

ABSTRACT
If there is a great new hope in the treatment of cancer, the immune system is it. Innate and adaptive immunity either promote or attenuate tumorigenesis and so can have opposing effects on the therapeutic outcome. Originally described as potent antivirals, Type-I interferons (IFNs) were quickly recognized as central coordinators of tumor-immune system interactions. Type-I-IFNs are produced by, and act on, both tumor and immune cells being either host-protecting or tumor-promoting. Here, we discuss Type-I-IFNs in infectious and cancer diseases highlighting their dichotomous role and raising the importance to deeply understand the underlying mechanisms so to reshape the way we can exploit Type-I-IFNs therapeutically.
KEYWORDS: Anticancer therapy, cancer stem cells, IFNs, immunotherapy, tumor immunity
Abbreviations
- AIM2
absent in melanoma 2
- AP-1
activated protein-1
- ATM
ataxia-telangiectasia mutated
- CARD
caspase activation and recruitment domain
- CARDIF
CARD adaptor-inducing IFN-β
- CDKN1A
cyclin-dependent kinase inhibitor 1A
- cGAMP
cyclic guanosine monophosphate-adenosine monophosphate
- cGAS
cyclic GMP-AMP synthase
- CSC
cancer stem cell
- CSF1
colony stimulating factor 1
- CTL
cytotoxic T lymphocyte
- CXCL10
C-X-C motif chemokine ligand 10
- DAI
DNA-dependent activator of IRFs
- DAMP
damage-associated molecular pattern
- DC
dendritic cell
- DDX
DExD/H-box helicase
- EGFR
epidermal growth factor receptor
- EMT
epithelial-to-mesenchymal transition
- FDA
Food and Drug Administration
- FASLG
FAS ligand
- HER2
human EGFR 2
- HLA
human leucocyte antigen
- HSPC
haematopoietic stem/progenitor cell
- ICD
immunogenic cell death
- IFI16
IFN-γ-inducible 16
- IFN
interferon
- IFNAR
IFN-α/β receptor
- IFNGR
IFN-γ receptor
- IKKε
IkB kinase ε
- IL
interleukin
- IPS-1
IFN-β promoter stimulator-1
- IRF
IFN regulatory factor
- ISG
IFN-stimulated gene
- ISGF3
IFN-stimulated gene factor 3
- JAK
Janus kinase
- LGP2
laboratory of genetics and physiology 2
- LPS
lipopolysaccharide
- Mal
MyD88 adaptor-like
- MAPK14
mitogen-activated protein kinase 14
- MAVS
mitochondrial antiviral signaling adaptor
- MCA
3′-methylcholanthrene
- MDA5
melanoma differentiation-associated protein 5
- MDSC
myeloid-derived suppressor cell
- MHC-I
major histocompatibility complex-I
- MyD88
myeloid differentiation primary response gene 88
- MX1
MX dynamin-like GTPase 1
- NF-κB
nuclear factor-κB
- NK
natural killer
- NLR
NOD-like receptor
- NOD2
NOD-containing protein 2
- OAS
2′-5′-oligoadenylate synthetase
- PAMP
pathogen-associated molecular pattern
- pDC
plasmacytoid DC
- PD-L1
programmed death-ligand 1
- PKR
protein kinase R
- POLR3
RNA polymerase-III
- PRR
pathogen recognition receptor
- p53/TP53
tumor protein p53
- RANK
receptor activator of NF-κB ligand
- RIG-I
retinoic acid-inducible gene-I
- RLR
RIG-I-like receptor
- ROS
reactive oxygen species
- SARM
sterile armadillo-motif-containing protein
- SOCS
suppressor of cytokine signaling
- STAT
signal transducer and activator of transcription
- STING
stimulator of IFN genes
- TAA
tumor-associated antigen
- TBK1
TANK-binding kinase 1
- TLR
Toll-like receptor
- TME
tumor microenvironment
- TMEM173
transmembrane protein 173
- TNF
tumor necrosis factor
- TRAIL
TNF-related apoptosis-inducing ligand
- TRAM
TRIF-related adaptor molecule
- Treg
regulatory T cell
- TREX1
3 prime repair exonuclease 1
- TRIF
TIR-domain containing adaptor protein-inducing IFN-β
- TYK2
tyrosine kinase-2
- VEGF
vascular endothelial growth factor
- VISA
virus-induced signaling adaptor.
Introduction
The sensing of altered-self, such as changes in tissue/organ homeostasis or integrity, and hence the need to detect and protect against potential danger (e.g., cellular stress, damage, or abnormal death), is upsetting the traditional view of immunity as a response to solely alien microbes and molecules.1 In particular, it is now clear that cancer cells, either transformed by foreign pathogens (e.g., human papillomavirus, hepatitis-B virus, Epstein-Barr virus, human T-lymphotropic virus-I, hepatitis-C virus, Kaposi's sarcoma herpesvirus or Helicobacter pylori) or totally aseptic, differ antigenically from their normal counterparts and, similar to virus-infected cells, emit danger signals to license the immune system. Such signals, best known as damage-associated molecular patterns (DAMPs), de facto favor the establishment of a productive and long-lasting immune response allowing to clear virus-infected cells (because they express virus-encoded proteins) and tumor cells (because they express tumor-associated antigens, TAAs). Intriguingly, antiviral and antitumor immune responses share common DAMPs, among which Type-I-interferons (IFNs) emerge as the primum movens for the sequential events bridging innate and cognate immunity.2
IFNs and their receptors are a subset of the class-2 α-helical cytokines that have been found in all vertebrates, although a systemic phylogenetic knowledge is lagging behind. Based on criteria such as their cellular source, their general biologic properties, their gene structure and the receptor through which they signal, IFNs have been categorized into three distinct families: Type-I, Type-II and Type-III. In humans, Type-I-IFNs consist of 13 partially homologous IFN-α cytokines, a single IFN-β and several not yet well characterized single gene products (IFN-ε, IFN-τ, IFN-κ, IFN-ω, IFN-δ and IFN-ζ) all of which are mostly non-glycosylated proteins of 165–200 aminoacids.3 The reason for the existence of multiple subtypes may be ascribed to differences in tissue-specific expression, the kinetic of production and the spectrum of biologic activities.4 Almost all cells in the body can produce Type-I-IFNs following the recognition of molecules, such as foreign and self-nucleic-acids and a minority of other non-nucleic-acids (collectively known as pathogen-associated molecular patterns, PAMPs), by the so-called pathogen recognition receptors (PRRs) located in the plasma membrane, cytosol or endosomal compartments.5 In the canonical Type-I-IFN signaling, Type-I-IFNs bind to a heterodimeric transmembrane receptor termed IFN-α/β receptor (IFNAR), in turn activating the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathway. This cascade induces the transcription of few hundreds of IFN-stimulated genes (ISGs), which steer the multiple facets of the cellular response.6 The Type-II-IFN family consists of a single IFN-γ glycosylated protein of 140 aminoacids, which is produced exclusively by cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells under immune and inflammatory stimuli. IFN-γ signals through the heterodimeric IFN-γ receptor (IFNGR), composed of IFNGR1 and IFNGR2 and characterized by a JAK1 binding domain and a STAT1 docking site.7 The Type-III-IFN family consists of the subtypes IFN-λ1, IFN-λ2, IFN-λ3 [also known as interleukin (IL)-29, IL-28A and IL-28B, respectively] and the newly identified IFN-λ4.8,9 Type-III-IFNs are structurally similar to IFN-γ but functionally identical to IFN-α/β. Only epithelial-like cells and, to a lesser extent, some immune cells respond to IFN-λs. Type-III-IFNs engage a receptor complex composed of the IFN-λR1 (or IL-28AR) and IL-10R2 chains to induce signaling pathways similar to those of Type-I-IFNs.8
This Review focuses on Type-I-IFNs and how pathogens and danger signals cross-regulate IFNAR signaling to mount immune defenses against virus-related and -unrelated diseases such as cancer. We conclude with open questions, future perspectives and implications for new clinical uses of Type-I-IFNs in oncology.
