

Summary
Background
Congenital pulmonary airway malformation (CPAM) is a relatively rare congenital anomaly with a wide spectrum of ultrasound features depending on the specific variety of CPAM. Antenatal ultrasound is a valuable, safe, nonionizing, cost-effective, widely available and easily reproducible imaging tool and is indispensable in the diagnosis of CPAM. In this paper, we aimed to report an atypical imaging presentation of CPAM type II in the second trimester, extensively involving all lobes of the left lung.
Case Report
A 25-year-old G1P0A0 woman with a gestational age of around 22 weeks was referred for an anomaly scan. The antenatal ultrasound scan showed a single, live, intrauterine foetus corresponding to a gestational age of around 22 weeks and 4 days. There were multiple, anechoic structures noted within the pulmonary tissue in the left hemithorax, each measuring around 3 to 4 mm in diameter. The lesion was extending from the left lower lobe up to the apical (apicoposterior) segment of the left upper lobe. The ultrasound diagnosis of congenital pulmonary airway malformation type II was made. After explaining the condition and the poor prognosis to the patient, an informed consent was obtained after she opted for medical termination of pregnancy.
Conclusions
Congenital pulmonary airway malformation (CPAM) is an uncommon foetal anomaly with a very wide range of ultrasound appearances depending on the specific type of CPAM. CPAM also has a wide spectrum of differential diagnoses and a variable prognosis. Antenatal ultrasound should always be the primary mode of diagnosis in CPAM.
MeSH Keywords: Congenital Abnormalities; Cystic Adenomatoid Malformation of Lung, Congenital; Ultrasonography
Background
Congenital pulmonary airway malformation (CPAM) is a relatively rare congenital anomaly with a wide spectrum of ultrasound features depending on the specific variety of CPAM. Antenatal ultrasound is a valuable, safe, nonionizing, cost-effective, widely available and easily reproducible imaging tool and is indispensable in the diagnosis of CPAM. Despite the rarity of CPAM, it is imperative for a radiologist to be aware of its wide spectrum of presentation on antenatal ultrasound. In this paper, we aimed to report an atypical imaging presentation of CPAM type II in the second trimester, extensively involving all lobes of the left lung. We also discuss the role of ultrasound and other imaging modalities in CPAM as well as the differential diagnoses which can mimic CPAM.
Case Report
A 25-year-old, G1P0A0, (Gravida 1, para 0, abortion 0) woman with a gestational age of around 22 weeks was referred to our Department of Radiodiagnosis for an anomaly scan. She had been in a non-consanguineous marriage for 4 years and 3 months. The patient and her husband had no present or past history of any significant medical illness. The patient denied exposure to any form of teratogenic substances such as ionizing radiation, medications, alcohol, tobacco, cocaine, lead, lithium, phenytoin, warfarin or valproic acid during present gestation. This pregnancy was a spontaneous conception. Clinically, the course of her first trimester was uneventful. General physical examination revealed a regular pulse of 84 beats per minute and blood pressure of 120/90 mm Hg. Haemoglobin was 12.9 gm%. She was negative for HIV and HBsAg serology.
B-mode, real-time transabdominal sonography and Doppler evaluation of the foetus was performed on GE VOLUSON 730 PRO machine (GE healthcare, Milwaukee, USA) equipped with a 5 MHz curvilinear array, transabdominal transducer. All sonograms obtained were saved in a picture archiving and communication system.
