

Summary
Hematopoietic stem cells (HSCs) are mobilized from niches in the bone marrow (BM) to the blood circulation by the cytokine granulocyte-colony stimulating factor (G-CSF) through complex mechanisms. Among these, signals from the sympathetic nervous system regulate HSC egress via its niche, but how the brain communicates with BM remains largely unknown. Here, we show that muscarinic receptor type-1 (Chrm1) signaling in the hypothalamus promotes G-CSF-elicited HSC mobilization via hormonal priming of the HPA axis. Blockade of Chrm1 in the CNS, but not the periphery, reduces HSC mobilization. Mobilization is impaired in Chrm1−/− mice, and rescued by parabiosis with wild-type mice, suggesting a relay by a blood borne factor. We have identified the glucocorticoid (GC) hormones as critical for optimal mobilization. Physiological levels of corticosterone promote HSC migration via the GC receptor Nr3c1-dependent signaling and upregulation of actin-organizing molecules. These results uncover long-range regulation of HSC migration emerging from the brain.
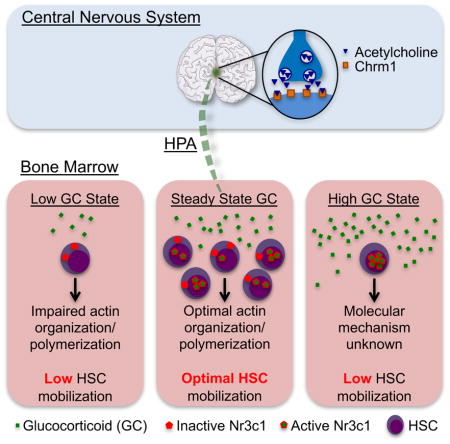
Graphical Abstract
Introduction
Hematopoietic stem cells (HSCs) are multipotent self-renewing units mediating the generation of all blood constituents after transplantation. HSCs can home to the bone marrow (BM) following intravenous injection through the coordinated actions of adhesion receptors and the chemokine CXCL12 expressed within in the BM microenvironment (Magnon and Frenette, 2008). Mobilized, HSC-enriched, mononuclear cells have emerged as the most common means to obtain HSCs for transplantation, but the procedure remains ineffective in a subset of patients (Levesque and Winkler, 2008; To et al., 2011).
Granulocyte colony-stimulating factor (G-CSF), the hematopoietic cytokine most commonly used to mobilize progenitors, can elicit circulating HSCs in the bloodstream over a period of ~5 days (Bensinger et al., 1993; Duhrsen et al., 1988; Lane et al., 1995). The delayed response suggests that the mechanisms likely involve the orchestration of a complex cascade of events, which culminate at least in part in the reduced expression of CXCL12 that retains HSCs in the BM (Petit et al., 2002). At the top of the cascade is the G-CSF receptor (Csf3r) whose expression by a transplantable hematopoietic cell, distinct from HSCs, is required (Liu et al., 2000). Further studies indicate that selective expression of Csf3r in mononuclear phagocytes can rescue the defective mobilization of Csf3r−/− mice (Christopher et al., 2011), implicating these cells as important intermediary cells. Among mononuclear phagocytes, macrophages have been found to promote HSC retention (Chow et al., 2011). However, robust G-CSF-elicited HSC mobilization occurs upon macrophage depletion (Chow et al., 2011), suggesting the participation of other pathways.
Neural signals from the sympathetic nervous system (SNS) have also been shown to promote G-CSF-induced HSC egress through β-adrenoreceptor–mediated regulation of CXCL12 (Katayama et al., 2006). Since intracerebral administration of G-CSF did not elicit HSC mobilization from the BM (Katayama et al., 2006), its effects on the SNS tone are thought to be mediated via the peripheral nervous system. Indeed, norepinephrine reuptake in superior cervical sympathetic ganglion neurons is modulated by G-CSF exposure (Lucas et al., 2012). In addition, neurotoxic chemotherapy or selective sympathectomy impairs HSC mobilization, suggesting that lesioned peripheral nerves may contribute to the poor stem cell yield of cancer patients previously treated with cytotoxic therapies (Lucas et al., 2013). Under steady state, physiological release of HSCs follows circadian oscillations of CXCL12 expression mediated by adrenergic innervation of Nestin+ mesenchymal stem cell niche (Mendez-Ferrer et al., 2008; Mendez-Ferrer et al., 2010). Homing of HSC and progenitors to the BM and expression of endothelial adhesion molecules are also under similar, albeit inverted, SNS-mediated circadian influence (Scheiermann et al., 2012). Additional studies have suggested that circadian oscillation of glucocorticoids (GCs) may regulates CXCL12 circadian expression and hematopoietic Nestin+ progenitor expansion via Notch1 signaling (Kollet et al., 2013). However, whether the central nervous system (CNS) can influence HSC migration in the BM via a humoral circuit, has not been demonstrated.
Understanding how cancer stem cells migrate may provide insight into mechanisms that regulate HSC mobilization. In this regard, we have previously tested the idea that nerves contributed to the cancer microenvironment by promoting cancer cell migration, and found that prostate cancer was infiltrated by adrenergic and cholinergic fibers which enhanced cancer progression in distinct ways (Magnon et al., 2013). Adrenergic fibers played a prominent role in cancer initiation via stromal β2 and β3 adrenergic receptors, whereas cholinergic fibers acting on type-1 muscarinic receptor (Chrm1) of the stroma, promoted cancer cell migration, lymph node invasion and metastasis (Magnon et al., 2013). Based on these observations, we investigated the possibility that Chrm1 signaling in the BM microenvironment might also influence the G-CSF-induced HSC migration. Here, we report that Chrm1 is required for efficient HSC mobilization but, unexpectedly, it acts on the CNS, rather than peripheral cholinergic nerves, to modulate HSC mobilization from the BM via the HPA axis, which primes HSC migration through the GC receptor Nr3c1.
Results
Cholinergic signals are required for robust HSC mobilization
To evaluate the role of muscarinic receptors in HSC mobilization, we treated mice with the pan-muscarinic receptor antagonist Scopolamine hydrobromide. Wild-type mice subjected to Scopolamine treatment exhibited significant reductions in G-CSF-induced phenotypic HSC mobilization, compared to vehicle-treated mice (Figure 1A). We next induced HSC mobilization with G-CSF in mice deficient in Chrm1 (Chrm1−/−; Figure 1B) which also revealed a significant and reproducible reduction of mobilized phenotypic HSCs (Figure 1C) and colony-forming progenitors (Figure 1D), compared to wild-type controls. However, we found no hematopoietic deficits in circulating blood counts (Supplemental Figure 1A) or progenitor numbers in the BM (Supplemental Figure 1B and Figure 1E) at steady state. In addition, we evaluated HSC relationships with arterioles by tridimensional BM imaging (Kunisaki et al., 2013), and found no alteration in HSC distribution in the BM (Supplemental Figure 1C). Thus, these results suggest that HSC numbers and distribution in the BM are not altered, indicating the possibility of specific defect of HSC migration in Chrm1−/− mice.
Figure 1. G-CSF-induced HSC mobilization requires signals from the muscarinic receptor type-1 (Chrm1).

(A) HSCs/mL following G-CSF mobilization in wild-type mice treated with saline or Scopolamine hydrobromide (Scop 1mg/kg; n = 5–8). HSCs defined as Lineage− Sca1+ cKit+ Flt3−.
(B) Schematic of G-CSF-induced mobilization and analyses. Wild-type mice are denoted by black bars and Chrm1−/− mice by white bars.
(C) HSCs/ml of peripheral blood (Lineage− Sca1+ cKit+ Flt3−; n = 12–16)
(D) Colony-forming units/ml of peripheral blood determined in vitro (CFU-C; n = 6–13).
(E) Lineage− Sca1+ cKit+ (LSK) cells/femur at steady state (n = 9).
(F) Reconstitution after competitive BM transplantation of mobilized blood into irradiated hosts
(G) Tri-lineage engraftment of donor cells 16 weeks post transplantation: B220+ cells (B) CD4/CD8+ cells (T) and Mac1+ cells (M; n= 6–7).
* p < 0.05, ** p < 0.01 determined by Student’s t test. Data are represented as mean ± SEM. See also Figure S1.
To assess whether the mobilization defect involved functional HSCs, we transplanted blood from mobilized wild-type or Chrm1−/− (CD45.2+) mice along with wild-type CD45.1+ competitor BM cells into lethally irradiated recipients. Mice reconstituted with Chrm1−/− mobilized blood consistently exhibited lower than wild-type CD45.2+ donor trilineage (B cell, T cell, myeloid) reconstitution in the blood 16 weeks after transplantation, confirming reduced numbers of mobilized HSCs in Chrm1−/− blood (Figure 1F and 1G). By contrast, no difference in engraftment was observed compared to wild-type controls when steady-state Chrm1−/− BM was competitively transplanted into irradiated recipients, indicating that BM HSC numbers and function in Chrm1−/− mice were unaltered (Supplemental Figure 1D and 1E). Thus, Chrm1 signals are not required for healthy steady state maintenance of HSCs within the BM niche but contribute specifically to the mobilization of these cells from their niche to peripheral blood.