Pathways triggering production of Type-I-IFNs
As reported in the introduction, Type-I-IFNs can be produced by all nucleated cells in the body. The production of Type-I-IFNs is transient and occurs upon stimulation of an array of transmembrane and cytosolic PRRs with viral or other xenogeneic or autologous nucleic acids (Fig. 1). Currently identified PRRs include Toll-like receptors (TLRs), RIG-I-like receptor (RLRs), NOD-like receptors (NLRs) and DNA sensors.10 Although viral nucleic acids are the predominant ligands, other molecules, including viral proteins, bacterial lipopolysaccharide (LPS), lipoproteins, or endogenous ectopic proteins, can bind PRRs ultimately leading to Type-I-IFN production and innate immune responses.10
Figure 1.
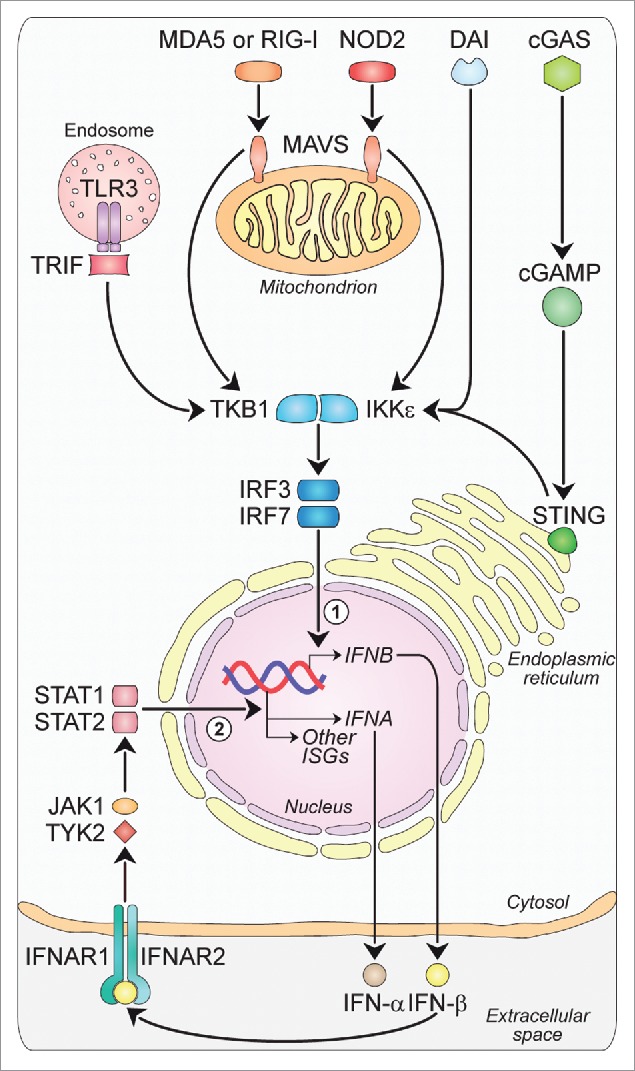
Major intracellular pathways leading to Type-I-IFN production. Families of sensors, known as PRRs, are available in the cells to detect viral and danger products, and induce the expression of Type-I-IFNs. One set of PRRs is localized in endosomal vesicles, while another set senses components in the cytoplasm. The endosome-associated TLR3 and the cytosolic MDA5, RIG-I and NOD2 sense double-stranded and single-stranded RNAs through the activation of adaptor molecules such as TRIF and MAVS, respectively. TRIF and MAVS in turn converge to activate the TBK1-IKKε kinase complex. This culminates in the activation of the transcription factors IRF3 and IRF7, which translocate to the nucleus and participate in the induction of a first wave of IFN-β production (1). IFN-β in turn acts in an autocrine/paracrine manner binding to the heterodimeric receptor IFNAR1-IFNAR2. This is followed by the activation of a JAK-STAT signaling pathway leading to a second wave of IFN-α production as well as to the transcription of other antiviral genes (2). Other PRRs sensing DNA are DAI and cGAS, with this last catalyzing the formation of ligands for STING upstream of the TBK1-IKKε complex, which finally drives the expression of IFNA and IFNB. cGAMP, cyclic guanosine monophosphate-adenosine monophosphate; cGAS: cyclic GMP-AMP synthase; DAI: DNA-dependent activator of IRFs; IFNs: interferons; IFNAR: IFN-α/β receptor; IKKε: IkB kinase ε; IRF: IFN regulatory factor; ISGs: IFN-stimulated genes; JAK: Janus kinase; MAVS: mitochondrial antiviral signaling adaptor; MDA5: melanoma differentiation-associated protein 5; NOD2: nucleotide oligomerization domain 2; PRRs: pathogen recognition receptors; RIG-I: retinoic acid-inducible gene-I; STAT: signal transducer and activator of transcription; STING: stimulator of IFN genes; TBK1: TANK-binding kinase 1; TLR3: Toll-like receptor 3; TRIF: TIR-domain containing adaptor protein-inducing IFN-β; TYK2, tyrosine kinase-2.
