RESULTS
While running the analysis, the output should include the number of variants removed through the filters:
Filtering removed 36528 variants. Returning 1181 filtered variants from initial list of 37709
For Step 2A, the output settings create Pfeiffer-phive.genes.tsv, Pfeiffer-phive.variants.tsv, Pfeiffer-phive.vcf and Pfeiffer-phive.html in the results directory. Pfeiffer-phive.genes.tsv has a row for each of the 892 postfiltered genes containing one of these 1,181 variants, sorted by the Exomiser combined score and then alphabetically by gene symbol for ties. Each row contains the gene symbol, EntrezGene ID, gene score from the prioritization, variant score based on allele frequency and pathogenicity, and the Exomiser combined score (the remaining columns contain intermediate, internal scores that can be ignored). Note that the variant score is that of the rarest and most pathogenic variant for each gene, or, in the case of a compound heterozygous scoring, the average of the top two scores.
The known causative gene (FGFR2) is the top hit in the output having a phenotype score of 0.8764 and a variant with a maximum predicted pathogenicity of 1 and a combined score of 0.9933. Exomiser outputs a TSV file with the variant score, the phenotype scores for human, mouse and fish comparison, the ExomeWalker score, as well as the combined hiPHIVE score, and other information. The header of the file specifies the meaning of the fields, and explanations of the algorithms can be found in the original publications5,21.
The Pfeiffer-phive.vcf file is equivalent to the original Pfeiffer.vcf input file but ordered by the best gene candidates and their variants. Extra information in the filter column to indicate which variants passed or failed particular filters and the Exomiser annotation and scoring in the INFO column is also included. For example, for the causative FGFR2 variant chr10:123256215T>G, Exomiser will add the following information to the INFO column of the output VCF file:
GENE=FGFR2; INHERITANCE=AD; MIM=101600; EXOMISER_GENE=FGFR2; EXOMISER_VARIANT_SCORE=1.0; EXOMISER_GENE_PHENO_SCORE=0.87642866; EXOMISER_GENE_VARIANT_SCORE=1.0; EXOMISER_GENE_COMBINED_SCORE=0.99334514
The Pfeiffer-phive.variants.tsv file contains every variant with its annotations in a tab-separated format. This format can easily be used to integrate Exomiser in pipelines. Printed annotations of variants are the Human Genome Variation Society (HGVS) annotation, the pathogenicity scores, frequencies in the population databases, genotype, functional class of the variant and if the variant passed the filter settings. The Pfeiffer-phive.html file can be opened in a browser for a visual representation of the analysis that is largely equivalent to that seen on the Exomiser website (Fig. 3). In particular, the evidence for the variant scoring based on allele frequency and predicted pathogenicity and the gene score based on the phenotype matching can be explored.
Step 2B produces equivalent results and files to those in Step 2A, but here the patient phenotypes have only been compared with human phenotype data using a different algorithm (Phenomizer). Again, in Pfeiffer-phenix.genes.tsv, the causative mutation in FGFR2 is the top hit, but this time the phenotype score is the maximum of 1, and the combined score is 0.9979.
Step 2C produces equivalent results and files to those in Step 2A, but here, instead of ranking the candidates based partially on phenotypic similarity, proximity in the interactome to FGFR1, FGF1 and FGF8 is used to assess candidacy. FGFR2 is the top hit in the TSV output file with an ExomeWalker gene score of 0.0210 and a combined score of 0.9887.
In Step 2D, no phenotype information has been entered, none of the genes get scored and therefore ranking is purely based on the variant scores. Hence, in Pfeiffer-no-patient-data.tsv the known causative gene (FGFR2) is on row 31, and it is one of 95 top-ranked candidates, with a variant with a maximum predicted pathogenicity of 1.
Acknowledgments
This project was supported by the Bundesministerium für Bildung und Forschung (BMBF; project no. 0313911), the European Community’s Seventh Framework Programme (grant agreement no. 602300; SYBIL) and NIH grant no. 5R24OD011883 (Monarch Initiative).
Footnotes
AUTHOR CONTRIBUTIONS P.N.R. and D.S. conceived of the project, programmed the prototype code, and wrote the manuscript. J.O.B.J., M.J., S.K., M.S., N.L.W. and E.S. developed software. T.Z., O.J.B. and W.P.B. tested code and contributed to the development of analysis strategies. S.K., M.A.H. and P.N.R. developed the phenotype analysis framework. M.A.H. helped develop the ontologies and the HPO curation standard. All authors reviewed and approved of the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details are available in the online version of the paper.