The antenatal ultrasound scan showed a single, live, intrauterine foetus corresponding to a gestational age of around 22 weeks and 4 days (Biparietal diameter=5.5 cm; Head circumference=19.8 cm; Abdominal circumference=18.0 cm; Femoral length=3.8 cm). The foetal heart rate was 158 beats per minute. There was no evidence of congenital cardiac anomalies. However, the cardia appeared to be mildly pushed to the right side. There was evidence of multiple, anechoic structures noted within the pulmonary tissue in the left hemithorax (Figures 1, 2). These cystic structures, each measuring around 3 to 4 mm in diameter, were communicating with each other. There was no communication of these structures with the proximal pulmonary airways. There was no single dominant cyst. The conglomerate size of this multicystic lesion was around 40×25×14 mm (craniocaudal x anteroposterior x transverse dimensions). This lesion was not restricted to a single segment or lobe as it could be visualized extending from the left lower lobe up to the apical (apicoposterior) segment of the left upper lobe (Figure 3). There was no evidence of any echogenic or hyperechoic structures within these cysts. The adjacent pulmonary tissue did not show sonological signs of sequestration. However, the volume of the left pulmonary tissue was significantly decreased due to the extensive compression by this multicystic lesion. The foetal thoracic circumference (12.9 cm) was reduced. The CPAM mass-thorax ratio was found to be 0.37, and the CPAM volume ratio (volume of the mass divided by head circumference) was found to be 1.42. On colour Doppler examination, there was no internal or peripheral vascularity of these lesions. There was no obvious ultrasound evidence of a systemic arterial supply to this lesion. The stomach bubble and the liver including the branches of the portal vein were normal in location (Figure 4). The diaphragm was intact and continuous along its complete extent. There was no inversion of the left hemidiaphragmatic contour due to the multicystic mass lesion. The remaining foetal parts including both kidneys were normal. There was mild polyhydramnios indicating early sequelae related to mediastinal herniation. There was, however, no evidence of hydrops fetalis. The foetal right lung appeared to be unremarkable on ultrasound. The ultrasound diagnosis of congenital pulmonary airway malformation type II (small cyst CPAM) was made.
Figure 1.
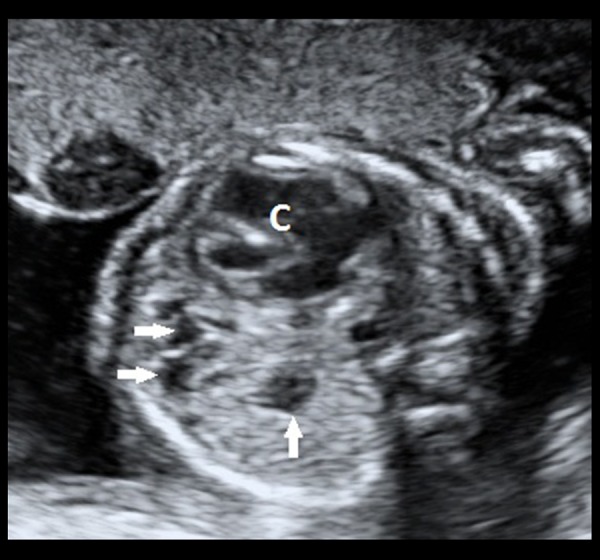
Grey-scale transabdominal ultrasound image of the foetus in a transverse section showing multiple, anechoic structures (white, filled arrows) within the lower lobe of the left lung. The cardia (denoted by ‘C’) appears to be mildly pushed to the right side.
Figure 2.
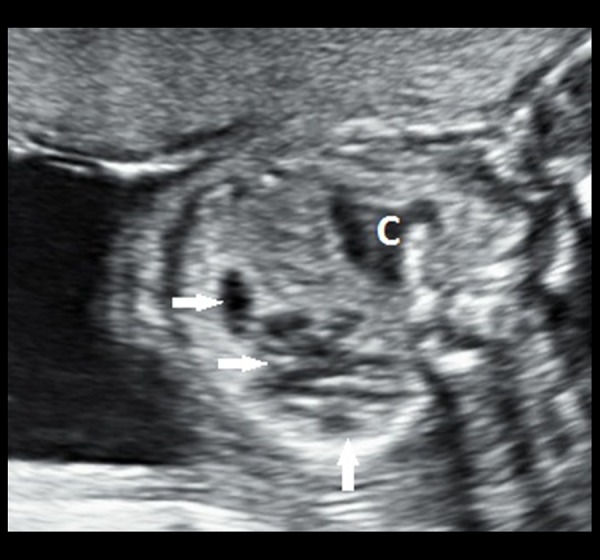
Grey-scale transabdominal ultrasound image of the foetus in a transverse section showing multiple, anechoic structures (white, filled arrows) within the upper lobe of the left lung. These cystic structures, each measuring around 3 to 4 mm in diameter, are communicating with each other. There is no single dominant cyst. The cardia (denoted by ‘C’) appears to be mildly pushed to the right side.