TLRs, the first PRRs identified, are transmembrane receptors either expressed on the cell surface or associated with intracellular vesicles.11 To date, 10 functional TLRs have been identified in humans, each of them detecting specific PAMPs. Briefly, lipoproteins are recognized by TLR1, TLR2 and TLR6; double-stranded and single-stranded RNAs by TLR3, TLR7 and TLR8; LPS by TLR4; flagellin by TLR5; and DNA by TLR9.11 Although recent evidence suggests that TLR10 could have either immune-stimulatory12 or immune-suppressive13 properties, its exact activating ligand(s) and function are not yet known. TLRs signal through five different adaptor molecules: myeloid differentiation primary response gene 88 (MyD88), MyD88 adaptor-like (Mal), TIR-domain containing adaptor protein-inducing IFN-β (TRIF), TRIF-related adaptor molecule (TRAM) and sterile armadillo-motif-containing protein (SARM).14 The association with these proteins recruits and activates the IkB kinase ε (IKKε)/TANK-binding kinase 1 (TBK1) complex (Fig. 1). This, in turn, is responsible for the phosphorylation and activation of the IFN regulatory factor (IRF)3, nuclear factor (NF)-κB, and activated protein (AP)1, all of them leading to the first-wave of IFN-β production. IFN-β then triggers the autocrine and paracrine expression of a related factor, IRF-7, which is responsible for a positive feedback loop initiating the synthesis of several IFN-α subtypes as the second wave of Type-I-IFNs.15
Among the cytosolic PRRs, RLRs are a family of DExD/H box RNA helicases (DDXs) sensing PAMPs within viral RNA. To date, three RLR members have been identified: (1) retinoic acid-inducible gene (RIG)-I, (2) melanoma differentiation-associated protein (MDA)5 and (3) laboratory of genetics and physiology (LGP)2. RIG-I and MDA5 detect a variety of viruses and share several structural similarities, including their organization into three domains: a tandem caspase activation and recruitment domain (CARD) to the N-terminal, a central DDX helicase region, and a repressor domain to the C-terminal that, in the case of RIG-I, is involved in autoregulation.16 Although presenting a similar organization, LGP2 lacks the N-terminal CARD and is currently thought to be a regulator of RIG-I and MDA5 rather than a bona fide PRR.17 Upon binding to double-stranded-RNAs, RLRs directly interact with a downstream molecule named independently by four different groups as mitochondrial antiviral signaling adaptor (MAVS),18 IFN-β promoter stimulator (IPS)-1,19 virus-induced signaling adaptor (VISA),20 and CARD adaptor-inducing IFN-β (CARDIF).21 As for TLRs, the association with this mitochondrial-resident protein via CARD induces Type-I-IFN production by the IKKε/TBK1 complex (Fig. 1).
NLRs are cytoplasmic PRRs with a tripartite structure consisting of a variable N-terminal effector domain, a middle nucleotide-binding domain and a C-terminal leucine-rich repeat domain.22 Among the more than 20 NLRs identified in humans so far,22 only the cytosolic molecular sensor NOD-containing protein 2 (NOD2) was clearly shown to recognize single-stranded RNAs leading to Type-I-IFN production through a mechanism dependent on MAVS and IRF3 activation (Fig. 1).23 Other NLRs are mainly described as regulators of the major histocompatibility complex-I (MHC-I),24 the inflammasome multiprotein complex assembly25 and some regulated cell death pathways (apoptosis, pyroptosis and pyronecrosis22). All these functions go beyond their sensing of DAMPs and PAMPs, which instead remains largely unknown.
The first described PRR for DNA, and still the only known endosomal-based DNA sensor, was TLR9.26 TLR9 is expressed preferentially in plasmacytoid dendritic cells (pDCs) and acts as a potent inducer of IFN-α via a signaling network dependent on MyD88 and IRF7.26 Moreover, DNA can end-up in the cytosol through several routes (e.g., intracellular pathogens, lysosome-internalized exogenous DNA from dead cells, or endogenous DNA replication debris) where it can be recognized by more than 10 cytosolic receptors.27 The search for cytosolic DNA sensors first led to the identification of the DNA-dependent activator of IRFs (DAI) (Fig. 1).28 When exogenously expressed in L929 murine fibroblasts, DAI increased Type-I-IFN production in a dose-dependent manner following stimulation by both B- and Z-form DNA.28 Similarly, knockdown of DAI with specific siRNAs impaired Type-I-IFN production in response to cytosolic DNA.28 RNA polymerase-III (POLR3), the second cytosolic DNA sensor discovered, was reported to use AT-rich and herpesvirus DNA as a template to produce 5′-triphosphate RNAs, which then induce Type-I-IFNs by activating RIG-I.29 However, POLR3 could not account for DAI-independent sensing of non-AT-rich DNA suggesting the existence of additional cytosolic DNA sensors. Remarkably, an adaptor molecule referred to as stimulator of IFN genes (STING) was identified as being crucial for recognizing cytoplasmic DNA and inducing innate immune responses to a variety of DNA pathogens even including certain RNA viruses.30 Nonetheless, despite the wealth of recent information on the mechanisms whereby STING contributes to signal Type-I-IFN induction, the upstream DNA-sensing events remain largely unknown. Recent evidence suggests that cytosolic DNA is perceived by the cyclic GMP-AMP synthase (cGAS), which then becomes catalytically active and generates the second messenger cyclic guanosine monophosphate-adenosine monophosphate (cGAMP) (Fig. 1). cGAMP in turn binds to STING stimulating its transit from the endoplasmic-reticulum to perinuclear endosomes where it triggers IRF3 activation via TBK1.30,31 Of note, STING-dependent Type-I-IFN production can also be activated by single-stranded DNA resulting from DNA damage or replication stress,32 by mitochondrial DNA released following apoptotic mitochondrial outer membrane permeabilization33 and possibly by retroelements not properly metabolized by the three prime repair exonuclease (TREX)1.34
Two essential mediators of distinct DNA-activated innate responses seem to be the PYHIN proteins absent in melanoma (AIM)2 and IFN-γ-inducible (IFI)16.35,36 Moreover, the helicases DDX3, DDX41, DHX9, DDX60, DDX1 and DHX36 were recently involved in DNA immune sensing through a pathway dependent on STING and TBK1.37 In particular, Liu and coworkers found that, in mouse splenic myeloid DCs with limited basal IFI16 expression, DDX41 was the initial sensor of cytoplasmic DNA inducing Type-I-IFNs and the subsequent IFI16 expression, with this latter operating as an amplifier of innate responses.37
Along with PAMPs and DAMPs, Type-I-IFNs can also be produced in response to rare physiologic stimuli such as colony stimulating factor (CSF)1,38 receptor activator of NF-κB ligand (RANK)39 and estrogens.40 More recently, an intriguing correlation between Type-I-IFNs and tumor protein p53 (TP53/p53) was reported.41 In sum, the absence of p53 was associated with extensive DNA hypomethylation that resulted in a massive transcription of normally silent retroelements and satellite DNA. The subsequent accumulation of these newly generated double-stranded RNA species eventually triggered a “suicidal” Type-I-IFN response.41
Overall, Type-I-IFN production is tightly regulated by major families of heterologous receptors engaged by diverse ligands during infectious and cancerous diseases. Each of the Type-I-IFN subtypes then induces a unique and partially overlapping set of ISGs able to act at different steps of virus and cancer life cycle.