References
- 1.Ng SB, et al. Exome sequencing identifies the cause of a Mendelian disorder. Nat Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ng SB, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang Y, et al. Clinical whole-exome sequencing for the diagnosis of Mendelian disorders. N Engl J Med. 2013;369:1502–1511. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang Y, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312:1870–1879. doi: 10.1001/jama.2014.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zemojtel T, et al. Effective diagnosis of genetic disease by computational phenotype analysis of the disease-associated genome. Sci Transl Med. 2014;6:252ra123. doi: 10.1126/scitranslmed.3009262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Soden SE, et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med. 2014;6:265ra168. doi: 10.1126/scitranslmed.3010076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boycott KM, Vanstone MR, Bulman DE, MacKenzie AE. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat Rev Genet. 2013;14:681–691. doi: 10.1038/nrg3555. [DOI] [PubMed] [Google Scholar]
- 8.Robinson PN, Krawitz P, Mundlos S. Strategies for exome and genome sequence data analysis in disease-gene discovery projects. Clin Genet. 2011;80:127–132. doi: 10.1111/j.1399-0004.2011.01713.x. [DOI] [PubMed] [Google Scholar]
- 9.Gilissen C, Hoischen A, Brunner HG, Veltman JA. Disease gene identification strategies for exome sequencing. Eur J Hum Genet. 2012;20:490–497. doi: 10.1038/ejhg.2011.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–362. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- 11.Li MX, et al. Predicting Mendelian disease-causing non-synonymous single nucleotide variants in exome sequencing studies. PLoS Genet. 2013;9:e1003143. doi: 10.1371/journal.pgen.1003143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pelak K, et al. The characterization of twenty sequenced human genomes. PLoS Genet. 2010;6:e1001111. doi: 10.1371/journal.pgen.1001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.MacArthur DG, et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335:823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moreau Y, Tranchevent LC. Computational tools for prioritizing candidate genes: boosting disease gene discovery. Nat Rev Genet. 2012;13:523–546. doi: 10.1038/nrg3253. [DOI] [PubMed] [Google Scholar]
- 15.Shashi V, et al. The utility of the traditional medical genetics diagnostic evaluation in the context of next-generation sequencing for undiagnosed genetic disorders. Genet Med. 2014;16:176–182. doi: 10.1038/gim.2013.99. [DOI] [PubMed] [Google Scholar]
- 16.de Ligt J, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367:1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- 17.Oellrich A, et al. The influence of disease categories on gene candidate predictions from model organism phenotypes. J Biomed Semantics. 2014;5:S4. doi: 10.1186/2041-1480-5-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Köhler S, et al. Construction and accessibility of a cross-species phenotype ontology along with gene annotations for biomedical research. F1000Res. 2013;2:30. doi: 10.12688/f1000research.2-30.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Washington NL, et al. Linking human diseases to animal models using ontology-based phenotype annotation. PLoS Biol. 2009;7:e1000247. doi: 10.1371/journal.pbio.1000247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Köhler S, Bauer S, Horn D, Robinson PN. Walking the interactome for prioritization of candidate disease genes. Am J Hum Genet. 2008;82:949–958. doi: 10.1016/j.ajhg.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smedley D, et al. Walking the interactome for candidate prioritization in exome sequencing studies of Mendelian diseases. Bioinformatics. 2014;30:3215–3222. doi: 10.1093/bioinformatics/btu508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pippucci T, et al. A novel null homozygous mutation confirms CACNA2D2 as a gene mutated in epileptic encephalopathy. PLoS ONE. 2013;8:e82154. doi: 10.1371/journal.pone.0082154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Requena T, et al. Identification of two novel mutations in FAM136A and DTNA genes in autosomal-dominant familial Meniere’s disease. Hum Mol Genet. 2015;24:1119–1126. doi: 10.1093/hmg/ddu524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Farwell KD, et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med. 2015;17:578–586. doi: 10.1038/gim.2014.154. [DOI] [PubMed] [Google Scholar]
- 25.Markello T, et al. York platelet syndrome is a CRAC channelopathy due to gain-of-function mutations in STIM1. Mol Genet Metab. 2015;114:474–482. doi: 10.1016/j.ymgme.2014.12.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Danecek P, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jäger M, et al. Jannovar: a java library for exome annotation. Hum Mutat. 2014;35:548–555. doi: 10.1002/humu.22531. [DOI] [PubMed] [Google Scholar]
- 28.Ramu A, et al. DeNovoGear: de novo indel and point mutation discovery and phasing. Nat Methods. 2013;10:985–987. doi: 10.1038/nmeth.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith KR, et al. Reducing the exome search space for Mendelian diseases using genetic linkage analysis of exome genotypes. Genome Biol. 2011;12:R85. doi: 10.1186/gb-2011-12-9-r85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abecasis GR, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smedley D, et al. PhenoDigm: analyzing curated annotations to associate animal models with human diseases. Database. 2013;2013:bat025. doi: 10.1093/database/bat025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blake JA, Bult CJ, Kadin JA, Richardson JE, Eppig JT. The Mouse Genome Database (MGD): premier model organism resource for mammalian genomics and genetics. Nucleic Acids Res. 2011;39:D842–D848. doi: 10.1093/nar/gkq1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koscielny G, et al. The International Mouse Phenotyping Consortium Web Portal, a unified point of access for knockout mice and related phenotyping data. Nucleic Acids Res. 2014;42:D802–D809. doi: 10.1093/nar/gkt977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Köhler S, et al. Clinical diagnostics in human genetics with semantic similarity searches in ontologies. Am J Hum Genet. 2009;85:457–464. doi: 10.1016/j.ajhg.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oti M, Brunner HG. The modular nature of genetic diseases. Clin Genet. 2007;71:1–11. doi: 10.1111/j.1399-0004.2006.00708.x. [DOI] [PubMed] [Google Scholar]
- 36.Brown GR, et al. Gene: a gene-centered information resource at NCBI. Nucleic Acids Res. 2015;43:D36–D42. doi: 10.1093/nar/gku1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Slyke CE, Bradford YM, Westerfield M, Haendel MA. The zebrafish anatomy and stage ontologies: representing the anatomy and development of Danio rerio. J Biomed Semantics. 2014;5:12. doi: 10.1186/2041-1480-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Köhler S, et al. The Human Phenotype Ontology project: linking molecular biology and disease through phenotype data. Nucleic Acids Res. 2014;42:D966–D974. doi: 10.1093/nar/gkt1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM.org: Online Mendelian Inheritance in Man (OMIM(R)), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015;43:D789–D798. doi: 10.1093/nar/gku1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rath A, et al. Representation of rare diseases in health information systems: the Orphanet approach to serve a wide range of end users. Hum Mutat. 2012;33:803–808. doi: 10.1002/humu.22078. [DOI] [PubMed] [Google Scholar]
- 41.Robinson PN, et al. The Human Phenotype Ontology: a tool for annotating and analyzing human hereditary disease. Am J Hum Genet. 2008;83:610–615. doi: 10.1016/j.ajhg.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gene Ontology Consortium. Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015;43:D1049–D1056. doi: 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gkoutos GV, et al. Entity/quality-based logical definitions for the human skeletal phenome using PATO. Conf Proc IEEE Eng Med Biol Soc. 2009;2009:7069–7072. doi: 10.1109/IEMBS.2009.5333362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Franceschini A, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:D808–D815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bone WP, et al. Computational evaluation of exome sequence data using human and model organism phenotypes improves diagnostic efficiency. Genet Med. doi: 10.1038/gim.2015.137. in the press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gahl WA, et al. The National Institutes of Health Undiagnosed Diseases Program: insights into rare diseases. Genet Med. 2012;14:51–59. doi: 10.1038/gim.0b013e318232a005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2015;43:D6–D17. doi: 10.1093/nar/gku1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 49.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 51.Liu X, Jian X, Boerwinkle E. dbNSFP v2.0: a database of human non-synonymous SNVs and their functional predictions and annotations. Hum Mutat. 2013;34:E2393–E2402. doi: 10.1002/humu.22376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rosenbloom KR, et al. The UCSC Genome Browser database: 2015 update. Nucleic Acids Res. 2015;43:D670–D681. doi: 10.1093/nar/gku1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guo Y, Ye F, Sheng Q, Clark T, Samuels DC. Three-stage quality control strategies for DNA re-sequencing data. Brief Bioinform. 2014;15:879–889. doi: 10.1093/bib/bbt069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.O’Rawe J, et al. Low concordance of multiple variant-calling pipelines: practical implications for exome and genome sequencing. Genome Med. 2013;5:28. doi: 10.1186/gm432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cunningham F, et al. Ensembl 2015. Nucleic Acids Res. 2015;43:D662–D669. doi: 10.1093/nar/gku1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aleman A, Garcia-Garcia F, Salavert F, Medina I, Dopazo J. A web-based interactive framework to assist in the prioritization of disease candidate genes in whole-exome sequencing studies. Nucleic Acids Res. 2014;42:W88–W93. doi: 10.1093/nar/gku407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coutant S, et al. EVA: Exome Variation Analyzer, an efficient and versatile tool for filtering strategies in medical genomics. BMC Bioinformatics. 2012;13(Suppl 14):S9. doi: 10.1186/1471-2105-13-S14-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sifrim A, et al. Annotate-it: a Swiss-knife approach to annotation, analysis and interpretation of single nucleotide variation in human disease. Genome Med. 2012;4:73. doi: 10.1186/gm374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee IH, et al. Prioritizing disease-linked variants, genes, and pathways with an interactive whole-genome analysis pipeline. Hum Mutat. 2014;35:537–547. doi: 10.1002/humu.22520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li MX, Gui HS, Kwan JS, Bao SY, Sham PC. A comprehensive framework for prioritizing variants in exome sequencing studies of Mendelian diseases. Nucleic Acids Res. 2012;40:e53. doi: 10.1093/nar/gkr1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.He Z, et al. Rare-variant extensions of the transmission disequilibrium test: application to autism exome sequence data. Am J Hum Genet. 2014;94:33–46. doi: 10.1016/j.ajhg.2013.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ionita-Laza I, et al. Finding disease variants in Mendelian disorders by using sequence data: methods and applications. Am J Hum Genet. 2011;89:701–712. doi: 10.1016/j.ajhg.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yandell M, et al. A probabilistic disease-gene finder for personal genomes. Genome Res. 2011;21:1529–1542. doi: 10.1101/gr.123158.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Singleton MV, et al. Phevor combines multiple biomedical ontologies for accurate identification of disease-causing alleles in single individuals and small nuclear families. Am J Hum Genet. 2014;94:599–610. doi: 10.1016/j.ajhg.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sifrim A, et al. eXtasy: variant prioritization by genomic data fusion. Nat Methods. 2013;10:1083–1084. doi: 10.1038/nmeth.2656. [DOI] [PubMed] [Google Scholar]
- 69.Masino AJ, et al. Clinical phenotype-based gene prioritization: an initial study using semantic similarity and the human phenotype ontology. BMC Bioinformatics. 2014;15:248. doi: 10.1186/1471-2105-15-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Javed A, Agrawal S, Ng PC. Phen-Gen: combining phenotype and genotype to analyze rare disorders. Nat Methods. 2014;11:935–937. doi: 10.1038/nmeth.3046. [DOI] [PubMed] [Google Scholar]
- 71.Robinson PN. Deep phenotyping for precision medicine. Hum Mutat. 2012;33:777–780. doi: 10.1002/humu.22080. [DOI] [PubMed] [Google Scholar]
- 72.Petrovski S, Goldstein DB. Phenomics and the interpretation of personal genomes. Sci Transl Med. 2014;6:254fs35. doi: 10.1126/scitranslmed.3010272. [DOI] [PubMed] [Google Scholar]
- 73.Corpas M. Crowdsourcing the corpasome. Source Code Biol Med. 2013;8:13. doi: 10.1186/1751-0473-8-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wright CF, et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet. 2014;385:1305–1314. doi: 10.1016/S0140-6736(14)61705-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cote R, et al. The ontology lookup service: bigger and better. Nucleic Acids Res. 2010;38:W155–W160. doi: 10.1093/nar/gkq331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Whetzel PL, et al. BioPortal: enhanced functionality via new Web services from the National Center for Biomedical Ontology to access and use ontologies in software applications. Nucleic Acids Res. 2011;39:W541–W545. doi: 10.1093/nar/gkr469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Girdea M, et al. PhenoTips: patient phenotyping software for clinical and research use. Hum Mutat. 2013;34:1057–1065. doi: 10.1002/humu.22347. [DOI] [PubMed] [Google Scholar]
- 78.Washington NL, et al. How good is your phenotyping? Methods for quality assessment. Proceedings of Phenotype Day 2014@ISMB 2014. 2014 http://phenoday2014.bio-lark.org/pdf/6.pdf.