Chrm1 is not expressed in bone marrow
To investigate whether Chrm1 expression was required on the HSC or the stroma, we used chimeric mice generated by reciprocal BM transplantation in which donor HSC or recipient microenvironment were deficient or sufficient in Chrm1. We found that mobilization by G-CSF was rescued when stably engrafted (>16 weeks after engraftment) chimeric recipients were wild type. By contrast, chimeras in which wild-type donor HSCs engrafted Chrm1−/− recipients exhibited the same reduction in mobilization efficiency, suggesting a non-hematopoietic origin of the defect (Figure 2A).
Figure 2. Central cholinergic signals are required for mobilization.

(A) G-CSF-induced mobilization after reciprocal BM transplantation (n = 6–13).
(B) qRT-PCR for Chrm1 mRNA transcripts in sorted BM cell populations. mRNA was undetectable in the BM (n = 3–13).
(C) HSCs mobilized to peripheral blood by G-CSF in mice treated peripherally with pirenzepine (PZP; n = 9).
(D) Experimental design and results of G-CSF-induced HSC mobilization when pirenzepine is administered centrally (n = 7–8).
** p > 0.01, *** p < 0.001 determined by Student’s t test. Data are represented as mean ± SEM.
We surveyed the expression levels of Chrm1 among FACS-purified hematopoietic and stromal cells. In contrast to the healthy prostate and brain tissue extracts where robust Chrm1 expression was detected, no expression was detectable in hematopoietic stem and progenitor cells or in various purified components of the stroma (Figure 2B). These unexpected results indicated that the inhibition of HSC mobilization was most likely mediated by an effector cell outside of the BM.
Central cholinergic signals regulate HSC mobilization elicited by G-CSF
Insights emerged from experiments using the selective Chrm1 antagonist pirenzipene (PZP). In our previous studies in prostate cancer, administration of either Scopolamine or PZP inhibited cancer cell migration induced by a cholinergic agonist (Magnon et al., 2013). However, in the hematopoietic system, we found that treatment of wild-type mice with PZP did not alter GSF-induced mobilization (Figure 2C), whereas Scopolamine treatment significantly reduced HSC egress (Figure 1A). Since Scopolamine, but not PZP, readily crosses the blood-brain barrier (Jaup and Blomstrand, 1980), we explored the possibility that the muscarinic receptors in the CNS might be controlling mobilization efficiency in the BM.
To evaluate directly that Chrm1 acts centrally, we administered PZP or vehicle control via third ventricle cannulation in wild-type animals, followed by G-CSF administration. Interestingly, mice treated centrally with PZP exhibited significantly reduced mobilization compared to controls in response to G-CSF (Figure 2D, Supplemental Figure 2A), very similar to that seen in Chrm1−/− mice challenged with G-CSF (Figure 1C). These data definitively identify a role for CNS signals in G-CSF-elicited mobilization of HSCs.
A blood-borne factor links CNS and bone marrow
To determine how signals from the CNS were relayed to the BM, we generated parabiotic mice, as depicted in Figure 3A, in which mice of the same and different genotypes (Chrm1−/− and wild type) were surgically joined. In this experimental strategy, if signals to the BM were directly transduced via a neural circuit, we would not expect any rescue from heterotypic parabionts. By contrast, if the relay was mediated by a blood-borne factor, we would expect that wild-type blood circulation might rescue the mobilization deficit in Chrm1−/− animals. Pairs were allowed to recover and blood chimerism the exchange of circulating leukocytes between partners (Supplemental Figure 2B). We found that Chrm1−/−: Chrm1−/− pairs retained the G-CSF-induced mobilization deficit, compared to the wild-type homotypic pairs (Figure 3B). In addition, the pairing of Chrm1−/− animals with wild-type mice rescued the mobilization defect in the knockout partner (Figure 3B), suggesting that a blood-borne soluble factor was linking the brain to the BM.
Figure 3. Glucocorticoids are necessary for enforced HSC mobilization by G-CSF.

(A) Schematic of parabiosis experimental design.
(B) HSCs/mL in blood of G-CSF-mobilized parabionts (n = 6–8).
(C) Plasma corticosterone levels measured by ELISA (n = 3–5)
(D) BMEF corticosterone levels measured by ELISA (n = 4–5).
(E) HSCs/ml in G-CSF-mobilized blood of Metyrapone-treated and control mice (n = 10).
(F) HSCs/ml in G-CSF-mobilized blood of vehicle-treated (Veh. 1.5% ETOH) or CORT-treated mice (n = 5–7).
G) Immunofluorescence images of sorted LSKF BM HSCs stained with anti-Nr3c1 antibody and DAPI. DAPI-stained nuclear border is denoted by yellow dashed outlines. Scale bar is 10 μm.
(H) Quantification of cytoplasmic staining in HSCs (n = 23–32).
* p < 0.05, ** p < 0.01 determined by Student’s t test. Data are represented as mean ± SEM.
We next examined the norepinephrine levels by HPLC in the plasma of mice centrally treated with PZP to investigate whether Chrm1 blockade could alter the SNS outflow. We found no significant alteration in norepinephrine levels (Supplemental Figures 2C), suggesting that the mechanism was distinct from a modulation of the SNS tone. In addition, we found that CXCL12 levels after G-CSF were comparable between Chrm1−/− and wild-type mice (Supplemental Figures 2D), arguing further that the mechanism was separate from SNS-mediated CXCL12 modulation.
Corticosterone deficiency impairs HSC mobilization
Since we determined that a blood borne factor links central Chrm1 signals to HSCs for robust mobilization, we hypothesized that a neuroendocrine factor controlled by the CNS may be at play. Since the primary source of neuroendocrine factors is the pituitary gland, we measured pituitary hormone levels in the plasma of wild-type and Chrm1−/− animals. Whereas the levels of vasopressin, oxytocin, thyroid-stimulating hormone (TSH) and growth hormone (GH) were not significantly changed between wild-type and Chrm1−/− mice (Supplemental Figure 3 A–D), levels of luteinizing hormone (LH) and adrenocorticotropic hormone (ACTH) were reduced significantly after G-CSF-induced mobilization (Supplemental Figure 3 E–F). To investigate whether the supplementation of either pituitary hormone would alter HSC migration, we administered them individually using subcutaneous osmotic pumps. We found that the administration of LH did not significantly alter HSC mobilization, and by contrast, ACTH dramatically reduced the number of mobilized cells in both WT and Chrm1−/− mice (Supplemental Figure 3 G–H).
We then measured the levels of corticosterone in plasma and found it to be significantly reduced in Chrm1−/− mice compared to controls (Figure 3C). Strikingly, corticosterone levels in BM extracellular fluids were significantly lower compared to the levels in plasma, and were dramatically reduced in Chrm1−/− animals (Figure 3D). Consistent with adrenal insufficiency in Chrm1−/− animals, a state clinically associated with serious morbidity and mortality after traumatic stress (Udelsman et al., 1986), we have observed that Chrm1−/− mice were susceptible to death after surgical procedures. For example, a large proportion (~55%) of Chrm1−/− mice did not recover the surgery for parabiosis (data not shown). In addition, we observed a significant perioperative mortality of adrenalectomized Chrm1−/− but not wild-type mice (Supplemental Figure 3I). To investigate the role of circulating GCs in HSC mobilization, we treated wild-type mice with metyrapone, an inhibitor of 11B-hydroxylase, the enzyme responsible for the conversion of precursor sterols into functional corticosterone (Hays et al., 1984). Animals treated with metyrapone phenocopied Chrm1−/− animals and exhibited significant reductions in mobilized HSCs while the baseline numbers HSCs in BM were not altered (Figure 3E and Supplemental Figure 3J). Importantly, administration of physiological amount of corticosterone to Chrm1−/− mice rescued the HSC mobilization defect to wild-type levels (Figure 3F). Taken together, these data strongly suggest that adrenal insufficiency is responsible for the mobilization deficit of Chrm1−/− mice and that GCs may play an important role in HSC egress.
Corticosterone typically binds to its receptor, Nr3c1, in the cytoplasm, and upon binding its ligand, Nr3c1 translocates into the nucleus (de Kloet et al., 2009). Thus, the activation state of the receptor can be determined by tracking the subcellular location of the receptor. Nr3c1 is expressed throughout the BM including on HSCs (Supplemental Figure 3K). To assess the activity of the GC receptor in HSCs, we sorted HSCs from wild-type or Chrm1−/− steady-state BM and imaged Nr3c1 using immunofluorescence analysis. Whereas the vast majority of Nr3c1 was located in the nucleus of wild-type HSCs, nuclear Nr3c1 was significantly reduced in HSCs isolated from Chrm1−/− animals (Figure 3G and H). Upon administration of corticosterone in Chrm1−/− mice at a dose allowing the rescue of mobilization, Nr3c1 nuclear translocation was also normalized (Figure 3H). Similar results were observed in whole-blood hematopoietic cells (Supplemental Figure 3L and M). These data are consistent with the marked reduction of corticosterone availability in the BM and plasma, supporting the conclusion that GCs play an important role in regulating HSC mobilization.
Role of hypothalamic Chrm1 in the HPA axis
Since the hypothalamus is known to orchestrate the HPA axis, we investigated the expression and function of Chrm1 in this region of the brain. In situ hybridization analyses revealed that Chrm1 was expressed in both the paraventricular nucleus (PVN) and the arcuate nucleus (ARC) of the hypothalamus (Figure 4A and Supplemental Figure 4A), which is consistent with results from the Gene Expression Nervous System Atlas Project (Gireesh et al., 2008; Heintz, 2004). Additionally, dissected hypothalamic regions from wild-type brains showed robust Chrm1 mRNA by RT-PCR analyses compared to Chrm1−/− hypothalamic tissue (Figure 4B).
Figure 4. Hypothalamic cholinergic signals are required for mobilization.

(A) In situ hybridization images of Chrm1 expression in wild-type brain. Hybridization probe is directed at Chrm1 mRNA and binds to cells localized in the paraventricular nucleus (PVN) and the arcuate nucleus (ARC) of the hypothalamus. i. 10X magnification, ii. 20X magnification. * Denotes the 3rd ventricle; white dashed outline indicates cells contained in the PVN or ARC nuclei.
(B) qRT-PCR of dissected hypothalamic regions of brains for Chrm1 transcripts (n = 7).
(C) Experimental design for ex vivo analysis of Crh production in wild-type hypothalamus tissue explants and Crh mRNA quantification relative to Gapdh (n = 7)
(D) Hypothalamic denervation by neonatal injection of MSG or control saline followed by mobilization by G-CSF at adulthood (n = 8–10).
** p < 0.01, *** p < 0.001 determined by Student’s t test. Data are represented as mean ± SEM.
Previous studies have suggested that cholinergic signals transduced via muscarinic receptors can activate the HPA axis (Hillhouse and Milton, 1989; Ohmori et al., 1995) through the production of corticotropin-releasing hormone (CRH) that can then triggers the release of ACTH in the pituitary. To test specifically if hypothalamic Chrm1 signaling regulate Crh expression, we stimulated cholinergic receptors in the hypothalamic regions using an ex vivo culture system (Figure 4C and Supplemental Figure 4B). To this end, wild-type hypothalamic regions of brains, were extracted, divided at the midline, and incubated in artificial cerebrospinal fluid, containing acetylcholine (Figure 4C). We found that in this system, the Chrm1 antagonist PZP significantly reduced Crh mRNA (Figure 4C and Supplemental Figure 4C). Thus, Chrm1+ cells in the hypothalamus region can activate the HPA axis by producing CRH.
To further evaluate the functional requirement of hypothalamic neurons in HSC mobilization, we treated neonatally wild-type animal with monosodium glutamate (MSG), which induces irreversible lesions in the mediobasal hypothalamus, mostly in the ARC region (Miquel et al., 2003). Mice were then aged to adulthood (6–8 weeks) and mobilized with G-CSF. MSG-treated mice exhibited significant reductions in HSC mobilization similar to those observed in Chrm1−/− mice, suggesting that hypothalamic neurons participate in the regulation of enforced HSC migration in the BM (Figure 4D).
Corticosterone regulates HSC mobilization via Nr3c1
To study how the deficit in GC signaling through Nr3c1 affect HSC migration capacity, we analyze the genome-wide changes in gene expression of purified HSCs from wild-type and Chrm1−/− mice. Hierarchical clustering of differentially expressed genes and subsequent gene set enrichment analyses revealed differential expression of motility set genes (Figure 5A and 5B). In particular, kinesin, microtubule motor activity, and Ras GTPase activator activity gene sets were significantly down-regulated. These results were confirmed by RT-PCR analyses of steady-state HSCs (Figure 5C). Using a transwell assay, we evaluated the effect of corticosterone on the migration of lineage-depleted hematopoietic progenitors towards the chemoattractant CXCL12. We found that after incubation with physiological concentrations of GCs the migration of colony-forming progenitors was significantly enhanced (Figure 5D–F). In addition, the effect of corticosterone on progenitor migration in vitro was reversed when the cells were treated with RU486, an inhibitor of the GC receptor (Figure 5F).
Figure 5. Altered expression of genes involved in actin polymerization and microtubule assembly in Chrm1−/− HSCs.

(A) Heat map of differentially expressed genes (by >2.0 fold and p <0.05) between wild-type and Chrm1−/− sorted HSCs.
(B) Gene set enrichment analyses showing significant alteration of gene sets involved in actin and microtubule assembly.
(C) qRT-PCR from sorted HSCs validating microarray data (n = 4–5).
(D) Transwell assay experimental design.
(E) CFU-C/well of migrated stem progenitor cells with and without CORT (n = 6).
(F) CFU-C/well of migrated stem-progenitor cells with and without CORT plus RU486 or vehicle (PEG; n = 8).
* p < 0.05, ** p < 0.01 determined by Student’s t test. * p < 0.05 determined by Student’s t test Data are represented as mean ± SEM.
To investigate further the effect of the GC receptor in a genetic model, we intercrossed Nr3c1fl/fl mice with Mx1-cre transgenic animals to conditionally delete the GC receptor. We investigated the effect of Nr3c1 deletion on actin organizing pathways in hematopoietic cells harvested from the blood of Nr3c1fl/fl; Mx1-cre+ and Nr3c1fl/fl control mice transplanted into wild-type recipients after poly (I:C) treatment, by staining blood leukocytes for phalloidin, which binds to polymerized actin. We found a marked reduction of polymerized actin in hematopoietic cells deficient in Nr3c1, compared to control cells (Figure 6A). We then evaluated actin polymerization by phalloidin staining in Chrm1−/− hematopoietic cells and observed a similar reduction, which was rescued by administration of a physiological dose of corticosterone (Figure 6B).
Figure 6. Nr3c1 expression on hematopoietic cells is required for enforced G-CSF-induced migration.

(A) Representative images of leukocytes stained for phalloidin (polymerized actin) and CD45.2 (donor cells) obtained from radiation chimeras generated by transplantation of control (Nr3c1fl/fl) or Nr3c1-deficient (Nr3c1fl/fl; Mx1-cre+) BM into lethally irradiated C57BL/6 mice and quantification of phalloidin staining normalized to control MFI (n=3).
(B) Quantification of phalloidin staining in Chrm1−/− cells and Chrm1−/− cells taken from animals treated with corticosterone normalized to control MFI (n-3).
(C) Schematic of experimental design for G-CSF-induced mobilization.
(D) G-CSF mobilized HSCs in control (Nr3c1fl/fl) and Nr3c1-deficient (Nr3c1fl/fl; Mx1-cre+) mice (n = 6–11).
(E) Model whereby finely tuned GC levels regulate HSC migration. States associated with low GCs (Chrm1−/− mice, Metyrapone administration, Nr3c1-deficient HSCs) or high GCs (administration of ACTH, Dexamethasone or corticosterone) lead to reduced mobilization efficiency.
* p < 0.05 ** p < 0.01 determined by Student’s t test. Data are represented as mean ± SEM.
We then subjected Nr3c1fl/fl; Mx1-Cre+ BM lineage-depleted cells to the transwell migration assay (Figure 5D) and found no effect of corticosterone in Nr3c1fl/fl; Mx1Cre+ progenitor migration, confirming that GCs were acting through Nr3c1 to elicit migration (Supplemental Figure 5A). We also assayed Chrm1−/− progenitor cells for transwell migration, and observed slight effect of GCs on these progenitors (Supplemental Figure 5A), which is consistent with the baseline impairment in the migration machinery in these cells. These data thus indicate that signaling through the GC receptor is critical to regulate hematopoietic cell motility.
To evaluate whether the deficiency of Nr3c1 in hematopoietic cells was sufficient to alter HSC mobilization, we transplanted the BM from Nr3c1fl/fl; Mx1-cre+ and Nr3c1fl/fl control animals into irradiated CD45.1 wild-type recipients. Four weeks after transplantation, mice were treated with poly (I:C), followed by a 10-day recovery to delete Nr3c1 in donor-derived cells (Figure 6C). Deletion of Nr3c1 in these chimeric mice did not lead to any significant change in BM cellularity or HSC numbers compared to control mice (Supplemental Figure 5B–D). Remarkably, the number of HSCs mobilized by G-CSF was reduced by 63% in Nr3c1fl/fl; Mx1-Cre+ mice compared to control animals (Figure 6D). These data thus strongly support the idea that corticosterone acts via its receptor, Nr3c1, on hematopoietic cells to promote migration.
To address if Nr3c1 signals are HSC autonomous, we generated mixed chimera mice in which CD45.1+ wild-type mice were transplanted with a mix of CD45.1+ wild-type BM and CD45.2+ Nr3c1fl/fl; Mx1-Cre+ BM (control group CD45.2+ Nr3c1fl/fl). Mice were allowed to reconstitute for 4 weeks, treated with Poly (I:C) to induce Nr3c1 gene recombination, and then mobilization was induced with G-CSF (Supplemental Figure 5E). Prior to inducing recombination, the BM was sampled in live mice to evaluate the possible bias in engraftment as Mx1-Cre driven recombination can be induced from interferon response during transplantation (Velasco-Hernandez et al., 2016). We observed an increased engraftment of CD45.2+ HSCs/progenitors (both Nr3c1fl/fl; Mx1-Cre+ and Nr3c1fl/fl) at steady state, consistent with reports suggesting a competitive advantage of CD45.2+ cells (Supplemental Figure 5F)(Mercier et al., 2016; Waterstrat et al., 2010). Interestingly, following gene recombination and G-CSF administration, we observed a significant expansion of the Nr3c1fl/fl; Mx1-Cre+ HSCs in the chimeric BM compared to the Nr3c1fl/fl control (Supplemental Figure 5G). Since we have not observed such HSC proliferation after G-CSF administration in native Nr3c1fl/fl;Mx1-Cre+ mice (Supplemental Figure 5B, C), these results suggest that Nr3c1 and G-CSF signals may act in HSCs and potentially other hematopoietic cell type(s)(Chow et al., 2011; Christopher et al., 2011) to regulate HSC proliferation after G-CSF treatment. Mobilization of Nr3c1fl/fl; Mx1-Cre+ HSCs in the chimeras was proportional to their numbers in the BM (Supplemental Figure 5H and I). However, the increased proportion of Nr3c13/3 HSCs in BM limits the interpretation of mobilization efficiency in this experimental context. These results thus suggest a complex signaling interplay between the G-CSF and GC receptors likely involving at least two cell types that will require further study to elucidate.
High doses of glucocorticoids negatively impact HSC mobilization
That steady-state GC signaling is required for robust HSC mobilization raised the possibility that pharmacological administration of GCs in healthy individuals might further enhance mobilization efficiency. To investigate this issue, we treated wild-type and Chrm1−/− mice with increasing concentrations of corticosterone in the drinking water (1 to 100 μg/ml) for two days prior to G-CSF treatment. Strikingly, we observed reductions in G-CSF-induced mobilization at supra-physiological doses of GCs (Supplemental Figure 5J). Similarly, the HSC mobilization deficit was only significantly rescued when a physiological dose of GCs was given (Supplemental Figure 5J), suggesting the existence of a “goldilocks zone” for GC signaling. In addition, we stimulated wild-type mice with a single dose of a powerful clinically used corticosteroid (dexamethasone) prior to G-CSF administration. Mice treated with dexamethasone also showed significant reductions in the number of circulating HSCs elicited by G-CSF (Supplemental Figure 5K). These results thus support a model whereby basal levels of GCs promote HSC egress, but that its levels must be tightly regulated within healthy margins (Figure 6E).
Discussion
In this study, we identified a mechanism by which HSCs undergo mobilization in response to the cytokine G-CSF. Using the Chrm1−/− mouse and chemical antagonism of the receptor, we show that central cholinergic signals act via Chrm1 to convey a hormonal relay that affects HSC’s ability to migrate upon enforced mobilization. Cholinergic signals trigger the HPA axis and the production of GCs which prime HSCs to migrate via Nr3c1 signaling and the upregulation of actin-organizing molecules.
G-CSF promotes HSC mobilization through a complex network of pathways leading to changes in the BM microenvironment that disrupt HSC retention in the BM (Bendall and Bradstock, 2014; Hoggatt et al., 2013; Lapidot and Kollet, 2010; Levesque and Winkler, 2008). Initial studies have suggested that the generation of protease activity (cathepsins, metalloproteinase, CD26) cleaves VCAM-1 or other key retention factors (e.g. CXCL12). However, the inactivation of all protease activity does not prevent efficient mobilization suggesting that protease activity is dispensable. Macrophages appear to play a critical role in retaining HSCs in the BM by promoting the synthesis of retention factors by the niche (Chow et al., 2011; Christopher et al., 2011; Winkler et al., 2010), possibly via the secretion of Oncostatin M (Albiero et al., 2015). G-CSF signaling in macrophage suppresses the retention influence of macrophage on the niche, thereby enhancing an opposing influence of the SNS to reduce CXCL12 synthesis and promote HSC release (Mendez-Ferrer et al., 2008). Whether GCs alter the macrophage regulation of the HSC niche is unknown. However, the finding that reduced mobilization in Chrm1−/− mice is not associated with dysregulation of CXCL12 (Supplemental Figure 2D), argues for a separate mechanism. Our results support the notion that GCs may act on the hematopoietic compartment to regulate HSC mobilization. The exact cell types targeted by GC signals will need to be further explored in future studies.
Hormonal regulation of the HSC niche was previously suggested by genetic manipulation of Parathormone (PTH) receptor signaling and PTH administration, which led to increased numbers of HSCs in BM (Calvi et al., 2003). Although initial studies suggested that this effect was mediated by osteoblast expansion, the PTH receptor is broadly expressed within the osteolineage, including in Nestin-GFP+ MSCs (Mendez-Ferrer et al., 2010). Administration of PTH to healthy mice can induce HSC mobilization possibly via the release of endogenous G-CSF (Brunner et al., 2008). Expression of the vitamin D receptor in the microenvironment is also required for G-CSF-induced mobilization (Kawamori et al., 2010). The present results uncover another hormonal class in regulating HSC behavior from afar. However, GCs’ mechanisms appear to differ in important ways to PTH and vitamin D in that they do not appear to promote HSC migration via regulation of the niche, but rather through the hematopoietic compartment. GCs may operate through a finely tuned Goldilocks zone where reduced mobilization is achieved when GC levels are either too low or too high (Figure 6E). The inhibition of HSC migration observed at high glucocorticoid dose is also consistent with the marked inhibitory effects observed with exogenous ACTH treatment (Supplemental Figure 3H). Since ACTH receptors are expressed throughout the BM (data not shown), whether ACTH directly regulates the BM cannot be excluded in our studies.
In response to a variety of stimuli including stress, GCs control the transcription of several genes to coordinate metabolic, immune, neural and endocrine responses (Kadmiel and Cidlowski, 2013). Deficiency in the receptor Nr3c1 leads to perinatal respiratory failure (Cole et al., 1995). In the hematopoietic system, GCs have been described as necessary for stress erythropoiesis (Bauer et al., 1999), and have powerful anti-inflammatory effects useful for the treatment of various autoimmune diseases, but equally devastating side effects associated with prolonged usage. GCs inhibit the immune response through various mechanisms, including NF-kB inhibition, Annexin I stimulation, and MAPK phosphatase stimulation that are at the nexus of critical pro-inflammatory pathways (Rhen and Cidlowski, 2005). Recently, GC receptor signaling was suggested to regulate HSC specification during development in the model of zebrafish (Kwan et al., 2016).
The involvement of GCs in HSC migration is supported by several lines of evidence: i) expression in HSC of actin organizing molecules is reduced in mice with low corticosterone in the BM microenvironment (Chrm1−/−); ii) acting assembly molecule expression is reduced in Chrm1−/− animals; iii) HSC mobilization deficits of Chrm1−/− mice are rescued by parabiosis or administration of GCs; iv) GCs can enhance the migration of hematopoietic progenitors in a transwell in vitro system; v) hematopoietic cells conditionally deficient in the GC receptor (Nr3c1fl/fl; Mx1-cre+) have severe reductions in polymerized actin; and finally, vi) chimeras in which hematopoietic cells, but not the microenvironment, are Nr3c1 deficient, exhibit marked reductions in HSC mobilization.
These results are in line with in vitro studies using cell lines that have revealed a contribution for GCs in actin remodeling. In a human lung carcinoma cell line, GCs enhanced the formation of stress fibers via up-regulation of CALD1, an actin stabilizing protein (Mayanagi et al., 2008). Other studies in AtT-20 cells have suggested that GC-induced actin stabilization may inhibit ACTH release and provide an explanation for the feedback loop of GCs in the pituitary (Castellino et al., 1992). More recent studies in hippocampal slices have suggested that GCs can engage signaling pathways that regulate dendritic spine actin networks (Jafari et al., 2012). In the BM, the loss of actin polymerization may decrease cells readiness for mobilization by reducing their ability to integrate external signals such as adhesion to extracellular matrix or endothelium which is likely important to intravasate from the marrow parenchyma. It is also notable that OPHN1 and RASGRP3, whose expression in Chrm1−/− is altered, are up-regulated in an experimental models of human prostate cancer (Goto et al., 2014; Zeng et al., 2014), suggesting that selective targeting of GC signaling in tumors should be given future consideration in the cancer treatment armamentarium.
The clinical yield of stem cells harvested in blood after G-CSF-induced mobilization is variable and can be particularly low in patients that have received prior anti-cancer therapies (Levesque and Winkler, 2008; To et al., 2011). BM neurotoxicity from these procedures and impaired sympathetic tone contribute to the blunted response (Lucas et al., 2013), but other defects may be at play. Since the chemotherapy regimen against acute lymphoblastic leukemias, lymphomas, and multiple myeloma includes high-dose GC treatment that ultimately may result in adrenal insufficiency (Gordijn et al., 2015; Ng et al., 2009; Park et al., 2007), our data raise the possibility that adrenal insufficiency may contribute to the poor mobilization observed in these patients.
STAR METHODS
KEY RESOURCES TABLE
| Reagent or Resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Anti-human/mouse CD45R (B220) | eBioscience | Cat# 47-0452-82 |
| Anti-mouse CD117 (c-Kit) | BioLegend | Cat# 105814 |
| Anti-mouse CD135 (Flt3) | eBioscience | Cat# 12-1351-83 |
| Anti-mouse CD150 (SLAM) | BioLegend | Cat# 115903 |
| Anti-mouse CD4 | eBioscience | Cat# 25-0041-82 |
| Anti-mouse CD41 | eBioscience | Cat# 13-0411-82 |
| Anti-mouse CD45.1 | eBioscience | Cat# 11-0453-85 |
| Anti-mouse CD45.2 | eBioscience | Cat# 45-0454-82 |
| Anti-mouse Cd48 | eBioscience | Cat# 13-0481-85 |
| Anti-mouse CD8a | eBioscience | Cat# 25-0081-82 |
| Anti-mouse Ly6A/E (Sca-1) | eBioscience | Cat# 17-5981-83 |
| Anti-mouse/human CD11b | Biolegend | Cat# 101218 |
| GR antibody (M20) | Santa Cruz | Cat# sc-1004 |
| Mouse lineage panel | BD Biosciences | Cat# 559971 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Scopolamine | Sigma-Aldrich | Cat# S0929 |
| Pirenzepine | Sigma-Aldrich | Cat# P7412 |
| Metyrapone | Tocris Bioscience | Cat# 3292 |
| Corticosterone | Sigma-Aldrich | Cat# H3035 |
| Dexamethasone (water soluble) | Sigma-Aldrich | Cat# D2915 |
| ACTH | Pheonix Pharmaceuticals | Cat# 001-06 |
| LH | National hormone and peptide program | |
| Recombinant murine SDF-1a (CXCL12) | PeproTech | Cat# 250-20A |
| Poly (I:C) | Invivogen | Cat# tlrl-pic |
| RU486 (Mifepristone) | Sigma-Aldrich | Cat# M8046 |
| MSG | Sigma-Aldrich | Cat# G1626 |
| Critical Commercial Assays | ||
| DIG RNA labeling Kit (SP6/T7) | Sigma-Aldrich | Cat# 11175025910 |
| Methocult | Stem Cell Technologies | Cat# M3534 |
| Corticosterone ELISA Kit | Abcam | Cat# ab108821 |
| Vasopressin ELISA Kit | Pheonix Pharmaceuticals | Cat# EK-065-07 |
| Oxytocin ELISA Kit | Pheonix Pharmaceuticals | Cat# EK-051-01 |
| Multiplex | EMD Millipore | Cat# MPTMAG-49K |
| Rneasy Plus Micro Kit | Qiagen | Cat# 74034 |
| Osmotic pumps | Alzet | Cat# 0004317 |
| LYMPHOLYTE-M | Cedarlane | Cat# CL5030 |
| Deposited Data | ||
| Microarray data | This paper | GEO: GSE76350 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Chrm1−/− | The Jackson Laboratory | Stock# 006468 |
| Mouse: Nestin-Gfp | Mignone et al., 2004 | |
| Mouse: Nr3c1fl/fl | The Jackson Laboratory | Stock# 021021 |
| Mouse: Mx1-Cre | The Jackson Laboratory | Stock# 003556 |
| Mouse: CD45.1 | National Cancer Institute | Stock# 564 |
| Sequence-Based Reagents | ||
| Chrm1 qPCR Forward | CAGAAGTGGTGATCAAGATGCCTAT | |
| Chrm1 qPCR Reverse | GAGCTTTTGGGAGGCTGCTT | |
| Nr3c1 qPCR Forward | CAAAGCCGTTTCACTGTCC | |
| Nr3c1 qPCR Reverse | ACAATTTCACACTGCCACC | |
| Chrm1 ISH Probe Sense Fwd | TGT GCA GTG GGT GTG GGT GTT T | |
| Chrm1 ISH Probe Sense Fwd (+Sp6) | att tag gtg aca cta tag a TGA TGT TGG GAC TGA CAG CAG | |
| Chrm1 ISH Probe Antisense Fwd (+Sp6) | att tag gtg aca cta tag a TGT GCA GTG GGT GTG GGT GTT T | |
| Chrm1 ISH Probe Antisense Rev | TGA TGT TGG GAC TGA CAG CAG | |
| Software and Algorithms | ||
| Slidebook | Intelligent Imaging Innovations (3i), version 6.0 | |
| OTSU algorithm for imaging processing | Intelligent Imaging Innovations (3i), version 6.0 | |
| Transcriptome analysis center | Affymetrix | |
| Prism 6 | GraphPad | |
| Gene Set Enrichment Analysis | Broad Institute | |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by the corresponding author Paul S. Frenette (paul.frenette@einstein.yu.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Chrm1−/− mice (C57BL/6-Chrm1tm1Stl/J, (Gerber et al., 2001) Nr3c1fl/fl mice, and Mx1-Cre mice were purchased from Jackson Laboratories. C57Bl/6 mice were purchased from the National Cancer Institute (Frederick National Laboratory for Cancer Research). Nestin-gfp transgenic mice were bred to Chrm1−/− mice for HSC localization studies (Mignone et al., 2004). Six to eight week old mice of both genders were used for experiments. For each condition, 3–16 mice were used in each experiment. All mice were housed in specific pathogen-free conditions and fed with autoclaved food, and experimental procedures performed on mice were approved by the Animal Care and Use Committee of Albert Einstein College of Medicine.
METHOD DETAILS
Surgical procedures
Cannulation of the 3rd ventricle was performed as previously described (Li et al., 2011). Briefly, mice were implanted with a 28G stainless steel cannula (Plastics One) targeting the 3rd ventricle (coordinates: A/P −1.6 mm posterior to bregma, D/V −4.7 mm). After one week, an osmotic pump (Alzet) was inserted subcutaneously on the back and connected to the 3rd ventricle cannula via a polyethylene tubing connector kit (Alzet). Pumps were preloaded with either pirenzepine (Sigma-Aldrich) solution (see below) or PBS.
Parabionts were generated by making an incision in the skin from the elbow to knee of mice on opposite sides of each mouse. The knees and elbows of mice were paired together by subcutaneous suturing. Skin was then matched from one mouse to the other, sutured together, and secured with wound clips as described previously (Wright et al., 2001). G-CSF was administered after weighing the pair and splitting the dose such that each mouse received the dose corresponding to half of the pair’s body weight. Adrenalectomy was performed by making small, bilateral incisions on the back of the mouse, exposing the adrenal glands and removing the gland completely under dissecting microscope. Subcutaneous implants of Alzet osmotic pumps were inserted by creating a small incision in the skin and opening a subcutaneous pocket for the pump. The skin was secured back together with wound clips.
Drug and chemical treatments
G-CSF was administered subcutaneously (s.c.) at a dose of 125 ug/kg twice a day beginning in the evening of the first day. Blood was harvested 1 hour after the final morning dose of G-CSF at the circadian time that is most optimized for HSC mobilization, zeitgeiber time 5, 5 hours after light onset for analysis. Scopolamine hydrobromide (1 mg/kg; Sigma-Aldrich) was administered by subcutaneous pump. Pirenzepine (Sigma-Aldrich) was given at 6mg/kg i.p. 30 min prior to G-CSF peripherally and 0.6 mg/kg/day i.c.v. delivered by ALZET osmotic pump centrally. Metyrapone (100 mg/kg/day; Tocris Bioscience) was given i.p. 30 min prior to evening G-CSF. ACTH (2.8 mg/kg/day; Pheonix) and LH (16 ug/day; National hormone and peptide program) were administered via osmotic pumps. MSG (2mg/g; Sigma-Aldrich) was administered neonatally beginning one day postnatally for 5 days every other day by subcutaneous injection (Caputo et al., 1996). Dexamethasone (5mg/kg; Sigma-Aldrich, water soluble) was given s.c. at ZT5 prior to beginning G-CSF treatment. Poly (I:C; 250ug/animal; Invivogen) was administered i.p. for 5 days, every other day. Animals rested for 10 days before experiments.
Progenitor and transplantation assays
Colony-forming units in culture (CFU-C) were assayed by processing blood over lympholyte-M (Cedarlane) to separate white and red cells as intended by the manufacturer. Resulting white blood cells were washed and plated in methylcellulose (Stem Cell Technologies, Cat#: 3534) and incubated for 7 days at 37°C in humid chambers when colonies were counted. BM transplantations were performed by retro-orbital injection of 106 competitor BM cells mixed with 10 μl of mobilized blood (competitive BMT to assay mobilized blood), 106 competitor cells mixed with 106 test cells (competitive BMT to assay BM HSCs), or 106 BM cells (reciprocal BMT) into lethally irradiated recipients (12 Gy).
Transwell migration assay
Wild-type femura, tibia, and humeri were flushed into sterile αMEM, erythrocytes were lysed, and the BM was immunomagnetically depleted of lineage-positive cells using a biotinylated lineage antibody cocktail (Benton Dickson) and subsequent streptavidin magnetic beads (Miltenyi Biotech). Lineage-depleted BM cells were transferred into the transwell plates (Corning Cat# 3421) containing αMEM + 10% FBS (Stem Cell Technologies) with or without 30 μg/ml corticosterone (Sigma). In experiments with RU486, cells were additionally incubated with 10−5 M RU486 or the equivalent volume of polyethylene glycol 400. The bottoms of transwells were filled with media and 100 ng/ml CXCL12 (Peprotech) and transwell plates were incubated for 3 h at 37°C. Migrated cells were then plated in CFU-C assays as described above.
RT-PCR
Isolation of RNA, subsequent cDNA amplification, and RT-PCR analysis of genes was performed as previously described (Chow et al., 2011). Whole tissue was first homoginized in Trizol (Life Technologies) and RNA was isolated. Subsequent cDNA was amplified as described above. Chrm1 mRNA was amplified by the primers: fwd- CAGAAGTGGTGATCAAGATGCCTAT and rev- GAGCTTTTGGGAGGCTGCTT. Nr3c1 mRNA was amplified by the primers: fwd- CAAAGCCGTTTCACTGTCC and rev- ACAATTTCACACTGCCACC.
In situ hybridization
The probe used for detection of Chrm1 mRNA was cloned from mouse Chrm1 cDNA and designed to bind to nucleotides 24–656 of the Chrm1 mouse coding sequence then subsequently labeled with digoxigenin for detection (Roche). The resulting antisense strand was used for positive detection of Chrm1 mRNA and the sense strand as a negative control. This probe was previously used to ascertain the deletion of Chrm1 (Gerber et al., 2001). ISH was performed as described previously, with incubation temperatures of 70°C (Oxtoby and Jowett, 1993).
ELISA
CXCL12 ELISA was prepared from individually purchased antibodies for capture and detection as previously described (Chow et al., 2011). In brief, ELISA plates were coated overnight at 4°C with 50 μl of 2 μg/ml anti-CXCL12 antibody (MAB350; R&D Systems). Plates were washed with 0.05% Tween 20 in PBS and incubated for 1 h at room temperature with 200 μl of blocking buffer (1% BSA, 5% D-Sucrose, and 0.05% NaN3 in PBS; all from Thermo Fisher Scientific). Next, 100 μl of sample diluent was added and incubated for 2 h at room temperature, followed by washing and incubation with 100 μl of 0.250 μg/ml polyclonal biotinylated anti–human/mouse SDF-1 (BAF310; R&D Systems) for 2 h at room temperature. After washing, 100 μl of 0.1 μg/ml Neutravidin-HRP (Thermo Fisher Scientific) was added and incubated for 30 min. Lastly, the reaction was developed by incubation for 20–30 min with 50 μl of TMB substrate solution (Sigma-Aldrich) and stopped by adding 50 μl of 1M HCl solution (Thermo Fisher Scientific). Optical density was determined with a microplate reader set at 450 nm. Optical density of PBS control wells was subtracted from optical density of samples. Recombinant mSDF-1α (PeproTech) was used to generate a linear standard curve.
Corticosterone ELISA kit was purchased from ABCAM (cat# ab108821) and used according to the manufacturer’s instructions. Multiplex hormone assays were purchased from EMD Millipore (cat# MPTMAG-49K) and used according to the manufacturer’s instructions. All BMEF used for ELISA were collected by flushing the BM of one femur into 1 mL of PBS and subsequently pelleting the cells by centrifugation. The resulting liquid phase was removed from the cell pellet and frozen at −800C for subsequent protein analysis.
Norepinephrine analysis
Plasma samples were rapidly harvested and frozen in liquid nitrogen. Norepinephrine levels in plasma were determined using HPLC at the Neurochemistry Core Laboratory, Vanderbilt University as previously reported (Hanoun et al., 2014; Katayama et al., 2006).
Immunofluorescence imaging
HSC distribution in whole-mount BM was analyzed as reported previously (Kunisaki et al., 2013). Briefly, sternal bones were collected and transected with a surgical blade into 2–3 fragments. The fragments were bisected sagittally, exposing the bone marrow cavity, and fixed in 4% PFA. Tissues were blocked/permeabilized in PBS containing 20% normal goat serum and 0.5% Triton X-100, and stained with primary antibodies for 1–3 days. The tissues were incubated with secondary antibodies for 2 h. Images were acquired using a ZEISS AXIO examiner D1 microscope (Zeiss) with a confocal scanner unit, CSUX1CU (Yokogawa), and reconstructed in three dimensions with Slide Book software (Intelligent Imaging Innovations).
Nr3c1 quantification was achieved by fixation (4% PFA), permeabilization/blocking (0.5% Triton-X, 20% goat serum), and staining with anti-Nr3c1 (Santa Cruz) or isotype control. Samples were imaged by ZEISS ZSIO examiner D1 microscope (Zeiss) with a confocal scanner unit, CSUX1CU (Yokogawa) then analyzed with Slide Book software (Intelligent Imaging Innovations). Localization analyses were performed by generating masks in each channel (405 nm, DAPI or 563 nm, anti-Nr3c1) using the OTSU algorithm and calculating the percent overlap between the channels.
Antibodies for flow cytometry and sorting
Cells were surface-stained in PEB buffer (PBS supplemented with 0.5% BSA and 2mM EDTA) for 20–30 min on ice. Multiparametric flow cytometric analyses were performed on a LSRII equipped with FACS Diva 6.1 software (BD Biosciences) and analysed with FlowJo software (Tree Star). Dead cells were excluded by FSC, SSC and 4’,6-diamino-2-phenylindole (DAPI, Sigma) staining. Cell sorting experiments were performed on Aria Cell Sorter (BD Biosciences). HSCs measured by flow cytometry were defined as: Lineage− Sca1+ cKit+ Flt3−. Antibodies used: anti-mouse mABs Sca1/Ly-6A/E (clone D7, ebioscience), CD117/cKit (clone ZB8, Biolegend), CD135/Flt3 (clone AZF10, ebioscience), lineage cocktail (BD Pharmingen cat# 559971), CD41 (clone RMV-7, ebiosicence), CD45 (clone 30-F11, ebioscience), Ter119 (clone TER-119, ebioscience), CD31 (clone MEC13.3, ebioscience), Gr-1 (clone RB6-8C5, ebioscience), CD11b (M1/70, ebioscience), CD115/c-fms (clone AFS98, ebioscience), F4/80 (BM8, ebioscience), and BrdU (clone BU20A, BD Pharmingen with BrdU flow kit reagents for fixation and permeabilization).
Microarray analysis
RNA for microarray analysis was isolated by sorting HSC (Lineage−Sca1+cKit+Flt3− directly into RLT buffer from the RNeasy Plus Micro kit (Qiagen) according to the manufacturercs instructions. Amplification and subsequent microarray was performed by the Genomics Core Facility at Albert Einstein College of Medicine using the One-Direct RNA amplification system (NuGEN) and GeneChip mouse gene 2.0 ST array (Affymetrix). Analysis was performed by first processing the chip through Transcriptome analysis center (TAC) software (Affymetrix) to determine fold changes and p-values which were then analyzed by gene set enrichment analysis (GSEA). GSEA analysis was performed against the C5: GO gene set ME: GO molecular function. Gene sets with p>0.05 and fold change greater than 2 were chosen for further validation.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are represented as ± SEM. n represents mouse number or cell number analyzed in each experiment, as detailed in figure legends. Statistical significance was determined by unpaired, two-tailed Student t test for most experiments, and unpaired, two-tailed Student t test with Welch’s correction for unequal standard deviations was used for Nr3c1 translocation assay. One-way ANOVA analysis was used to determine significant differences in Chrm1 expression. Prism 6 software (GraphPad) was used for all statistical analysis except microarray analysis.
DATA AND SOFTWARE AVAILABILITY
Data Resources
Raw data files for the microarray analysis have been deposited in the NCBI Gene Expression Omnibus under accession number GEO: GSE76350.
Supplementary Material
Acknowledgments
We are grateful to C. Prophete, P. Ciero, C. Cruz, and L. Wu for their technical assistance; D. Reynolds and W. Tran for the microarray analysis; Dr. Raymond Johnson for noradrenaline measurements; and L. Tesfa for cell sorting assistance. This work was supported by an R01 grant from the National Institutes of Health (DK056638, HL097700, HL069438 to P.S.F). H.P. is supported by the Training Program in Cellular, Molecular Biology, and Genetics (T32 GM007491). We are also grateful to the New York State Department of Health (NYSTEM Program) for shared facility (C029154) and research support (N13G-262).
Footnotes
Author Contributions
H.P. performed experiments, analyzed data, and wrote the manuscript. D.Z. C.M., D.L., J.C. and M.H. performed experiments and provided valuable input on the manuscript. G.J.S. designed central cannulation experiment, discussed data, and provided valuable input on the manuscript. P.S.F. designed and supervised the study, discussed data, and wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albiero M, Poncina N, Ciciliot S, Cappellari R, Menegazzo L, Ferraro F, Bolego C, Cignarella A, Avogaro A, Fadini GP. Bone Marrow Macrophages Contribute to Diabetic Stem Cell Mobilopathy by Producing Oncostatin M. Diabetes. 2015;64:2957–2968. doi: 10.2337/db14-1473. [DOI] [PubMed] [Google Scholar]
- Bauer A, Tronche F, Wessely O, Kellendonk C, Reichardt HM, Steinlein P, Schutz G, Beug H. The glucocorticoid receptor is required for stress erythropoiesis. Genes & development. 1999;13:2996–3002. doi: 10.1101/gad.13.22.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall LJ, Bradstock KF. G-CSF: From granulopoietic stimulant to bone marrow stem cell mobilizing agent. Cytokine & growth factor reviews. 2014;25:355–367. doi: 10.1016/j.cytogfr.2014.07.011. [DOI] [PubMed] [Google Scholar]
- Bensinger W, Singer J, Appelbaum F, Lilleby K, Longin K, Rowley S, Clarke E, Clift R, Hansen J, Shields T, et al. Autologous transplantation with peripheral blood mononuclear cells collected after administration of recombinant granulocyte stimulating factor. Blood. 1993;81:3158–3163. [PubMed] [Google Scholar]
- Brunner S, Zaruba MM, Huber B, David R, Vallaster M, Assmann G, Mueller-Hoecker J, Franz WM. Parathyroid hormone effectively induces mobilization of progenitor cells without depletion of bone marrow. Experimental hematology. 2008;36:1157–1166. doi: 10.1016/j.exphem.2008.03.014. [DOI] [PubMed] [Google Scholar]
- Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, Martin RP, Schipani E, Divieti P, Bringhurst FR, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–846. doi: 10.1038/nature02040. [DOI] [PubMed] [Google Scholar]
- Caputo FA, Ali SF, Wolff GL, Scallet AC. Neonatal MSG reduces hypothalamic DA, beta-endorphin, and delays weight gain in genetically obese (A viable yellow/alpha) mice. Pharmacology, biochemistry, and behavior. 1996;53:425–432. doi: 10.1016/0091-3057(95)02009-8. [DOI] [PubMed] [Google Scholar]
- Castellino F, Heuser J, Marchetti S, Bruno B, Luini A. Glucocorticoid stabilization of actin filaments: a possible mechanism for inhibition of corticotropin release. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:3775–3779. doi: 10.1073/pnas.89.9.3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow A, Lucas D, Hidalgo A, Mendez-Ferrer S, Hashimoto D, Scheiermann C, Battista M, Leboeuf M, Prophete C, van Rooijen N, et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J Exp Med. 2011;208:261–271. doi: 10.1084/jem.20101688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopher MJ, Rao M, Liu F, Woloszynek JR, Link DC. Expression of the G-CSF receptor in monocytic cells is sufficient to mediate hematopoietic progenitor mobilization by G-CSF in mice. J Exp Med. 2011;208:251–260. doi: 10.1084/jem.20101700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole TJ, Blendy JA, Monaghan AP, Krieglstein K, Schmid W, Aguzzi A, Fantuzzi G, Hummler E, Unsicker K, Schutz G. Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes & development. 1995;9:1608–1621. doi: 10.1101/gad.9.13.1608. [DOI] [PubMed] [Google Scholar]
- de Kloet ER, Fitzsimons CP, Datson NA, Meijer OC, Vreugdenhil E. Glucocorticoid signaling and stress-related limbic susceptibility pathway: about receptors, transcription machinery and microRNA. Brain research. 2009;1293:129–141. doi: 10.1016/j.brainres.2009.03.039. [DOI] [PubMed] [Google Scholar]
- Duhrsen U, Villeval JL, Boyd J, Kannourakis G, Morstyn G, Metcalf D. Effects of recombinant human granulocyte colony-stimulating factor on hematopoietic progenitor cells in cancer patients. Blood. 1988;72:2074–2081. [PubMed] [Google Scholar]
- Gerber DJ, Sotnikova TD, Gainetdinov RR, Huang SY, Caron MG, Tonegawa S. Hyperactivity, elevated dopaminergic transmission, and response to amphetamine in M1 muscarinic acetylcholine receptor-deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:15312–15317. doi: 10.1073/pnas.261583798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gireesh G, Kaimal SB, Kumar TP, Paulose CS. Decreased muscarinic M1 receptor gene expression in the hypothalamus, brainstem, and pancreatic islets of streptozotocin-induced diabetic rats. J Neurosci Res. 2008;86:947–953. doi: 10.1002/jnr.21544. [DOI] [PubMed] [Google Scholar]
- Gordijn MS, Rensen N, Gemke RJ, van Dalen EC, Rotteveel J, Kaspers GJ. Hypothalamic-pituitary-adrenal (HPA) axis suppression after treatment with glucocorticoid therapy for childhood acute lymphoblastic leukaemia. The Cochrane database of systematic reviews. 2015;8:CD008727. doi: 10.1002/14651858.CD008727.pub3. [DOI] [PubMed] [Google Scholar]
- Goto K, Oue N, Hayashi T, Shinmei S, Sakamoto N, Sentani K, Teishima J, Matsubara A, Yasui W. Oligophrenin-1 is associated with cell adhesion and migration in prostate cancer. Pathobiology. 2014;81:190–198. doi: 10.1159/000363345. [DOI] [PubMed] [Google Scholar]
- Hanoun M, Zhang D, Mizoguchi T, Pinho S, Pierce H, Kunisaki Y, Lacombe J, Armstrong SA, Duhrsen U, Frenette PS. Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell. 2014;15:365–375. doi: 10.1016/j.stem.2014.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hays SJ, Tobes MC, Gildersleeve DL, Wieland DM, Beierwaltes WH. Structure-activity relationship study of the inhibition of adrenal cortical 11 beta-hydroxylase by new metyrapone analogues. Journal of medicinal chemistry. 1984;27:15–19. doi: 10.1021/jm00367a004. [DOI] [PubMed] [Google Scholar]
- Heintz N. Gene expression nervous system atlas (GENSAT) Nat Neurosci. 2004;7:483. doi: 10.1038/nn0504-483. [DOI] [PubMed] [Google Scholar]
- Hillhouse EW, Milton NG. Effect of acetylcholine and 5-hydroxytryptamine on the secretion of corticotrophin-releasing factor-41 and arginine vasopressin from the rat hypothalamus in vitro. The Journal of endocrinology. 1989;122:713–718. doi: 10.1677/joe.0.1220713. [DOI] [PubMed] [Google Scholar]
- Hoggatt J, Speth JM, Pelus LM. Concise review: Sowing the seeds of a fruitful harvest: hematopoietic stem cell mobilization. Stem cells. 2013;31:2599–2606. doi: 10.1002/stem.1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafari M, Seese RR, Babayan AH, Gall CM, Lauterborn JC. Glucocorticoid receptors are localized to dendritic spines and influence local actin signaling. Mol Neurobiol. 2012;46:304–315. doi: 10.1007/s12035-012-8288-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaup BH, Blomstrand C. Cerebro-spinal fluid concentrations of pirenzepine after therapeutic dosage. Scandinavian journal of gastroenterology Supplement. 1980;66:35–37. [PubMed] [Google Scholar]
- Kadmiel M, Cidlowski JA. Glucocorticoid receptor signaling in health and disease. Trends in pharmacological sciences. 2013;34:518–530. doi: 10.1016/j.tips.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA, Frenette PS. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell. 2006;124:407–421. doi: 10.1016/j.cell.2005.10.041. [DOI] [PubMed] [Google Scholar]
- Kawamori Y, Katayama Y, Asada N, Minagawa K, Sato M, Okamura A, Shimoyama M, Nakagawa K, Okano T, Tanimoto M, et al. Role for vitamin D receptor in the neuronal control of the hematopoietic stem cell niche. Blood. 2010;116:5528–5535. doi: 10.1182/blood-2010-04-279216. [DOI] [PubMed] [Google Scholar]
- Kollet O, Vagima Y, D’Uva G, Golan K, Canaani J, Itkin T, Gur-Cohen S, Kalinkovich A, Caglio G, Medaglia C, et al. Physiologic corticosterone oscillations regulate murine hematopoietic stem/progenitor cell proliferation and CXCL12 expression by bone marrow stromal progenitors. Leukemia. 2013;27:2006–2015. doi: 10.1038/leu.2013.154. [DOI] [PubMed] [Google Scholar]
- Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, Mizoguchi T, Wei Q, Lucas D, Ito K, et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature. 2013;502:637–643. doi: 10.1038/nature12612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan W, Cortes M, Frost I, Esain V, Theodore LN, Liu SY, Budrow N, Goessling W, North TE. The Central Nervous System Regulates Embryonic HSPC Production via Stress-Responsive Glucocorticoid Receptor Signaling. Cell Stem Cell. 2016;19:370–382. doi: 10.1016/j.stem.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane TA, Law P, Maruyama M, Young D, Burgess J, Mullen M, Mealiffe M, Terstappen LW, Hardwick A, Moubayed M, et al. Harvesting and enrichment of hematopoietic progenitor cells mobilized into the peripheral blood of normal donors by granulocyte-macrophage colony-stimulating factor (GM-CSF) or G-CSF: potential role in allogeneic marrow transplantation. Blood. 1995;85:275–282. [PubMed] [Google Scholar]
- Lapidot T, Kollet O. The brain-bone-blood triad: traffic lights for stem-cell homing and mobilization. Hematology/the Education Program of the American Society of Hematology American Society of Hematology Education Program. 2010;2010:1–6. doi: 10.1182/asheducation-2010.1.1. [DOI] [PubMed] [Google Scholar]
- Levesque JP, Winkler IG. Mobilization of hematopoietic stem cells: state of the art. Curr Opin Organ Transplant. 2008;13:53–58. doi: 10.1097/MOT.0b013e3282f42473. [DOI] [PubMed] [Google Scholar]
- Li X, Wu X, Camacho R, Schwartz GJ, LeRoith D. Intracerebroventricular leptin infusion improves glucose homeostasis in lean type 2 diabetic MKR mice via hepatic vagal and non-vagal mechanisms. PloS one. 2011;6:e17058. doi: 10.1371/journal.pone.0017058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Poursine-Laurent J, Link DC. Expression of the G-CSF receptor on hematopoietic progenitor cells is not required for their mobilization by G-CSF. Blood. 2000;95:3025–3031. [PubMed] [Google Scholar]
- Lucas D, Bruns I, Battista M, Mendez-Ferrer S, Magnon C, Kunisaki Y, Frenette PS. Norepinephrine reuptake inhibition promotes mobilization in mice: potential impact to rescue low stem cell yields. Blood. 2012;119:3962–3965. doi: 10.1182/blood-2011-07-367102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas D, Scheiermann C, Chow A, Kunisaki Y, Bruns I, Barrick C, Tessarollo L, Frenette PS. Chemotherapy-induced bone marrow nerve injury impairs hematopoietic regeneration. Nat Med. 2013;19:695–703. doi: 10.1038/nm.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnon C, Frenette PS. Hematopoietic stem cell trafficking. StemBook; Cambridge (MA): 2008. [PubMed] [Google Scholar]
- Magnon C, Hall SJ, Lin J, Xue X, Gerber L, Freedland SJ, Frenette PS. Autonomic nerve development contributes to prostate cancer progression. Science. 2013;341:1236361. doi: 10.1126/science.1236361. [DOI] [PubMed] [Google Scholar]
- Mayanagi T, Morita T, Hayashi K, Fukumoto K, Sobue K. Glucocorticoid receptor-mediated expression of caldesmon regulates cell migration via the reorganization of the actin cytoskeleton. The Journal of biological chemistry. 2008;283:31183–31196. doi: 10.1074/jbc.M801606200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez-Ferrer S, Lucas D, Battista M, Frenette PS. Haematopoietic stem cell release is regulated by circadian oscillations. Nature. 2008;452:442–447. doi: 10.1038/nature06685. [DOI] [PubMed] [Google Scholar]
- Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, Scadden DT, Ma’ayan A, Enikolopov GN, Frenette PS. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466:829–834. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercier FE, Sykes DB, Scadden DT. Single Targeted Exon Mutation Creates a True Congenic Mouse for Competitive Hematopoietic Stem Cell Transplantation: The C57BL/6-CD45.1(STEM) Mouse. Stem cell reports. 2016;6:985–992. doi: 10.1016/j.stemcr.2016.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignone JL, Kukekov V, Chiang AS, Steindler D, Enikolopov G. Neural stem and progenitor cells in nestin-GFP transgenic mice. J Comp Neurol. 2004;469:311–324. doi: 10.1002/cne.10964. [DOI] [PubMed] [Google Scholar]
- Miquel M, Font L, Sanchis-Segura C, Aragon CM. Neonatal administration of monosodium glutamate prevents the development of ethanol-but not psychostimulant-induced sensitization: a putative role of the arcuate nucleus. The European journal of neuroscience. 2003;17:2163–2170. doi: 10.1046/j.1460-9568.2003.02646.x. [DOI] [PubMed] [Google Scholar]
- Ng AC, Kumar SK, Russell SJ, Rajkumar SV, Drake MT. Dexamethasone and the risk for adrenal suppression in multiple myeloma. Leukemia. 2009;23:1009–1011. doi: 10.1038/leu.2008.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmori N, Itoi K, Tozawa F, Sakai Y, Sakai K, Horiba N, Demura H, Suda T. Effect of acetylcholine on corticotropin-releasing factor gene expression in the hypothalamic paraventricular nucleus of conscious rats. Endocrinology. 1995;136:4858–4863. doi: 10.1210/endo.136.11.7588217. [DOI] [PubMed] [Google Scholar]
- Oxtoby E, Jowett T. Cloning of the zebrafish krox-20 gene (krx-20) and its expression during hindbrain development. Nucleic acids research. 1993;21:1087–1095. doi: 10.1093/nar/21.5.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CK, Miller C, Lawrence G. Addison’s disease from non-Hodgkin’s lymphoma with normal-size adrenal glands. J Clin Oncol. 2007;25:2322–2324. doi: 10.1200/JCO.2007.10.8159. [DOI] [PubMed] [Google Scholar]
- Petit I, Szyper-Kravitz M, Nagler A, Lahav M, Peled A, Habler L, Ponomaryov T, Taichman RS, Arenzana-Seisdedos F, Fujii N, et al. G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nat Immunol. 2002;3:687–694. doi: 10.1038/ni813. [DOI] [PubMed] [Google Scholar]
- Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids--new mechanisms for old drugs. The New England journal of medicine. 2005;353:1711–1723. doi: 10.1056/NEJMra050541. [DOI] [PubMed] [Google Scholar]
- Scheiermann C, Kunisaki Y, Lucas D, Chow A, Jang JE, Zhang D, Hashimoto D, Merad M, Frenette PS. Adrenergic nerves govern circadian leukocyte recruitment to tissues. Immunity. 2012;37:290–301. doi: 10.1016/j.immuni.2012.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- To LB, Levesque JP, Herbert KE. How I treat patients who mobilize hematopoietic stem cells poorly. Blood. 2011;118:4530–4540. doi: 10.1182/blood-2011-06-318220. [DOI] [PubMed] [Google Scholar]
- Udelsman R, Ramp J, Gallucci WT, Gordon A, Lipford E, Norton JA, Loriaux DL, Chrousos GP. Adaptation during surgical stress. A reevaluation of the role of glucocorticoids. The Journal of clinical investigation. 1986;77:1377–1381. doi: 10.1172/JCI112443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco-Hernandez T, Sawen P, Bryder D, Cammenga J. Potential Pitfalls of the Mx1-Cre System: Implications for Experimental Modeling of Normal and Malignant Hematopoiesis. Stem cell reports. 2016;7:11–18. doi: 10.1016/j.stemcr.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterstrat A, Liang Y, Swiderski CF, Shelton BJ, Van Zant G. Congenic interval of CD45/Ly-5 congenic mice contains multiple genes that may influence hematopoietic stem cell engraftment. Blood. 2010;115:408–417. doi: 10.1182/blood-2008-03-143370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler IG, Sims NA, Pettit AR, Barbier V, Nowlan B, Helwani F, Poulton IJ, van Rooijen N, Alexander KA, Raggatt LJ, et al. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood. 2010;116:4815–4828. doi: 10.1182/blood-2009-11-253534. [DOI] [PubMed] [Google Scholar]
- Wright DE, Wagers AJ, Gulati AP, Johnson FL, Weissman IL. Physiological migration of hematopoietic stem and progenitor cells. Science. 2001;294:1933–1936. doi: 10.1126/science.1064081. [DOI] [PubMed] [Google Scholar]
- Zeng X, Hu Z, Wang Z, Tao J, Lu T, Yang C, Lee B, Ye Z. Upregulation of RASGRP3 expression in prostate cancer correlates with aggressive capabilities and predicts biochemical recurrence after radical prostatectomy. Prostate Cancer Prostatic Dis. 2014;17:119–125. doi: 10.1038/pcan.2013.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.