ISGs: A complex net of host defenses
Type-I-IFN-mediated innate immune response is hardwired within genomes to provide a robust first-line of host defense and preserve homeostasis. Once secreted by cells, Type-I-IFNs bind to the same ubiquitous heterodimeric IFNAR1-IFNAR2 receptor.42 The assembly of IFNAR1, Type-I-IFN and IFNAR2 in a 1:1:1 stoichiometry seems to occur via a two-step process whereby Type-I-IFN first binds to one IFNAR and then promotes the recruitment of the second IFNAR without identified interactions between the two IFNARs.42 Once assembled, this ternary complex promotes the phosphorylation and activation of IFNAR1-associated tyrosine kinase (TYK)2 and IFNAR2-associated JAK1, which, in turn, phosphorylate cytosolic STAT1 and STAT2 (Fig. 1). This results in the formation of STAT1-STAT2 heterodimers that dissociate from receptors and migrate into the nucleus where they bind IRF9 to form the heterotrimeric transcriptional complex IFN-stimulated gene factor (ISGF)3. In the final step, ISGF3 binds to specific DNA response elements transactivating hundreds of ISGs.6 The nature and precise mechanisms through which ISGs prime cells for enhanced pathogen/danger detection and clearance, and then allow them to recover to normal function are not entirely elucidated. Recent evidence, reviewed in ref. 4, showed that Type-I-IFNs lead to cell-type and context-dependent patterns of ISG expression through a complex modulation of all seven STAT family members and other kinases (e.g., PI3K, p38, ERK and JNK) in addition to JAK. This may explain the complexity to regulate the pattern and magnitude of so many different biologic functions in so many different cells during infection, cancer and inflammation.4 For more insights in these issues refer to databases on signaling pathways and immune cell types such as Interferome (Interferome.org), Innate DB (http://www.innatedb.com) and the NIAIDs Systems Biology (http:/www.niaid.nih.gov/labsansresources/laboratories/about-laboratories/lsb/Pages/).
Similar to most cytokines, Type-I-IFN cascade is tightly regulated by positive and negative feedforward and feedback loops, which collectively ensure that the strength and duration of the response are effective yet limited, thereby preventing the toxic consequences of excessive/prolonged signaling.43 This balance is finely tuned by host factors operating at multiple levels, including signaling, transcription and translation. To give an example, many components of upstream PRR pathways (including receptors and IRFs) are ISGs.44 Type-I-IFNs are also reported to induce a network of inhibitors of their own signaling, such as members of the suppressor of cytokine signaling (SOCS) protein family.45 Overall, a complex net of signaling pathways makes proper use of the Type-I-IFN-ISG system to induce host protection while limiting tissue damage and preventing responses to self. Accumulating evidence indeed suggests that an aberrant activation of immunity by high levels of Type-I-IFNs contributes to the development of autoimmune diseases, such as systemic lupus erythematosus.46 This observation highlights the importance of understanding the mechanisms maintaining strict control over Type-I-IFN signaling to support the development of smart therapies for eradicating the danger and alleviate autoimmune diseases.
Type-I-IFNs in cancer
Type-I-IFNs are back in the oncological spotlight due to a greater understanding of their role in tumor generation, pathogenesis and treatment. Regardless of their source in the tumor microenvironment (TME), Type-I-IFNs have the potential to exert their opposed anti- and pro-tumorigenic effects acting directly on tumor cells and indirectly on immune infiltrating cells (Fig. 2).
Figure 2.
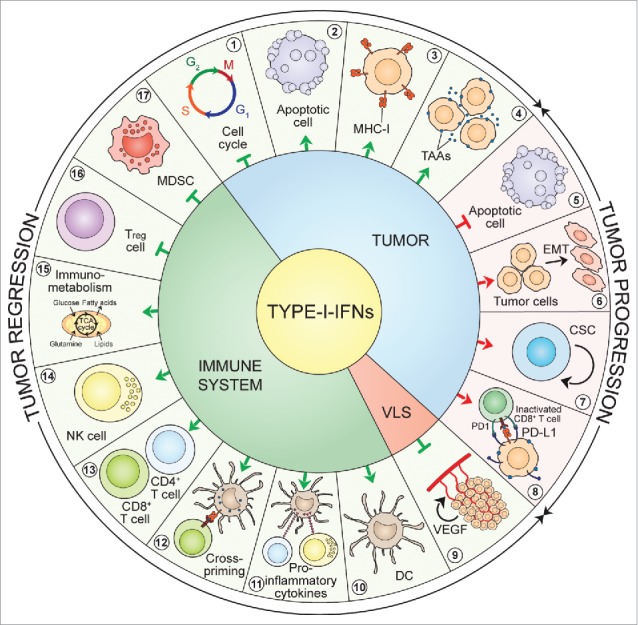
Type-I-IFN-triggered signals. Type-I-IFNs may favor tumor regression and/or tumor progression by acting on tumor cells, immune cells and endothelial cells via various mechanisms. First, acting on tumor cells Type-I-IFNs may promote either tumor regression, by inducing cell-cycle arrest (1), apoptosis (2) and enhanced immunogenicity through cell surface expression of MHC-I (3) and TAAs (4), or tumor progression by inducing resistance to apoptosis (5), EMT (6), tumor-cell stemness (7), and the upregulation of immune-inhibitory signals such as PD-L1 (8). Second, acting on the vascular and lymphatic system Type-I-IFNs inhibit angiogenesis through VEGF downregulation (9). Finally, acting on the immune system Type-I-IFNs may favor tumor regression by stimulating the maturation of DCs (10), promoting the release of pro-inflammatory cytokines (11), favoring CTL cross-priming (12), fostering the activation and survival of CD8+ and CD4+ T cells (13) and of NK cells (14), having a crucial role on core energetic metabolism regulation (15), and negatively regulating immune suppressive Treg cells (16) and MDSCs (17). CSC: cancer stem cell; DC: dendritic cell; EMT: epithelial-to-mesenchymal transition; IFNs: interferons; MDSC: myeloid-derived suppressor cell; MHC-I: major histocompatibility complex-I; NK: natural killer; PD1: programmed death 1; PD-L1: programmed death-ligand 1; TAAs: tumor-associated antigens; TCA: tricarboxylic acid; Treg: regulatory T cell; VEGF: vascular endothelial growth factor; VLS: vascular and lymphatic system.
Cancer-intrinsic effects of Type-I-IFNs
The cancer cell-intrinsic effectiveness of Type-I-IFNs is well documented in experimental animal systems and is reported to depend on specific cellular effects such as growth inhibition,47 modulation of apoptosis,48 differentiation,49 migration,49 alteration of cell surface expression of TAAs50 and promotion of the epithelial-to-mesenchymal transition (EMT).51 Type-I-IFNs are known to affect different phases of the mitotic cell-cycle (panel 1, Fig. 2) with the most common perturbation being the G1 arrest.51 In a seminal work, Balkwill et al. showed that in vitro treatment of human breast cancer cell lines with exogenous crude preparations of Type-I-IFNs had a direct anti-proliferative effect that was attributed to the prolongation of the cell cycle.52 Accordingly, observations from two independent studies showed that IFN-α inhibited the growth of human prostatic cancer cells and murine macrophages stalling the G1-S transition through the increased expression of the cyclin-dependent kinase inhibitor (CDKN)1A, best known as p21.53,54 Type-I-IFNs are also reported to induce other CDK inhibitors, including CDKN1B and CDKN2B (best known as p27 and p15, respectively), whose upregulation leads to cell-cycle blockade at the G1 phase.55 More recently, Katayama and colleagues provided evidence that, in human colon cancer cells, the anti-proliferative action of Type-I-IFNs relied on a p21-dependent prolongation of the S phase rather than a G1 block.56 Yet other nets involved in Type-I-IFN-induced cell-cycle arrest are believed to include the downregulation of the transcription factor MYC and the activation of mitogen-activated protein kinase (MAPK)14 or the adapter molecule CRK.57,58 Contrasting experimental findings indicate that Type-I-IFNs can either induce tumor cell death59 or protect cancer cells from chemical-induced apoptosis60 (panels 2 and 5, Fig. 2). This discrepancy may be ascribed to the degree of cellular differentiation, tumor-related factors and differences in the TME. Indeed, the administration of Type-I-IFNs was reported to modulate the two major apoptotic responses: the extrinsic or death receptor-mediated pathway and the intrinsic or mitochondrial pathway.48 Briefly, the former cascade requires ligation of cell-surface death receptors, such as the tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL), to activate the initiator caspase-8, whereas the latter requires the release of apoptotic factors such as cytochrome c1 from the mitochondria to activate other cytoplasmic initiator caspases. The ISGs involved in apoptosis include (but are not limited to) FAS, FAS ligand (FASLG), protein kinase R (PKR) and oligoadenylate synthetase (OAS), particularly the 9–2 isozyme (extensively reviewed in ref. 61).
The in vitro modulation of cultured tumor cells by Type-I-IFNs has also been documented. Some early reports showed that IFN-β has the ability to boost human leucocyte antigen (HLA)-class-I expression62 (panel 3, Fig. 2) and modulate the antigenic landscape of cultured melanoma cells63 (panel 4, Fig. 2). More recently, these discoveries were characterized by Dunn et al., who showed that IFN-β simultaneously augments TAAs (e.g., Melan-A/MART-1, gp100 and MAGE-A1) and HLA-class-I thus increasing the likelihood of improved immune recognition and cytotoxic killing of tumor targets, respectively.64
The EMT is a process by which epithelial cells lose their polarization and cell-cell contacts and undergo remarkable morphologic changes switching from an epithelial cobblestone phenotype to an elongated fibroblastic phenotype.65 The EMT provides for the evolution of cancer cells to the metastatic phenotype and contributes to their invasiveness, stemness and drug resistance.65 In a recent study, the IFN-α-inducible protein-27 was associated with the EMT marker vimentin in ovarian cancer66 (panel 6, Fig. 2). This phenomenon finally led to chemoresistant cells with a cancer stem cell (CSC) phenotype.66 CSCs are defined as the reservoir of a chemoresistant niche within the tumor and the driving force for tumor relapse.67,68 Mounting observations indicate a potential contribution of Type-I-IFN signaling in the generation and/or maintenance of CSCs (panel 7, Fig. 2). Indeed, IFN-α was reported to affect the migration and invasion of pancreatic ductal adenocarcinoma cells through the upregulation of specific CSC markers such as CD24, CD44 and CD133.69 In addition, it was recently shown that TLR3 stimulation on somatic cells caused global changes in the expression of epigenetic modifiers leading to enhanced chromatin remodeling, nuclear reprogramming, cell plasticity, pluripotentiality, transdifferentiation and even malignant transformation.70 In line with these data, experiments in breast cancer cells put in evidence that NF-κB and β-catenin signaling downstream of TLR3 promoted the enrichment of a subset of cells with CSC phenotype.71 Similarly, in the haematopoietic stem/progenitor cell (HSPC) compartment, chronic Type-I-IFN stimulation resulted in HSPC loss of quiescence and dysfunction.72 This phenomenon was mainly due to Type-I-IFN-induced accumulation of reactive oxygen species (ROS).73 Additional indirect proofs of the tumor growth promoting role of Type-I-IFNs come from recent studies showing that, in cancer cells, Type-I-IFNs upregulated the ISG programmed death-ligand (PD-L)174 (panel 8, Fig. 2). PD-L1 is a cell-surface molecule expressed by most tumor cells that mediates inhibitory signals toward CTLs and thus plays a major role in cancer immune-evasion through CTL exhaustion.75 It is tempting to speculate that sustained therapeutic responses could rely on the combination of Type-I-IFNs or Type-I-IFN-inducing therapies with antibodies targeting the PD1-PD-L1 axis. Accordingly, a recent study from Shen et al. demonstrated that the oncolytic vesicular stomatitis virus engineered to constitutively express IFN-β had significant anti-leukemia activity, which was further enhanced when combined with an anti-PD-L1 antibody.76 These observations lend further support to the double-edge sword of Type-I-IFNs in controlling tumor growth and promoting tumor escape. Further insights are needed to decipher the mechanisms through which Type-I-IFNs may paradoxically favor tumor progression, which will certainly have a great impact in the clinical use of Type-I-IFNs.
Cancer-extrinsic effects of Type-I-IFNs
In addition to the direct impact on cancer cells, Type-I-IFNs have extrinsic effects on tumors by regulating processes such as angiogenesis and immunity.77 Type-I-IFNs have been long recognized as powerful angiogenesis inhibitors. The effects of Type-I-IFNs on the vasculature have been mainly attributed to the downregulation of vascular endothelial growth factor (VEGF) expression as well as to the impairment of endothelial cell proliferation and migration78 (panel 9, Fig. 2). Seminal experimental findings from Schreiber's group strongly suggest that, although the immune system plays a major part in restraining the development of cancer, it may also promote the emergence of tumors that escape immune control.79 According to the immune-editing model, malignant cells, initially held in check by immune-surveillance means, can grow into clinically manifest tumors provided that (1) they lose the cancer molecular determinants that make them recognizable by immune-effectors (immune-selection), or (2) they actively counteract immune responses (immune-suppression).79 Immuno-editing consists of three phases: first, at an early stage, malignant cells are recognized and eradicated by immune-effector cells (elimination); second, at a later stage small tumors are still held in check by increasingly less proficient immune responses (equilibrium); and finally, neoplastic cells lose their antigenic properties or establish potent immune-suppressive networks, thus avoiding any control (escape).79 Most noteworthy, Dunn et al. proved that Type-I-IFNs intervene in all these three phases.80 They demonstrated that endogenously produced Type-I-IFNs were required, in immunocompetent mice, to reject highly immunogenic 3′-methylcholanthrene (MCA)-induced sarcomas and to prevent the outgrowth of primary carcinogen-induced tumors. Furthermore, they observed that several MCA-induced sarcomas from Ifnar1−/− mice were rejected in a T cell-dependent manner in wild-type mice, which suggests that tumors arising in the absence of Type-I-IFN responsiveness are more immunogenic than tumors growing in IFNAR competent hosts.80
The earliest indication that Type-I-IFNs could stimulate extrinsic antitumor effects was reported in a mouse model of lymphocytic leukemia, in which it was shown that survival rates were increased by administering crude (mixed-type) IFN preparations, irrespective of whether tumor cells themselves were intrinsically sensitive to the anti-proliferative actions of these IFN preparations.81 From then, an impressive number of instrumental studies in both mice and humans unveiled the plethora of mechanisms by which Type-I-IFNs act on immune cells to mount a strong antitumor response. In the early 1990s, Ferrantini and colleagues showed that highly metastatic Friend leukemia cells genetically modified to secrete IFN-α1 exhibited a marked loss of their tumorigenic potential when injected into syngeneic immunocompetent mice,82 and inhibited the growth of metastatic parental cells in transplantation assays mainly through CD8+ CTLs.83 Despite these encouraging data, the clinical development of Type-I-IFNs remained underappreciated for many years. In the past two decades, the findings that IFN-α induced the differentiation/activation of DCs (panel 10, Fig. 2) in both mice84 and humans85 have spurred the ideation of new immunotherapeutic regimens. Today, new attention is given to Type-I-IFNs as crucial factors bridging innate and adaptive immunity. Several studies support the importance of Type-I-IFNs as a stimulus for the production of various cytokines (e.g., TNF, IL-1, IL-6, IL-8, IL-12 and IL-18) by macrophages86 (panel 11, Fig. 2), and as factors that markedly affect DC-mediated TAA retention and cross-priming87 (panel 12, Fig. 2) and stimulate antibody-dependent cellular cytotoxicity on established B16 murine melanoma liver micrometastases.88 Furthermore, Type-I-IFNs were reported to play a major role in the development and differentiation of the Th1 subset, as well as in the generation, activity, expansion and long-term survival of CTLs89 (panel 13, Fig. 2). Type-I-IFNs are also responsible for the activation of tumoricidal NK cells (panel 14, Fig. 2), which represent one of the host key mechanisms to preempt tumor growth.90 More recently, the role of Type-I-IFNs in the immunometabolism - which is an emerging field investigating the interplay between immunological and metabolic processes91 - gained increasing appreciation (panel 15, Fig. 2). A substantial number of evidence indicates that signaling downstream of PRRs induces changes in core metabolism of DCs and macrophages, which are crucial in shaping their function and fate.91 In macrophages, Type-I-IFNs downstream of TLR3 induced a shift in the balance of lipid metabolism away from de novo cholesterol and fatty-acid synthesis in favor of the uptake of exogenous lipids.92 This immunometabolic circuit is critical for host immune responses. In line with this discovery, TLR9 stimulation in pDCs led to an autocrine IFNAR signaling resulting in an increased fatty-acids oxidation and oxidative phosphorylation, which is key for pDC immune functions.93 In addition, fasting or the administration of caloric restriction mimetics has been shown to improve the efficacy of immunogenic chemotherapy correlating with the depletion of immunosuppressive regulatory T (Treg) cells from the TME.94 Notably, Type-I-IFNs are known to negatively regulate the proliferation and activity of Treg cells (panel 16, Fig. 2) and other immune-suppressive cells such as myeloid-derived suppressor cells (MDSCs; panel 17, Fig. 2).77 Undoubtedly, understanding the multilevel interactions between metabolic, immunologic and Type-I-IFN nets will offer additional tools to manage beneficial and detrimental Type-I-IFN immune effects and reshape the way Type-I-IFN-IFNAR axis can be exploited therapeutically during infection and cancer.
The role of Type-I-IFNs in anticancer therapy
Although soon after their discovery the antiviral activity of Type-I-IFNs attracted widest interest, the first US Food and Drug Administration (FDA) approval for IFN-α2, in 1986, was for cancer treatment (Fig. 3). Even before recombinant IFNs were available, reduction of disease morbidities with partially purified IFN-α was reported in several studies performed in patients with hairy-cell leukemia and chronic myelogenous leukemia.95,96 In both cases, however, over time more effective therapeutic regimens than IFN have been devised (e.g., the targeted inhibitor of the activated BCR-ABL tyrosine kinase Imatinib97). In following clinical studies, the therapeutic effectiveness of IFN-α2-either as unmodified recombinant proteins or pegylated variants-in inducing at least partial disease regression was reported for other hematological and solid tumors including myelomas, lymphomas, melanomas, Kaposi's sarcoma and renal-cell and bladder carcinoma.98 To date, IFN-α2 is still commonly used combined with IL-2 in immunotherapeutic regimens for metastatic renal-cell carcinomas and cutaneous melanoma.99,100 In addition, more than 100 clinical trials are currently underway worldwide using IFN-α2 as monotherapy or in combination regimens for both hematological and solid malignancies (for further details, please refer to https://clinicaltrials.gov/ and ref. 101).
Figure 3.

Timeline of IFN discovery and clinical use. The discovery of IFNs evolved from studies of viral interference beginning in 1950. Since then, great attention has been devoted to the molecular understanding and clinical use of IFNs for virus-related and -unrelated malignancies. DC: dendritic cell; FDA: Food and Drug Administration; HBV: hepatitis B virus; HCV: hepatitis C virus; HIV: human immunodeficiency virus; ICD: immunogenic cell death; IFNs: interferons; IFNAR: IFN-α/β receptor; IFNGR: IFN-γ receptor; IPS-1, IFN-beta promoter stimulator-1; IRF: IFN regulatory factor; ISG: IFN-stimulated gene; ISGF3: IFN-stimulated gene factor 3; JAK: Janus kinase; MDA5: melanoma differentiation-associated protein 5; RIG-I: retinoic acid-inducible gene-I; SCID: severe combined immunodeficiency; STAT: signal transducer and activator of transcription; TLR3: Toll-like receptor 3.
A wide range of conventional chemotherapy, radiotherapy and immunotherapy, including oncolytic virotherapy, currently licensed for use in humans are particularly successful if they induce tumor-targeting immune responses.102,103 The current view is that therapeutic agents must induce a sort of “viral mimicry”, i.e., a combination of stress signals that are usually linked to viral infection, such as Type-I-IFNs, and are believed to contribute to their clinical effectiveness. We recently showed that Type-I-IFNs lie at the nexus that controls immunogenic cell death (ICD) and constitutes a hallmark of successful chemotherapy.2 In particular, we showed that the treatment of various tumor types (e.g., MCA205-fibrosarcomas and AT3-breast carcinoma) with anthracyclines or oxaliplatin gave rise to the rapid production of Type-I-IFNs, thus mimicking the immune reactions evoked by viruses. We also elucidated the mechanism of Type-I-IFN-mediated ICD demonstrating that (1) hit dying cancer cells emit self-nucleic-acids (especially single-stranded RNAs) in the TME, which are sensed by TLR3 on surrounding yet viable cells and (2) released Type-I-IFNs act as the primum movens for the sequential events bridging innate and cognate antiviral immunity through a specific ISG signature that includes soluble chemotactic mediators such as the C-X-C motif chemokine ligand (CXCL)10. This is crucial for the recruitment, selection and differentiation/maturation of engulfing cells thus dictating the immunogenic outcome of cell death. Corroborating this evidence, the efficacy of anthracyclines was lost upon co-administration of anti-IFNAR or anti-IFN-α/β neutralizing antibodies.2 Importantly, in breast cancer patients, increased expression levels of the ISG MX dynamin-like GTPase (MX)1 predicted the likelihood of response to anthracycline-based treatment in neoadjuvant and adjuvant settings.2 In previous studies, Type-I-IFNs were described as crucial mediators of the off-target immunomodulatory effects of cyclophosphamide, an alkylating agent inducing ICD104 responsible for the expansion of memory CD4+ and CD8+ T cells105 as well as of DCs.104 In patients with hematological malignancies, the administration of high-dose cyclophosphamide induced a rapid, transient and broad transcriptional modulation on peripheral blood mononuclear cells resulting in DNA damage, cell death and, noticeably, a Type-I-IFN signature.106 This promoted the establishment of a systemic sterile inflammatory response characterized by the release of endogenous adjuvant signals able to enhance the efficacy of immunotherapy.106 Similar to chemotherapy, radiation therapy was also reported to increase the levels of Type-I-IFNs and CXCL10 in the TME.107 In this study, CXCL10 was shown to promote tumor CD8+ T cell-homing and cytolytic activity. Subsequent observations revealed that radiation-mediated antitumor immunity in immunogenic tumors requires a functional cytosolic DNA-sensing pathway upstream of Type-I-IFNs.108 Accordingly, Hartlova and colleagues recently found that in the absence of ataxia-telangiectasia mutated (ATM, which is an apical component of the DNA damage response), the accumulation of DNA lesions generated spontaneously or provoked by irradiation induced Type-I-IFNs via STING-mediated signaling.32 Type-I-IFNs in turn primed the innate immune system for a rapid and amplified response to microbial and environmental threats. In addition, Type-I-IFNs boosted the antineoplastic activity of antibodies specific for oncogenic receptors, such as epidermal growth factor receptor (EGFR) and human EGFR (HER)2, mobilizing DCs to cross-present TAAs to CTLs.74 However, despite these observations strongly support the antitumor and immune-stimulatory effects of Type-I-IFNs, paradoxical proofs of a dichotomous, detrimental tumor growth-promoting role for these cytokines are also reported. In this context, some harmful effects seem to depend on the ability to induce immune-checkpoint pathways as a major mechanism of immune resistance, particularly against CTLs specific for TAAs. As reported above, Type-I-IFNs upregulate PD-L1 in tumor cells,2, 74 which can lead to T cell exhaustion.109 It remains a central goal of studies on tumor immunity to elucidate the multitude of molecular nets activated by Type-I-IFNs. Big issues to solve are when and through which pathways Type-I-IFNs counteract or promote tumor growth. These insights will likely pave the way to more effective IFN-based immunotherapies.
Conclusions and perspectives
Type-I-IFNs are among the most pleiotropic cytokines and are produced and sensed by almost every cell type in the body. The discovery of Type-I-IFN role in cancer immune-surveillance at first, and cancer immune-editing later, made these cytokines and the immune sensing networks that drive their production very attractive for deeper investigation in preclinical and clinical contexts. As cancer-related genomic information is constantly published, it is emerging that Type-I-IFNs can be produced by, and act on, both malignant and immune cells, thus eliciting immune responses via tumor cell-intrinsic or -extrinsic means. Type-I-IFNs, either naturally produced, exogenously administered or induced by chemotherapy, radiotherapy or oncolytic virotherapy exert all biologic effects through the action of ISGs. Therefore, efforts to decipher the specific functions of individual ISGs on the reciprocal crosstalk between cancer cells and immune cells may likely help fulfill IFN therapeutic efficacy and identify predictive biomarkers of response. Taken into account the dual role of Type-I-IFNs in containing and favoring tumor growth, it will be important to understand which subtype of, at which time point and through which mechanisms Type-I-IFNs cease to be immune effectors and flip to become immune suppressors and CSC promoters. The limited efficacy of Type-I-IFNs in cancer medicine may likely reflect this gap of knowledge.
Matter-of-factly, Type-I-IFNs have more than reached the potential envisioned by early discovering virologists, however answering these questions will certainly have a tremendous impact on tumor immunology and biomedicine.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
RDM is supported by the AIRC (5 per Mille #9979), and the Fondo per gli Investimenti della Ricerca di Base, (FIRB #RBAP11WCRZ-005 U54 2010). IV is supported by the AIRC (MFAG 2013 #14641), Ministero Italiano della Salute (RF_GR-2011–02351355), and the Programma per i Giovani Ricercatori “Rita Levi Montalcini” 2010. AS is supported by the AIRC (Start-Up 2016 #18418) and Ministero Italiano della Salute (RF_ GR-2013–02357273).
References
- 1.Matzinger P. The danger model: a renewed sense of self. Science 2002; 296:301-5; PMID:11951032; https://doi.org/ 10.1126/science.1071059 [DOI] [PubMed] [Google Scholar]
- 2.Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remédios C et al.. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med 2014; 20:1301-9; PMID:25344738; https://doi.org/ 10.1038/nm.3708 [DOI] [PubMed] [Google Scholar]
- 3.Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev 2004; 202:8-32; PMID:15546383; https://doi.org/ 10.1111/j.0105-2896.2004.00204.x [DOI] [PubMed] [Google Scholar]
- 4.van Boxel-Dezaire AH, Rani MR, Stark GR. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity 2006; 25:361-72; PMID:16979568; https://doi.org/ 10.1016/j.immuni.2006.08.014 [DOI] [PubMed] [Google Scholar]
- 5.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell 2010; 140:805-20; PMID:20303872; https://doi.org/ 10.1016/j.cell.2010.01.022 [DOI] [PubMed] [Google Scholar]
- 6.Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol 2014; 32:513-45; PMID:24555472; https://doi.org/ 10.1146/annurev-immunol-032713-120231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bach EA, Aguet M, Schreiber RD. The IFN gamma receptor: a paradigm for cytokine receptor signaling. Annu Rev Immunol 1997; 15:563-91; PMID:9143700; https://doi.org/ 10.1146/annurev.immunol.15.1.563 [DOI] [PubMed] [Google Scholar]
- 8.Lazear HM, Nice TJ, Diamond MS. Interferon-lambda: immune functions at barrier surfaces and beyond. Immunity 2015; 43:15-28; PMID:26200010; https://doi.org/ 10.1016/j.immuni.2015.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O'Brien TR, Prokunina-Olsson L, Donnelly RP. IFN-lambda4: the paradoxical new member of the interferon lambda family. J Interferon Cytokine Res 2014; 34:829-38; PMID:24786669; https://doi.org/ 10.1089/jir.2013.0136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol 2011; 30:16-34; PMID:21235323; https://doi.org/ 10.3109/08830185.2010.529976 [DOI] [PubMed] [Google Scholar]
- 11.Beutler BA. TLRs and innate immunity. Blood 2009; 113:1399-407; PMID:18757776; https://doi.org/ 10.1182/blood-2008-07-019307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee SM, Kok KH, Jaume M, Cheung TK, Yip TF, Lai JC, Guan Y, Webster RG, Jin DY, Peiris JS. Toll-like receptor 10 is involved in induction of innate immune responses to influenza virus infection. Proc Natl Acad Sci U S A 2014; 111:3793-8; PMID:24567377; https://doi.org/ 10.1073/pnas.1324266111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oosting M, Cheng SC, Bolscher JM, Vestering-Stenger R, Plantinga TS, Verschueren IC, Arts P, Garritsen A, van Eenennaam H, Sturm P et al.. Human TLR10 is an anti-inflammatory pattern-recognition receptor. Proc Natl Acad Sci U S A 2014; 111:E4478-84; PMID:25288745; https://doi.org/ 10.1073/pnas.1410293111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O'Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol 2007; 7:353-64; PMID:17457343; https://doi.org/ 10.1038/nri2079 [DOI] [PubMed] [Google Scholar]
- 15.Sato M, Hata N, Asagiri M, Nakaya T, Taniguchi T, Tanaka N. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett 1998; 441:106-10; PMID:9877175; https://doi.org/ 10.1016/S0014-5793(98)01514-2 [DOI] [PubMed] [Google Scholar]
- 16.Loo YM, Gale M Jr. Immune signaling by RIG-I-like receptors. Immunity 2011; 34:680-92; PMID:21616437; https://doi.org/ 10.1016/j.immuni.2011.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo YM, Jr Gale M, Akira S et al.. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol 2005; 175:2851-8; PMID:16116171; https://doi.org/ 10.4049/jimmunol.175.5.2851 [DOI] [PubMed] [Google Scholar]
- 18.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005; 122:669-82; PMID:16125763; https://doi.org/ 10.1016/j.cell.2005.08.012 [DOI] [PubMed] [Google Scholar]
- 19.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol 2005; 6:981-8; PMID:16127453; https://doi.org/ 10.1038/ni1243 [DOI] [PubMed] [Google Scholar]
- 20.Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell 2005; 19:727-40; PMID:16153868; https://doi.org/ 10.1016/j.molcel.2005.08.014 [DOI] [PubMed] [Google Scholar]
- 21.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005; 437:1167-72; PMID:16177806; https://doi.org/ 10.1038/nature04193 [DOI] [PubMed] [Google Scholar]
- 22.Ting JP, Willingham SB, Bergstralh DT. NLRs at the intersection of cell death and immunity. Nat Rev Immunol 2008; 8:372-9; PMID:18362948; https://doi.org/ 10.1038/nri2296 [DOI] [PubMed] [Google Scholar]
- 23.Sabbah A, Chang TH, Harnack R, Frohlich V, Tominaga K, Dube PH, Xiang Y, Bose S. Activation of innate immune antiviral responses by Nod2. Nat Immunol 2009; 10:1073-80; PMID:19701189; https://doi.org/ 10.1038/ni.1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kobayashi KS, van den Elsen PJ. NLRC5: a key regulator of MHC class I-dependent immune responses. Nat Rev Immunol 2012; 12:813-20; PMID:23175229; https://doi.org/ 10.1038/nri3339 [DOI] [PubMed] [Google Scholar]
- 25.Ogura Y, Sutterwala FS, Flavell RA. The inflammasome: first line of the immune response to cell stress. Cell 2006; 126:659-62; PMID:16923387; https://doi.org/ 10.1016/j.cell.2006.08.002 [DOI] [PubMed] [Google Scholar]
- 26.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K et al.. A Toll-like receptor recognizes bacterial DNA. Nature 2000; 408:740-5; PMID:11130078; https://doi.org/ 10.1038/35047123 [DOI] [PubMed] [Google Scholar]
- 27.Ishii KJ, Coban C, Kato H, Takahashi K, Torii Y, Takeshita F, Ludwig H, Sutter G, Suzuki K, Hemmi H et al.. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat Immunol 2006; 7:40-8; PMID:16286919; https://doi.org/ 10.1038/ni1282 [DOI] [PubMed] [Google Scholar]
- 28.Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K et al.. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007; 448:501-5; PMID:17618271; https://doi.org/ 10.1038/nature06013 [DOI] [PubMed] [Google Scholar]
- 29.Chiu YH, Macmillan JB, Chen ZJ. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 2009; 138:576-91; PMID:19631370; https://doi.org/ 10.1016/j.cell.2009.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Corrales L, McWhirter SM, Dubensky TW Jr., Gajewski TF. The host STING pathway at the interface of cancer and immunity. J Clin Invest 2016; 126:2404-11; PMID:27367184; https://doi.org/ 10.1172/JCI86892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013; 339:786-91; PMID:23258413; https://doi.org/ 10.1126/science.1232458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hartlova A, Erttmann SF, Raffi FA, Schmalz AM, Resch U, Anugula S, Lienenklaus S, Nilsson LM, Kröger A, Nilsson JA et al.. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 2015; 42:332-43; PMID:25692705; https://doi.org/ 10.1016/j.immuni.2015.01.012 [DOI] [PubMed] [Google Scholar]
- 33.White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, van Delft MF, Bedoui S, Lessene G, Ritchie ME et al.. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 2014; 159:1549-62; PMID:25525874; https://doi.org/ 10.1016/j.cell.2014.11.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 2008; 134:587-98; PMID:18724932; https://doi.org/ 10.1016/j.cell.2008.06.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009; 458:509-13; PMID:19158676; https://doi.org/ 10.1038/nature07710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS et al.. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol 2010; 11:997-1004; PMID:20890285; https://doi.org/ 10.1038/ni.1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Z, Yuan B, Bao M, Lu N, Kim T, Liu YJ. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol 2011; 12:959-65; PMID:21892174; https://doi.org/ 10.1038/ni.2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hamilton JA, Whitty GA, Kola I, Hertzog PJ. Endogenous IFN-alpha beta suppresses colony-stimulating factor (CSF)-1-stimulated macrophage DNA synthesis and mediates inhibitory effects of lipopolysaccharide and TNF-alpha. J Immunol 1996; 156:2553-7; PMID:8786318 [PubMed] [Google Scholar]
- 39.Takayanagi H, Kim S, Matsuo K, Suzuki H, Suzuki T, Sato K, Yokochi T, Oda H, Nakamura K, Ida N et al.. RANKL maintains bone homeostasis through c-Fos-dependent induction of interferon-beta. Nature 2002; 416:744-9; PMID:11961557; https://doi.org/ 10.1038/416744a [DOI] [PubMed] [Google Scholar]
- 40.Fung KY, Mangan NE, Cumming H, Horvat JC, Mayall JR, Stifter SA, De Weerd N, Roisman LC, Rossjohn J, Robertson SA et al.. Interferon-epsilon protects the female reproductive tract from viral and bacterial infection. Science 2013; 339:1088-92; PMID:23449591; https://doi.org/ 10.1126/science.1233321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leonova KI, Brodsky L, Lipchick B, Pal M, Novototskaya L, