

Abstract
The 3 human RAS genes play pivotal roles regulating proliferation, differentiation, and survival in normal cells and become mutated in 15% to 20% of all human tumors and amplified in many others. In this report, we examined data from The Cancer Genome Atlas to investigate the relationship between RAS gene mutational status and messenger RNA expression. We show that all 3 RAS genes exhibit increased expression when they are mutated in a context-dependent manner. In the case of KRAS, this increase is manifested by a larger proportional increase in KRAS4A than KRAS4B, although both increase significantly. In addition, the mutational status of RAS genes can be associated with expression changes in other RAS genes, with most of these cases showing decreased expression. The mutational status associations with expression are recapitulated in cancer cell lines. Increases in expression are mediated by both copy number variation and contextual differences, including mutational status of epidermal growth factor receptor (EGFR) and BRAF. These findings potentially reveal an adaptive response during tumor evolution that is dependent on the mutational status of proximal genes in the RAS pathway and cellular context. Cell contextual differences in these adaptations may influence therapeutic responsiveness and alternative resistance mechanisms.
Keywords: TCGA, KRAS, RAS Pathway
Introduction
Since their discovery as virally transduced oncogenes and transforming genes in human tumor DNA, RAS genes have been shown to regulate many cellular processes including cell growth, differentiation, and survival.1,2 The KRAS gene produces 2 spliced isoforms, KRAS4A and KRAS4B,3 so that there are 4 primary protein products of these genes (HRAS, NRAS, KRAS4A, and KRAS4B). The Ras proteins are nearly identical except for differences in their carboxy-termini that give rise to differential posttranslational processing and trafficking mechanisms.4
Ras proteins act as molecular switches: the guanosine diphosphate (GDP)–bound form is inactive and guanosine triphosphate (GTP)–bound form is the active state. Guanosine triphosphatase (GTPase)–activating enzymes (GAPs) stimulate low-level intrinsic GTPase activity and convert Ras proteins to their inactive GDP-bound forms, whereas corresponding exchange enzymes (guanine nucleotide exchange factors [GEFs]) catalyze the replacement of GDP with GTP to activate the switch.5 In cancer, mutations (primarily in codons 12, 13, and 61) render Ras proteins resistant to GAP-assisted GTP hydrolysis resulting in a constitutively active RAS molecule.6,7 Less frequently, mutations occur (eg, at alanine 146) that lower the affinity of Ras proteins for GDP, allowing exchange of GDP for GTP without upstream signaling through GEFs.7 Importantly, these activating mutations in KRAS affect both the KRAS4A and KRAS4B isoforms.
Mutations in the KRAS gene occur in most pancreas and many lung, colorectal and uterine corpus endometrial cancers. Less frequent KRAS mutations have been observed in cancers of the breast, cervix, hepatobiliary duct and testicular germ cell. Although less frequent, NRAS and HRAS mutations are also seen in many tumor types especially thyroid and melanoma (NRAS) and bladder cancer HRAS.7
In addition to point mutations that increase RAS activity, RAS genes are often amplified in human cancers. For example, KRAS is frequently amplified in ovarian, lung squamous cell, uterine, adrenocortical, and esophageal tumors. This suggests that both mutation and expression levels contribute to the roles that RAS genes play in oncogenesis. Given renewed efforts underway to target mutant RAS cancers8–11 and the observation that inhibitors of downstream effectors show context-dependent therapeutic efficacies and alternative resistance mechanisms, efforts to better understand the relationships between genes within the RAS pathway could improve our ability to predict clinical outcomes and anticipate the development of resistance.
As one of the largest public repositories of cancer genomic data, The Cancer Genome Atlas (TCGA) project has produced a wealth of new insights into cancer biology and the underlying driver events that give rise to the disease (see http://cancerge-nome.nih.gov/abouttcga/overview). The Cancer Genome Atlas data are publicly available and contain very broad data modalities spanning many different genomic platforms and associated patient clinical information and as such represent a considerable data mining resource. To date, several detailed integrative analyses of data from many of the individual tumor types have been reported (see https://tcga-data.nci.nih.gov/docs/publications/ and http://cancergenome.nih.gov/publications). In addition, a series of reports have detailed “pan-cancer” analyses where data aggregated across many tumor types and genomics platforms were used to identify similarities and differences among and between different tumor types (http://www.nature.com/tcga/). To more directly benefit the Ras community, a Ras gene–specific survey is desperately needed.
In this report, we take advantage of the wealth of clinical specimens available in TCGA across multiple tumor types where mutation information derived from either whole-exome or whole-genome sequencing and RNASeq–based expression analysis data are available. We analyzed these data to determine the relationship between RAS gene mutation status and expression levels. We show that mutation-associated expression increases occur with all 4 RAS isoforms and also correlates with the mutational status of other RAS pathway genes. These findings suggest that tumor type–specific gene-gene co-expression networks can be heavily influenced by gene mutational status.
Methods
Data preparation
The Cancer Genome Atlas data were obtained from the standard release folder from the Broad DCC firehose (http://gdac.broadinstitute.org/runs/stddata__latest/) for each available tumor type. In addition, the transcript-level messenger RNA expression data were obtained from the TCGA data portal (https://cancergenome.nih.gov/publications) for each individual sample for each tumor type and merged into a single matrix file per tumor type using R (https://www.r-project.org). Following download, the level 3 data were parsed into a table format where each sample/attribute/value becomes a separate database row. These files were then directly loaded into our Oracle 11g database. Additional tables corresponding to summary information derived during the parsing process were also prepared to facilitate the querying process. The data were produced using the July 2016 data release. The tumor type codes used throughout this article can be found on the TCGA Web site (https://tcga-data.nci.nih.gov/tcga/). Tumor purity data were obtained from the synapse portal (https://www.synapse.org) that was made available through a collaboration with TCGA/Broad Institute. The copy number variation (CNV) data were obtained from the DCC analysis page (https://confluence.broadinstitute.org/display/GDAC/Dashboard-Analyses) and we loaded the GISTIC (Genomic Identification of Significant Targets in Cancer)12 data into oracle using the method outlined above (see https://www.biostars.org/p/133927/). The downloaded RNA-Seq expression data used were derived using the RSEM package13 and all expression data were log2 transformed.
Cancer Cell Line Encyclopedia (CCLE) data were downloaded from the following Web site: http://www.broadinsti-tute.org/ccle/home, and the mutation and expression files were then analyzed using standard R analysis workflows (see below). The CCLE RNASeq data were downloaded from CGHub and processed through the Cufflinks suite14 to quantitate genes and transcripts using standard RNA-Seq workflows. The resulting gene and transcript expression levels, as RPKM (reads per kilobase per million), were then loaded into our Oracle database as described above.
Data analysis
We used the ROracle Database interface package (https://cran.r-project.org/web/packages/ROracle/index.html) to connect from R to the Oracle TCGA database instance and a series of wrapper functions to facilitate querying. For example, a function to extract a matrix of gene expression data for all samples given and input gene list, tumor type, and RNA-Seq platform was called for each tumor type, and then, a second function was called to subset the samples into tumor-normal pairs. The resulting list of data matrices is subsequently subdivided by mutation or CNV status (using the GISTIC values12) and then passed to plotting functions for the production of box plots, barplots, and other distribution plots. All plots and statistical tests were produced and performed using built-in R functions. Figures were produced interactively in R using the data matrices produced by those functions and manually subsetting samples.
Results
KRAS expression is increased in samples harboring KRAS mutations
We first focused on KRAS and its KRAS4A and KRAS4B isoforms within the TCGA tumor types in which KRAS is frequently mutated. Figure 1A shows expression levels for KRAS in lung adenocarcinoma (LUAD), pancreatic ductal adenocarcinoma (PAAD), and colon adenocarcinoma (COAD) samples. In each tumor type, we observed statistically significant increases in expression of mutant KRAS using the t test. The values are shown on the figure along with the numbers of samples for each condition.
Figure 1.

(A) KRAS expression is elevated in KRAS-mutant samples from lung pancreatic, and colon adenocarcinomas relative to WT samples. Sample subsets from each tumor type were produced according to their KRAS mutation status and the log2-normalized RSEM13 data were plotted. The P values from t test between sample groups and the number of samples in each sample group are shown on the figure. Lane 1—KRAS WT samples from LUAD, lane 2—KRAS-mutant samples from LUAD, lane 3—KRAS WT samples from PAAD, lane 4—KRAS-mutant samples from PAAD, lane 5—KRAS WT samples from COAD, and lane 6—KRAS-mutant samples from COAD. (B) KRAS4A and KRAS4B expression is increased in KRAS-mutant samples from lung, pancreatic, and colon adenocarcinoma samples. The same sample subgroups from Figure 1A were used to derive the KRAS4A and KRAS4B expression. Lane 1—KRAS4A expression in KRAS WT LUAD samples, lane 2—KRAS4A expression in KRAS-mutant LUAD samples, lane 3—KRAS4B expression in KRAS WT samples from LUAD, lane 4—KRAS4 expression in KRAS-mutant samples from LUAD, lane 5—KRAS4A expression in KRAS WT PAAD samples, lane 6—KRAS4A expression in KRAS-mutant PAAD samples, lane 7—KRAS4B expression in KRAS WT samples from PAAD, lane 8—KRAS4B expression in KRAS-mutant samples from PAAD, lane 9—K4BRAS4BA expression in KRAS WT COAD samples, lane 10—KRAS4A expression in KRAS-mutant COAD samples, lane 11—KRAS4B expression in KRAS WT samples from COAD, lane 12—KRAS4B expression in KRAS-mutant samples from COAD. Sample numbers are shown at the bottom of the figure along with the P values from a t test performed between the sample groups. (C) The proportion of KRAS4A is also increased in KRAS-mutant tumors. Median group expression is indicated by the green/yellow border in each bar. The bars correspond to the data groups from Figure 1B. The additional lines represent the 25th and 75th percentiles, respectively, of the sample proportion of KRAS4A and KRAS4B. All 3 tissues show an increase in the proportion of KRAS4A in mutant tumors. The lung and colon sets were significantly different using the raw expression values, but only the lung was significant using the log2-transformed values. The numbers of samples in each group are the same as in Figure 1B. The P values are as follows: LUAD (339/152): raw: 2e−07, log2: 2e−10; PAAD (45/133): raw: .034, log2: .009; and COAD (105/59): raw: .019, log2: .097. COAD indicates colon adenocarcinoma; LUAD, lung adenocarcinoma; PAAD, pancreatic ductal adenocarcinoma; WT, wild type.
Close examination of the plots also shows that while the overall separation between KRAS-mutant and wild type (WT) tumors in expression is significant, the distributions do overlap. To delineate this further, we plotted the combined WT and mutant samples for LUAD ordered by KRAS expression and then colored the bars by their mutation status (Figure S1). As expected, there are KRAS WT samples showing higher expression than some KRAS-mutant samples and vice versa.
We also explored whether mutations in other frequently mutated genes tracked with KRAS expression levels. In the bars at the bottom of the plot, the positions of samples with mutations in TP53, epidermal growth factor receptor (EGFR), KEAP1, and NF1 are indicated. Of these, the EGFR mutations do seem to be enriched toward the left side of the panel, indicating lowered KRAS expression. Although the TP53 mutations are enriched in the non-KRAS–mutated samples (data not shown), that relationship is not evident on the plot. We did not see any enrichment of either KEAP1 or NF1 in terms of KRAS expression levels. The EGFR relationship is expected as KRAS mutations and EGFR mutations are mutually exclusive.15,16
KRAS4A and KRAS4B isoforms both show increased expression in the context of KRAS gene mutations
Because the biological significance of these differentially trafficked isoforms is the focus of intensive study, and it has been difficult to tease apart their separate function, we next looked at KRAS isoform expression in samples harboring KRAS mutations. Figure 1B shows the expression of KRAS4A and KRAS4B across the same sample subsets presented in Figure 1A. Both isoforms track with the increase in mutated samples; however, the overall magnitude of the KRAS4A increase is slightly larger in all 3 tissues (about 1 log2 unit compared with about 0.5 for KRAS4B). The differences were significant in both the lung and pancreas samples, but not in the colon samples, although the KRAS4A is borderline significant in that tissue (.055). To illustrate this point further, we plotted the proportions of the 2 isoforms using their median expression values in the WT and mutant samples (Figure 1C). In all 3 tumor types, there is an increase in the proportion of KRAS4A in the KRAS-mutant samples relative to that in the corresponding KRAS WT samples. The difference is significant in both the lung and colon samples (see figure legend for details). The biological significance of this is unclear, but it is consistent with a role for KRAS4A in tumorigenesis or maintenance of KRAS mutation–driven tumors as has been suggested in mouse studies.17,18
Median values of the KRAS4A and KRAS4B expression values for each of the same sample subsets were converted to the proportional values and plotted. The additional lines in the bars represent the 25th and 75th percentiles, respectively, from bottom to top.
Beyond the 3 tumor types profiled above, we also evaluated the impact of KRAS mutation on its expression in other tumor types with at least 5 KRAS-mutant samples (including any nonsynonymous mutations, Figure S2A and B). We also observe significantly increased expression in the mutated samples relative to WT KRAS counterparts in uterine carcinosarcoma, stomach adenocarcinoma, and skin cutaneous melanoma (SKCM). Kidney renal papillary cell carcinoma (KIRP), uterine corpus endometrial cancers (UCECs), rectal adenocarcinoma (READ), cervical squamous cell cancers, breast cancers (BRCA) and liver hepatocellular carcinomas also all show a trend toward increased expression in mutant samples with KIRP displaying near significance (Figure S2A and B). Testicular germ cell tumors (TGCT), bladder cancer (BLCA), and thyroid cancer (THCA) fail to show the trend and thus appear to be exceptions (Figure S2A and B).
Increased expression is associated with mutation of other RAS genes
KRAS is the most frequently mutated RAS gene in cancer. However, both HRAS and NRAS genes can have driver roles, although usually in different tumor types than those in which KRAS mutations are most frequent. Therefore, we wanted to determine whether the observed increase in KRAS expression associated with its mutation could be observed in HRAS or NRAS. As with KRAS, we assessed the potential expression shift for any tumor type where 5 or more HRAS-mutant or NRAS-mutant samples existed.
For HRAS, there are a total of 7 tumor types that meet these criteria—bladder (BLCA), head and neck squamous cell (HNSC), pheochromocytoma and paraganglioma (PCPG), lung squamous cell (LUSC), SKCM, thymoma (THYM), and THCA. Of these, we observed a statistically significant increase in HRAS in BLCA, HNSC, and PCPG tumors with HRAS mutations (Figure 2A). In SKCM, THYM, and THCA (Figure 2A), the trend was apparent, but the number of samples was too low for significance (values indicated on figure). As with the exceptions noted for KRAS, there was no evidence of the trend in LUSC (data not shown).
Figure 2.

(A) HRAS expression also associates with mutation status. Sample subsets for each of the tumor types were prepared based on the HRAS mutation status. HRAS expression values were then plotted for each sample group. Sample numbers and P values are indicated on the plot. Lane 1—HRAS expression in HRAS WT samples from BLCA, lane 2—HRAS expression for HRAS-mutant samples from BLCA, lane 3—HRAS expression in HRAS WT samples from HNSC, lane 4—HRAS expression for HRAS-mutant samples from HNSC, lane 5—HRAS expression in HRAS WT samples from PCPG, lane 6—HRAS expression in HRAS-mutant samples from PCPG, lane 7—HRAS expression in HRAS WT samples from SKCM, HRAS expression in HRAS-mutant samples from SKCM. Lane 9—HRAS expression for HRAS WT samples from THYM, lane 10—HRAS expression for HRAS-mutant samples from THYM, lane 11—HRAS expression for HRAS WT samples from THCA, lane 12—HRAS expression from HRAS-mutant samples from THCA. (B) NRAS expression in SKCM sample subsets. Sample subsets were prepared for the total tumor set, the primary tumor set, and the metastatic sample set for both the NRAS WT and NRAS-mutant sample groups. Sample numbers for each group and t test P values are shown on the plot. Lane 1—NRAS expression in total NRAS WT subset of SKCM, lane 2—NRAS expression in NRAS-mutant samples from SKCM, lane 3—NRAS expression in NRAS WT primary tumor samples from SKCM, lane 4—NRAS expression in NRAS-mutant primary tumor samples from SKCM, lane 5—NRAS expression in NRAS WT metastatic tumor samples (mets) from SKCM, lane 6—NRAS expression in NRAS-mutant metastatic tumor samples (mets) from SKCM. BLCA indicates bladder cancer; HNSC, head and neck squamous cell; PCPG, pheochromocytoma and paraganglioma; SKCM, skin cutaneous melanoma; THCA, thyroid cancer; THYM, thymoma; WT, wild type.
For NRAS, there are 8 tumor types that meet our selection criteria (5 or more mutated samples). Because of a historical issue, the data for COAD and READ are divided into 2 sequencing platforms for expression analysis (Illumina HiSeq [HS] and Illumina GA). They are as follows: SKCM, COAD (GA and HS), READ (HS and GA), acute myelogenous leukemia (LAML), TGCT, THCA, BLCA, and UCEC. Of these tumor types, SKCM (where NRAS has a presumed driver role) and READ (both in GA and HS) showed a significant increase in the NRAS-mutant sample subset relative to the WT subset (Figure S3A). Although not significant, the trend is also apparent in the COAD samples from both platforms. A weaker trend is present in the BLCA and UCEC tumor types (Figure S3B), but not in the TGCT, THCA (where NRAS mutation is considered a driver event) and LAML sample subsets (Figures S3A and B).
In the case of SKCM, the samples consist of both primary tumors and metastatic samples. Therefore, we can also examine the mutational effect against these different sample backgrounds. In both cases, the increase in NRAS expression in mutant samples relative to WT samples is observed, although it is only significant in the metastatic samples. Interestingly, the overall level of NRAS also appears to increase between the primary and metastatic samples, both in mutant and WT samples suggesting continued selective/adaptive forces influencing RAS gene expression levels.
The significant RAS gene expression differences between WT and mutated samples that we observed across the 3 RAS genes are summarized in Table S1. Across the 3 RAS genes, there were 34 possible settings where we evaluated the mutation-associated expression differences. Of these, we observed significant increases in 12 settings—7 for KRAS, 3 for HRAS, and 2 for NRAS (Table S1). Using a 1-tailed Fisher ’s exact test with 12 of 34 possible observations produces a P value of .003 suggesting that this observation would not be expected by chance. As this analysis involves multiple tests, a false discovery rate value was also computed for each of the observations. The complete set of results for all tumor types with 5 or more mutations in any of the RAS genes is shown in Table S2.
Mutation status of RAS genes can be associated with expression changes in other RAS genes
The recurrent association between RAS gene mutation status and increased expression (self-adaptation) and the highly redundant function of the RAS genes led us to evaluate the possibility that mutation in one type of RAS gene might be associated with expression changes in other RAS genes (non-self-adaptation). Therefore, we examined the tumor types outlined above to identify expression changes in other RAS genes against the backdrop of mutated RAS in each tumor type. There were a total of 68 tumor type settings in which we examined the association between mutation status and expression of other RAS genes. We observed a significant difference in 17 cases (6 cases with HRAS as the mutated gene, 4 with NRAS, and 7 with KRAS) suggesting that their adaptive influence extends beyond the mutated gene itself (Table 1). Of these settings, there were 7 effecting NRAS expression, 5 effecting KRAS expression, and 5 effecting HRAS expression. Using the same criteria as above for the Fisher’s exact statistical test, here, the P value is .0006 of this observation occurring by chance. As can be seen from Table 1, when the mutation of one RAS gene is significantly associated with a change in the expression of another WT RAS gene, the direction of the shift is downward (the WT RAS gene is lower) with the exceptions of NRAS expression in KRAS-mutant PAAD tumor samples and HRAS expression in NRAS-mutant UCEC samples which both increase. As with the “self ” mutation associations (Table S1), we suspect that tumor/cellular context plays a large role in determining whether these shifts are observed, their extent, and their direction (see Table S2 for complete data).
Table 1.
RAS mutations and other RAS genes—assessment of mutational impact on RAS gene expression.

CCLE cell lines with KRAS mutations also show an associated increase in KRAS expression
The tumor purity of the samples from TCGA has been estimated using copy number and other data19 and is typically in the range of 50% to 70%. Therefore, the expression data reflect the expression of both tumor and surrounding contaminating cells. We therefore analyzed expression data from cancer cell lines to verify our observations derived from TCGA data. We used the chip-based expression data from the CCLE Web site and cross-checked using the RNA-Seq data from caHUB (Cancer Human Biobank) for the same lines (data not shown). Figure 3 shows that KRAS expression is significantly higher in KRAS-mutant lung-derived and also intestine (colon)-derived cell lines using the per-gene aggregated affy data (see Figure 3 for P values and sample numbers for each sample group). The trend is also present in the pancreas-derived lines, but there are very few KRAS WT lines available and so this was borderline significant (.054; Figure 3). A more direct comparison of the relative extent of these changes between the TCGA and CCLE samples is not permitted as the data are derived from different platforms and using different methods.
Figure 3.

KRAS expression in KRAS-mutant CCLE cell lines. The Affymetrix expression data (gene level) were used and cell lines were divided by tissue source. Sample subsets were prepared from each of intestine (colon), lung, and pancreas tissues and separating KRAS-mutant and KRAS WT lines. Only protein-affecting mutations were included in the mutant lines. The numbers of lines and the t test P values are shown at the bottom of the plot. Lane 1—KRAS WT lung cell lines and KRAS-mutant lung lines, lane 3—KRAS WT intestine lines, lane 4—KRAS-mutant intestine lines, lane 5—KRAS WT pancreatic lines, and lane 6—KRAS-mutant pancreatic lines. CCLE indicates Cancer Cell Line Encyclopedia, WT, wild type.
The CCLE collection is more limited in terms of mutations in HRAS and NRAS, which made validation on the other genes more difficult. This limitation also made the identification of examples where the non–self associated expression change was observed (ie. mutation of one RAS gene was associated with an expression change in another RAS gene) more difficult. We did observe trends in the non–self associated expression changes, but the differences were not significant. However, we do take this overall to show that our observations using tumor samples are reproducible in tissue culture systems making it possible to experimentally perturb cells and evaluate cellular responses at the transcriptional level. Such experiments will allow further delineation of the underlying mechanisms responsible for our observations to be made.
KRAS expression and CNV
Given our observations showing frequent associations between RAS gene mutation status and expression, we wanted to pursue possible explanations for these effects.
We divided the LUAD samples into subsets based both on their KRAS mutational status and their KRAS copy number status. The results of this sample breakdown are shown in Figure 4. The first pair (all) shows the aggregate of all CNV states for either the WT or KRAS-mutant samples and shows the previously described expression increase in the mutant-harboring samples. The additional pairs represent 1-allele loss (Loss=1), neutral (N=2), 1-allele gain (Gain=1), and 2-allele gain (Gain=2) for the WT and mutant samples (there are not adequate 2 allele loss samples available). As expected, CNV affects KRAS expression levels: there is a gradient of increased expression across the increasing CNV states, but in each pair, the difference between the KRAS-mutant and KRAS WT samples remains significant except for the 2-allele gain state where we suspect that the combination of low sample number and higher variance confounds the data. The P values and the sample numbers are indicated at the base of the figure. This finding confirms the contribution of CNV to KRAS expression increases, but as the KRAS-mutant samples consistently express a higher KRAS levels across the CNV continuum, there must be additional contributing factors. This result also confirms that at least in LUAD, for KRAS, there is a good correlation between KRAS copy number and expression using the derived GISTIC values. We also observed the expected increases in KRAS expression in pancreas and colon samples as a function of copy number, but they were not significant (Table S3).
Figure 4.
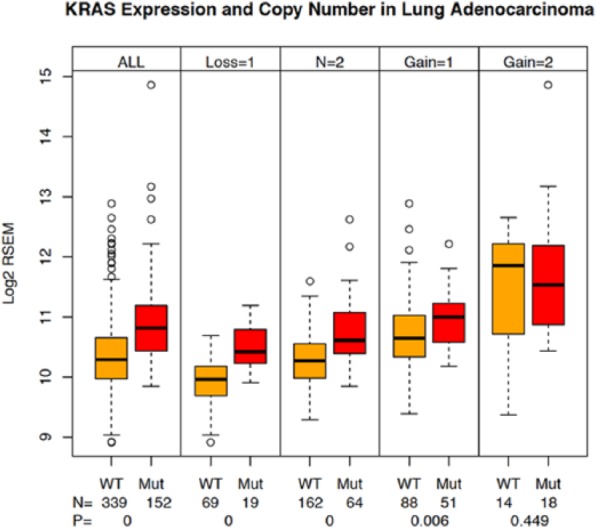
KRAS expression and CNV in lung adenocarcinoma samples. Sample subsets were prepared from the lung adenocarcinoma tumor samples to capture pairs (KRAS wild type and KRAS mutant) of samples in each of the 5 possible CNV states (−2, −1, 0, +1, +2) using the GISTIC scores.12 KRAS expression was plotted for each sample subset. The numbers of samples in each group and the associated t test P values are shown on the plot. Lane 1—all KRAS WT samples, lane 2—all KRAS-mutant samples, lane 3—KRAS WT samples with CNV = −1, lane 4—KRAS-mutant samples with CNV = −1, lane 5—KRAS WT samples with CNV = 0, lane 6—KRAS-mutant samples with CNV = 0, lane 7—KRAS WT samples with CNV = +1, lane 8—KRAS-mutant samples with CNV = +1, lane 9—KRAS WT samples with CNV = +2, lane 10—KRAS-mutant samples with CNV = +2. CNV indicates copy number variation; GISTIC, Genomic Identification of Significant Targets in Cancer; WT, wild type.
The CNV analysis can also be used to address the question of whether CNV gains (or losses) are more common in the KRAS-mutated samples than in the WT samples. For this, we produced a frequency display of each CNV state shown in Figure S4 for the lung, pancreas, and colon samples. We applied a χ2 test as well as visual inspection to evaluate the data. Of the 3 tumor types, both the lung and colon samples showed a significant enrichment for amplification in the mutant samples relative to their WT counterparts (.002 and 0, respectively). We did not see enrichment for deletion events in any of the tumor types between the mutant and WT samples. The finding that amplification is associated with mutation status in colon and lung samples, but not pancreas may simply result from low sample numbers in this analysis, or could be related to other pancreatic-specific tumor attributes (eg, context).
EGFR mutational status is also associated with KRAS expression
Mutation of KRAS and EGFR in LUAD samples is mutually exclusive.15,16 As a result, in the LUAD samples above (Figures 1, S1, and S2), the samples labeled as WT KRAS actually contain all of the EGFR-mutated samples. To probe the connection between background mutational status and KRAS expression further, we separated the EGFR-mutant samples from the double WT samples and again assessed KRAS expression. In Figure 5A, the expected elevated KRAS expression in the KRAS-mutant samples is shown (eg, compare lanes 2 and 4). However, we also see a trend toward diminished expression of KRAS in the EGFR-mutant samples, even relative to the double WT samples. Both the difference between KRAS-mutant samples and the WT/WT samples and between the WT/WT samples and the EGFR-mutant samples is significant. When we examine the expression in a uniform CNV neutral background, the EGFR-associated decrease can also be observed, although it loses significance (see Figure 5A). This observation suggests possible cross talk between KRAS and its immediate upstream pathway elements that through the adaptive process selects for decreased expression in a mutation selective manner. This observation extends the reported mutational exclusivity and crosstalk16 to include possible impacts of even WT co-expression of these genes.
Figure 5.

(A) KRAS-mutant and EGFR-mutant influences on KRAS expression in lung adenocarcinoma samples. Sample subsets were prepared by dividing samples by their mutation status (lanes 2–4) or by mutation status and CNV state = 0 (lanes 5–7) and KRAS expression was plotted. The numbers of samples in each group and the t test P values from the class comparisons are shown on the plot. Lane 1—pooled tumor samples (KRAS mutant and WT), lane 2—pooled KRAS-mutant samples, lane 3—pooled EGFR-mutant samples, lane 4—pooled EGFR + KRAS WT samples, lane 5—CNV = 0 KRAS-mutant samples, lane 6—CNV = 0 EGFR-mutant samples, lane 7—CNV = 0 KRAS/EGFR WT samples. (B) NRAS and BRAF mutations impact on both BRAF and NRAS expression in melanoma samples. Sample subsets were prepared using either BRAF mutation status or NRAS mutation status from all metastatic melanoma tumor samples (TCGA barcode = 06) and either NRAS expression was plotted (top panel) or BRAF expression was plotted (lower panel). The sample counts and t test P values are shown on the plot. Lane 1—BRAF WT samples, lane 2—BRAF-mutant samples, lane 3—NRAS WT samples, lane 4—NRAS-mutant samples. EGFR indicates epidermal growth factor receptor; TCGA, The Cancer Genome Atlas; WT, wild type.
To further explore this possible interaction, we then examined the expression of EGFR in the same sample subsets (Figure S5). As can be seen, the samples with mutated EGFR show elevated EGFR expression. However, in this case, the reciprocal decrease in KRAS-mutant samples was not observed as these show the same expression levels for EGFR as the double WT samples. In both EGFR-mutant and KRAS-mutant samples, it appears that more of an activated oncogene is selected for in a tumor and the possible cross talk extends this selective response to include additional pathway elements.
NRAS and BRAF mutual exclusivity in SKCM tumor samples
The LUAD, EGFR-mutant observation with KRAS suggests that the connections between mutational status and expression between genes within the RAS signaling node (KRAS, NRAS, and HRAS) extend to other RAS pathway genes. We evaluated whether this observation could be extended to other tumor types where mutational exclusivity has been demonstrated. For this, we applied a similar analysis to the BRAF-mutant and NRAS-mutant subsets of the metastatic melanoma tumor type. The mutual exclusivity of the NRAS and BRAF mutations applies uniformly to fully activating mutations; however, in a limited number of cases, there are overlaps (eg, (NRAS/BRAF): G12D/G466E, Q61R/P348T and HRAS: Q61K/G469R, P34L/N581S). As can be seen in Figure 5B, NRAS expression is higher in samples where it is mutated and also diminished when BRAF is mutated, consistent with the mutual exclusivity observations made with EGFR (Figure 5A). As in lung, a subset of the NRAS WT samples overlaps the BRAF-mutant samples and vice versa. In both cases, these differences were significant. We also looked at BRAF expression between these sample classes and those results are shown in Figure 5B (lower panel). Although the difference is only significant in the BRAF-mutant samples for expression of BRAF (see Figure 5B), the results remain consistent with cross talk between these proximal pathway nodes.
Together, these results suggest a complex interaction between different genes within the RAS pathway, whereby mutation in a gene can be associated with differences in its own expression as well as changes in the expression in other genes in the pathway, both within the same node as with the RAS genes, but also extending both upstream (EGFR) and downstream (BRAF) in the pathway. Importantly, there must also be an additional context component because we did not observe this same pattern for BRAF and NRAS in the THCA tumor type samples (data not shown).
Discussion
Mutation-associated expression changes
Using data from TCGA, we have confirmed and extended the early observation that KRAS gene expression in samples harboring KRAS mutations is elevated relative to their WT KRAS counterparts in many, but not all tumor types. Notably, the increase is significant in the principal tumor types where KRAS mutation is thought to have a driver role (LUAD, PAAD, and COAD) (Figure 1A). We extended this to show that the same principle applies to the 2 other RAS genes and that it also applies to the 2 major spliced isoforms of KRAS, KRAS4A, and KRAS4B (Figure 1B and C). We also demonstrated that the mutation-associated expression increase could be reproduced in cell lines (Figure 3), providing a convenient experimental paradigm for further experiments.
Although selection for increased expression of an oncogenic driver appears intuitively obvious, the precise mechanism is unclear. Increased signal output may be necessary to overcome feedback mechanisms, consistent with the possibility that the feedback itself can be overwhelmed. Alternatively, increased levels of oncogenic Ras proteins might engage effectors that are not engaged during normal signal transduction and thus provide a selective advantage through phenotypes that modulate malignant transformation. If so, this could offer novel therapeutic opportunities. Increased expression might also be necessary to overcome the suppressive effects of WT Ras proteins, which are known to interfere20 with oncogenic signaling, at least for KRAS and HRAS. The KRAS expression increase is also consistent with the reported rewiring of the cellular circuitry to effect metabolic changes to manage increased reactive oxygen species.21 It is also important to mention that although our results show an increased KRAS expression in the KRAS-mutant harboring samples, the analysis does not distinguish which allele is being adjusted. Thus, the WT allele could also be compensating in some way as well.
Factors influencing expression changes
We also showed evidence of RAS signaling cross talk, whereby mutation in one RAS gene is associated with changes in the expression of other RAS genes. This observation is pertinent to the role of the WT RAS isoforms when one becomes mutated because it is possible that through cross talk, adjustments in expression could influence overall signal output as well as the ratios of outputs from multiple RAS gene–specific signaling cascades. Previously, a dependence on HRAS and NRAS in KRAS-driven cell proliferation has been described20 consistent with the proposed interdependency.
Mechanistically, our analysis revealed good correlation between copy number and KRAS gene expression in LUAD samples (Figure 4). However, we showed that across different CNV states, the KRAS-mutated samples consistently expressed even higher levels of KRAS consistent with involvement of one or more additional contributing factors. Referring to Figure S3, we observed a significant increase in KRAS amplification in mutant samples in the COAD data set, but not in the LUAD and PAAD data sets, suggesting an additional contextual impact at play. Similarly, as there are KRAS WT samples that express higher KRAS levels than some of the KRAS-mutant lung samples (Figure S1), there are also influences stemming from the individual—presumably including germline single-nucleotide polymorphisms and somatic mutation burdens.
Cross talk between RAS pathway genes
Because we observed gene expression changes in many but not all tumor types with sufficient sample size (Figures 1A and 2A, B; Tables 1 and S1), we evaluated the possibility that the changes are tissue dependent. We used LUAD samples and showed that mutually exclusive mutations in EGFR and KRAS appear to be diametrically opposed. When EGFR is mutated, there is an increase in EGFR expression and a corresponding decrease in KRAS expression, and in KRAS-mutant samples, KRAS expression is elevated with no apparent effect on EGFR expression levels (Figure 5A). These observations are consistent with the previously reported analysis of the KRAS-EGFR mutual exclusivity.14,15 In this report, attempts to introduce mutant KRAS into EGFR-mutant expression cell lines and introduce mutated EGFR into mutant KRAS–expressing lines failed to produce lines co-expressing these 2 products. The authors interpreted this as a sensing mechanism that detected too much signal resulting in the elimination of the co-expressing cells from the population that was selected. Our observations are consistent with this result but suggest that even the balance between a mutated EGFR or KRAS protein and the WT counterparts requires adjustment at the expression level, consistent with the sensing mechanism, but extending it to include mutant-WT interactions.
This possibility was further investigated in the analysis of SKCM tumors where a comparable association was observed between the mutation status of NRAS and BRAF and their expression demonstrating the ability of both upstream and downstream pathway genes to participate in the cross talk process (Figure 5B). We did not resolve why the observed expression changes were not observed in all cases; there could be confounding mutations or alternatively expressed genes that can more or less substitute for the increase in RAS gene expression.
Our data support a model where RAS gene expression in tumors is shaped by a number of complex factors including context, mutant status, and mutant status of both upstream and downstream regulatory molecules. Ideally, going forward, the data collected from clinical trials that are increasingly guided by tumor genotype information will allow for deeper analysis of possible outcome-genomic connectivity and more conclusive determinations of the role of cellular context and adaptive responses to tumor evolution, as well as be used to guide therapeutic intervention to improve patient survival and outcome.
Acknowledgments
The authors would like to thank Brad Hollinger and Henri Tuthill for support configuring and loading the Oracle tables with TCGA data. Peter Johnson for helpful suggestions about the manuscript. They also thank Jim Hartley, Dan Soppett, Matthew Holderfield, and Tommy Turbyville for many discussions about the data analysis and figures.
Footnotes
PEER REVIEW: Five peer reviewers contributed to the peer review report. Reviewers’ reports totaled 1393 words, excluding any confidential comments to the academic editor.
FUNDING: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is supported by a contract with the National Cancer Institute HHSN261200800001E.
DECLARATION OF CONFLICTING INTERESTS: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
RS and MY performed the analysis of TCGA and CCLE data sets. RS wrote the first draft of the manuscript. MY and BK provided important suggestions for the revisions of the manuscript. FM and DN provided scientific guidance and conceived the hypothesis pursued in the analysis and also provided critical suggestions for the revision of the manuscript.
REFERENCES
- 1.Downward J. The ras superfamily of small GTP-binding proteins. Trends Biochem Sci. 1990;15:469–472. doi: 10.1016/0968-0004(90)90300-z. [DOI] [PubMed] [Google Scholar]
- 2.Barbacid M. ras oncogenes: their role in neoplasia. Eur J Clin Invest. 1990;20:225–235. doi: 10.1111/j.1365-2362.1990.tb01848.x. [DOI] [PubMed] [Google Scholar]
- 3.Tsai FD, Lopes MS, Zhou M, et al. K-Ras4A splice variant is widely expressed in cancer and uses a hybrid membrane-targeting motif. Proc Natl Acad Sci U S A. 2015;112:779–784. doi: 10.1073/pnas.1412811112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Castellano E, Santos E. Functional specificity of ras isoforms: so similar but so different. Genes Cancer. 2011;2:216–231. doi: 10.1177/1947601911408081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129:865–877. doi: 10.1016/j.cell.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 6.Trahey M, McCormick F. A cytoplasmic protein stimulates normal N-ras p21 GTPase, but does not affect oncogenic mutants. Science. 1987;238:542–545. doi: 10.1126/science.2821624. [DOI] [PubMed] [Google Scholar]
- 7.Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72:2457–2467. doi: 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ostrem JM, Shokat KM. Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat Rev Drug Discov. 2016;15:771–785. doi: 10.1038/nrd.2016.139. [DOI] [PubMed] [Google Scholar]
- 9.Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging Ras back in the ring. Cancer Cell. 2014;25:272–281. doi: 10.1016/j.ccr.2014.02.017. [DOI] [PubMed] [Google Scholar]
- 10.Adjei AA. K-ras as a target for lung cancer therapy. J Thorac Oncol. 2008;3:S160–S163. doi: 10.1097/JTO.0b013e318174dbf9. [DOI] [PubMed] [Google Scholar]
- 11.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov. 2014;13:828–851. doi: 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beroukhim R, Getz G, Nghiemphu L, et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci U S A. 2007;104:20007–20012. doi: 10.1073/pnas.0710052104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cancer Genome Atlas Research Network Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Unni AM, Lockwood WW, Zejnullahu K, Lee-Lin SQ, Varmus H. Evidence that synthetic lethality underlies the mutual exclusivity of oncogenic KRAS and EGFR mutations in lung adenocarcinoma. Elife. 2015;4:e06907. doi: 10.7554/eLife.06907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.To MD, Rosario RD, Westcott PM, Banta KL, Balmain A. Interactions between wild-type and mutant Ras genes in lung and skin carcinogenesis. Oncogene. 2013;32:4028–4033. doi: 10.1038/onc.2012.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Westcott PM, Halliwill KD, To MD, et al. The mutational landscapes of genetic and chemical models of Kras-driven lung cancer. Nature. 2015;517:489–492. doi: 10.1038/nature13898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carter SL, Cibulskis K, Helman E, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grabocka E, Pylayeva-Gupta Y, Jones MJ, et al. Wild-type H- and N-Ras promote mutant K-Ras-driven tumorigenesis by modulating the DNA damage response. Cancer Cell. 2014;25:243–256. doi: 10.1016/j.ccr.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kerr EM, Gaude E, Turrell FK, et al. Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature. 2016;531:110–113. doi: 10.1038/nature16967. [DOI] [PMC free article] [PubMed] [Google Scholar]