Figure 3.
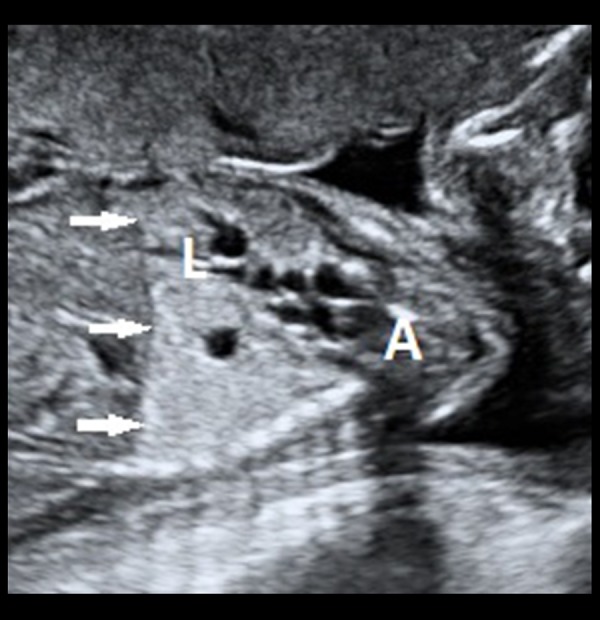
Grey-scale transabdominal ultrasound image of the foetus in a sagittal section showing the multicystic lesion extending from the left lower lobe (denoted by ‘L’) up to the apical (apicoposterior) segment (denoted by ‘A’) of the left upper lobe. The diaphragm (white, filled arrows) is continuous along its complete extent. There is no inversion of the left hemidiaphragmatic contour due to the multicystic mass lesion.
Figure 4.
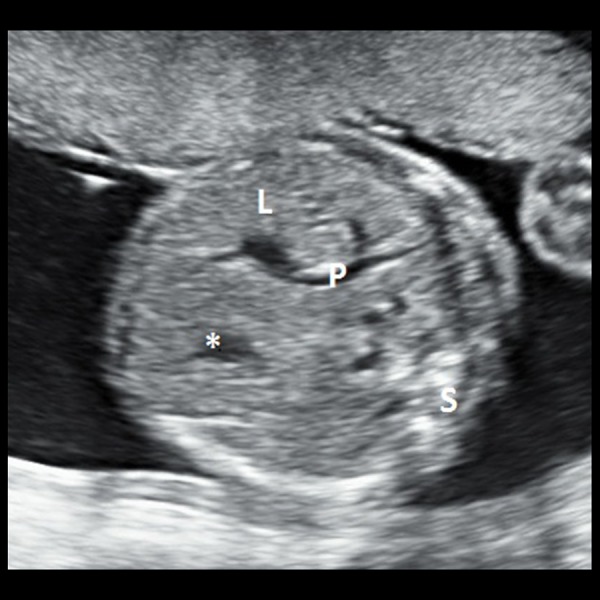
Grey-scale transabdominal ultrasound image of the foetus in a transverse section showing normal position of the gastric bubble (asterix) and the liver (denoted by ‘L) including the branches of the portal vein (denoted by ‘P’). Foetal spine is denoted by ‘S’ in the image.
After explaining the condition and the poor prognosis to the patient, in view of pulmonary hypoplasia, extensive involvement of the left lung and the mediastinal herniation, an informed consent was obtained after she opted for medical termination of pregnancy. The autopsy confirmed all the above-mentioned ultrasound features.
Discussion
Congenital pulmonary airway malformation (CPAM) is a relatively rare congenital anomaly with an incidence of around 1 per 25,000 to 35,000 live births. It was first described by Ch’In and Tang in 1949 [1]. It is a hamartomatous, dysplastic developmental abnormality of the lung characterized by abnormal airway patterning during lung branching morphogenesis and is formed by abnormal branching of the immature bronchioles [2,3]. CPAM occurs sporadically with a slight male predilection. CPAM was earlier referred to as CCAM (Congenital cystic adenomatoid malformation). However, the term ‘CCAM’ has become obsolete in the current context as these lesions are not always cystic in 2 (types 0 and III) out of 5 types and also are adenomatoid in only one (type III) out of 5 types [4].
Histopathologically, CPAM is classified into 5 types depending on the stage of developmental arrest of the tracheobronchial tree. Type 0 lesions (<2% of CPAM cases) have a tracheal or bronchial origin, type 1 lesions (60–70% of CPAM cases) have a bronchial or bronchiolar origin, type 2 lesions (15–20% of CPAM cases) have a bronchiolar origin, type 3 lesions (5–10% of CPAM cases) have a bronchiolar–alveolar duct origin and type 4 lesions (around 10% of CPAM cases) have a distal acinar origin [4,5].
In 1977, Stocker et al. proposed a classification of CPAM based on clinical, gross and microscopic criteria. According to this classification, there were three types of CPAM. CPAM type I lesions comprised of single or multiple cysts, measuring more than 2.0 cm in diameter and lined by ciliated pseudostratified columnar epithelium. CPAM type II lesions comprised of multiple small cysts, measuring less than 1.0 cm in diameter and lined by ciliated cuboidal to columnar epithelium. CPAM type III lesions comprised of a single, large, bulky, solid lesion lined by ciliated cuboidal epithelium and separated by areas of alveolus-sized structures lined by non-ciliated cuboidal epithelium. In 1985, Adzick et al. studied the natural history and pathophysiology of CPAM detected prenatally by ultrasound. Based on gross anatomy, ultrasound findings and prognosis, they classified CPAM into microcystic lesions, which were measuring less than 5 mm and were associated with foetal hydrops and had a poor prognosis, and macrocystic lesions which were measuring more than 5 mm and were not usually associated with hydrops and had a favourable prognosis. In 1999, Morotti et al. evaluated the cellular composition of different CPAM types by immunohistochemistry and divided CPAM into two major subtypes – subtype one consisting of CPAM types 1, 2 and 3 with a bronchiolar type of epithelium, and subtype two consisting of CPAM type 4 with an acinar-alveolar type of epithelium. In 2001, Bush proposed a classification based on histologic differences and named cystic, intermediate and solid congenital thoracic malformations. In 2003, Langston classified cystic lung lesions based on pathologic features due to airway obstruction during development into a large cyst-type lesions and a small cyst-type lesions [1].
The aetiopathogenesis of CPAM is still not completely understood, but molecular studies have shown that bronchial atresia plays an important role in its pathogenesis [6]. It is hypothesized that defects in the HOXB5 gene partially mediated by glial cell-derived neurotrophic factor (GDNF) are implicated in the pathogenesis of few cases of CPAM [7]. Abnormalities involving the thyroid transcription factor 1 (TTF1), fibroblast growth factor-7 (FGF 7), fibroblast growth factor 9 (FGF 9), fibroblast growth factor 10 (FGF 10), fatty acid-binding protein 7 (FABP-7), Clara cell marker 10, SOX2 transcription factor and altered integrin cytoplasmic signalling have all been proposed as possible causative factors in the aetiopathogenesis of CPAM [1].
The natural course of CPAM varies from complete regression in utero to life-threatening hydrops fetalis. CPAM lesions are usually unilateral and involve a single lobe. Although there is no well-documented study regarding the lobar predilection, they appear to involve the middle lobe less frequently. Numerous co-morbidities associated with CPAM include pulmonary sequestration (hybrid lesion), renal agenesis, polyhydramnios, hydrops fetalis, bronchoalveolar carcinoma (with CPAM type I), pleuropulmonary blastoma (with CPAM type IV), mucoepidermoid carcinoma (with CPAM type I), tracheoesophageal fistula, agenesis of the corpus callosum, congenital cardiac defects including atrial septal defect (ASD), ventricular septal defect (VSD), truncus arteriosus, tetralogy of Fallot (TOF) and patent ductus arteriosus (PDA) [8–11]. A recent study has also shown CPAM type II to be associated with congenital nephrotic syndrome (diffuse mesangial sclerosis), cleft lip and cleft palate [12].
The case discussed in our report was CPAM type II as the cysts within the pulmonary tissue in the left hemithorax were each measuring around 3 to 4 mm in diameter. It was an atypical presentation, because contrary to the typical involvement of a single lobe in CPAM, there was extensive involvement of the whole left lung extending from the left lower lobe up to the apical (apicoposterior) segment of the left upper lobe.
Ultrasound is a valuable, safe, nonionizing, cost-effective, widely available and easily reproducible imaging tool and is indispensable in the diagnosis of CPAM [13]. CPAM appears as an isolated cystic or solid intrathoracic mass lesion. In type 0 CPAM, the cysts are usually very small and arise from the trachea or the bronchus. In type I CPAM, there might be visualization of a single or multiple cysts ranging from 2.0 cm to 10.0 cm in diameter with the presence of a dominant cyst. Sequelae related to significant mass effect are seen in these cases due to the large size of lesions. In type II CPAM, there are multiple cysts ranging from 0.5 to 2.0 cm in size and around 60% of CPAM II cases have associated anomalies. Type III CPAM lesions appear highly echogenic due to the very small size of these cysts. Type IV CPAM lesions are seen as large cysts, mostly unilocular and measuring around 10 cm in diameter. These cysts are indistinguishable from unilocular type I cysts on imaging. Therefore, the macrocystic variety of CPAM is seen in CPAM types I, II and IV. CPAM type III lesions are typically solid (microcystic CPAM). CPAM I is a large-cell CPAM and CPAM II is a small-cell CPAM variety. Antenatal ultrasound helps in throwing light on the anatomical location of the lesion, its relationship with the bronchial tree, number and size of the cysts and any coexisting pulmonary lesions such as pulmonary sequestration in case of a hybrid lesion. Colour Doppler helps in identifying the presence or absence of systemic arterial blood supply, aiding in prenatal differentiation of CPAM from bronchopulmonary sequestration (which shows a feeding systemic artery). Hydrops fetalis is seen in cases of large-sized CPAMs resulting in mediastinal shift and vena cava obstruction. There are three antenatal features that have been found to be highly specific for hydrops fetalis: CPAM mass-to-thorax ratio of 0.56, cystic predominance and diaphragmatic inversion [1]. Polyhydramnios is seen in cases of large CPAMs causing oesophageal compression. CPAM volume ratio (CVR) greater than 1.6 also helps in predicting an increased risk of hydrops fetalis. CVR is the ratio of CPAM volume to the foetal head circumference. Around 40% of CPAMs are known to increase as the gestational age advances until around 26 weeks of gestation, after which the growth plateaus. Microcystic CPAMs generally tend to regress while the macrocystic CPAMs do not regress [1].
Magnetic resonance imaging (MRI) helps in a better identification of the foetal anomalies. Unlike ultrasound, it is not operator dependent. Also, it can be very helpful in cases where there is severe oligohydramnios or inaccessible foetal position preventing adequate visualization of the foetal parts on ultrasonography. Foetal CPAMs manifest as unilocular or multilocular lesions which appear hypointense on T1WI and hyperintense on T2WI sequences. Chest radiography in the postnatal period shows CPAM as a soft tissue density lesion. There might be a mediastinal shift in cases of large CPAMs. Air fluid levels may be seen as there is a delay in the clearance of the fluid from the cysts due to abnormal airways. 3D CT reconstructions also aid in the postnatal evaluation of CPAMs by giving important structural information, measurements and visualization of critical anatomic details. In comparison to MRI, CT scan has a faster acquisition time, thus reducing the need for sedation and is less expensive. CT angiography can be useful in the evaluation of any systemic feeding arterial supply, which can be missed on ultrasound scans. However, CT scan exposes the child to ionizing radiation. Both MRI and CT are helpful in the appropriate planning of surgery.
The prognosis of CPAM depends mainly on the presence or absence of foetal hydrops. In CPAM cases without hydrops, a survival rate of more than 95% has been reported, while a survival rate of less than just 5% has been reported in CPAM cases with hydrops which have been managed expectantly [1]. Prognosis also depends on the size of the cysts, with larger cysts having a poorer prognosis. The prenatal management of CPAM mainly revolves around maternal steroid therapy (betamethasone, intramuscular) for the maturation of the foetal lungs, thoraco-amniotic shunting in severe cases and EXIT procedure (Ex utero intrapartum procedure) which allows controlled and monitored resection of large foetal lung lesions during delivery. Postnatally, in asymptomatic cases of CPAM, open lobectomy is the best option in case of unilobar involvement, while parenchyma saving resection is performed in case of bilobar or bilateral pulmonary involvement. In symptomatic cases of CPAM, open surgery after stabilization is the usual line of management [1].
The potential postnatal complications of CPAM include spontaneous pneumothorax, haemopneumothorax and pyopneumothorax. There is also an increased likelihood of several malignancies in CPAM such as bronchoalveolar carcinoma, bronchogenic carcinoma, pleuropulmonary blastoma and rhabdomyosarcoma.
The ultrasound differential diagnosis of CPAM includes bronchogenic cyst, pulmonary sequestration, congenital diaphragmatic hernia (CDH), congenital lobar emphysema, congenital cystic bronchiectasis and mediastinal masses such as cystic hygroma and teratoma. Macrocystic CPAM should be differentiated from a bronchogenic cyst which is usually isolated and originates from the upper airway with which a direct communication can be visualized in some cases. CPAM type III should be differentiated from pulmonary sequestration which appears as an echogenic portion of lung that is supplied by a feeding vessel from the aorta. CDH can be easily differentiated from a CPAM as the echo-free stomach and the small bowel will be visualized at the same transverse level as the heart on the four chamber view in case of left-sided CDH. Also, the stomach and the small bowel will be visualized at a level superior to the inferior margin of the scapula. There might be leftward displacement of the gallbladder. In case of a right-sided CDH, there will be leftward bowing of the umbilical segment of the portal vein, portal branches to the lateral segment of the left hepatic lobe coursing towards or above the diaphragm, gallbladder present above the diaphragm and an echodense space between the left heart border and the stomach representing the left hepatic lobe. CPAM type III should be differentiated from congenital lobar emphysema, which also will appear echogenic due to an excessive accumulation of fluid within the pulmonary alveoli. Similar to CPAM type III, there will not be any vascular supply from the aorta and there will be a mass effect on the mediastinal structures. However, CPAM type III is uniformly echogenic, while congenital lobar emphysema can present as a bright echogenic region of the lung with or without cystic areas. Congenital cystic bronchiectasis, also known as Williams-Cambell syndrome, is usually symmetric and bilateral. Teratomas are usually more vascular, heterogeneous and cast a distal acoustic shadowing as compared to CPAMs. Mediastinal cystic hygromas can be easily differentiated from CPAMs, because they are generally seen extending from the neck into the anterior mediastinum.
Conclusions
Congenital pulmonary airway malformation (CPAM) is an uncommon foetal anomaly with a very wide range of ultrasound appearances depending on the specific type of CPAM. CPAMs have a wide spectrum of differential diagnoses and a variable prognosis. Ultrasound is a valuable, safe, nonionizing, cost-effective, widely available and easily reproducible imaging tool for the diagnosis of CPAM. Antenatal ultrasound should always be the primary mode of diagnosis in CPAM. Although magnetic resonance imaging (MRI) can help in a better delineation of foetal anomalies, ultrasound helps in an easy and reproducible follow-up of patients with CPAM in addition to disclosing details regarding the anatomical location, number and size of the cysts.
References
- 1.David M, Lamas-Pinheiro R, Henriques-Coelho T. Prenatal and postnatal management of congenital pulmonary airway malformation. Neonatology. 2016;110(2):101–15. doi: 10.1159/000440894. [DOI] [PubMed] [Google Scholar]
- 2.Taştekın E, Usta U, Kaynar A, et al. Congenital pulmonary airway malformation type 2: A case report with review of the literature. Turkish Journal of Pathology. 2016;32(3):200–4. [Google Scholar]
- 3.Hellmund A, Berg C, Geipel A, et al. Prenatal diagnosis and evaluation of sonographic predictors for intervention and adverse outcome in congenital pulmonary airway malformation. PloS One. 2016;11(3):e0150474. doi: 10.1371/journal.pone.0150474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biyyam DR, Chapman T, Ferguson MR, et al. Congenital lung abnormalities: Embryologic features, prenatal diagnosis, and postnatal radiologic-pathologic correlation 1. Radiographics. 2010;30(6):1721–38. doi: 10.1148/rg.306105508. [DOI] [PubMed] [Google Scholar]
- 5.Tetsumoto S, Kijima T, Morii E, et al. Echinoderm microtubule-associated protein-like 4 (EML4)-anaplastic lymphoma kinase (ALK) rearrangement in congenital pulmonary airway malformation. Clin Lung Cancer. 2013;14(4):457–60. doi: 10.1016/j.cllc.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 6.Wu, Zhi J, et al. Congenital pulmonary airway malformation. Clinical Pulmonary Medicine. 2016;23(5):227–30. [Google Scholar]
- 7.Soriano V, Chia D, Aronin SI. Congenital pulmonary airway malformation (CPAM) with malignant transformation. D110. Unusual tumors of the chest. American Thoracic Society. 2016:A7752. [Google Scholar]
- 8.Harini N, Chakravarthy R, Archana L. Congenital pulmonary airway malformation with mucoepidermoid carcinoma: A case report and review of literature. Indian J Pathol Microbiol. 2012;55(4):540–42. doi: 10.4103/0377-4929.107807. [DOI] [PubMed] [Google Scholar]
- 9.Pizzi M, Fassan M, Ludwig K, et al. Congenital pulmonary airway malformation (CPAM)[congenital cystic adenomatoid malformation] associated with tracheoesophageal fistula and agensesis of the corpus callosum. Fetal Pediatr Pathol. 2012;31(3):169–75. doi: 10.3109/15513815.2012.659392. [DOI] [PubMed] [Google Scholar]
- 10.Chiluveru SA, Dave NM, Dias RJ, Garasia MB. Congenital pulmonary airway malformation with atrial septal defect and pulmonary hypertension for lobectomy-anesthetic considerations. Ann Card Anaesth. 2016;19(2):372–74. doi: 10.4103/0971-9784.179624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sfakianaki AK, Copel JA. Congenital cystic lesions of the lung: congenital cystic adenomatoid malformation and bronchopulmonary sequestration. Rev Obstet Gynecol. 2012;5(2):85–93. [PMC free article] [PubMed] [Google Scholar]
- 12.Millington KA, Mani H. Type 2 congenital pulmonary airway malformation and congenital nephrotic syndrome: Report of a new association. Pediatr Dev Pathol. 2013;16(3):210–13. doi: 10.2350/12-07-1226-CR.1. [DOI] [PubMed] [Google Scholar]
- 13.Srinivas MN, Amogh VN, Gautam MS, et al. A prospective study to evaluate the reliability of thyroid imaging reporting and data system in differentiation between benign and malignant thyroid lesions. J Clin Imaging Sci. 2016;6:5. doi: 10.4103/2156-7514.177551. [DOI] [PMC free article] [PubMed] [Google Scholar]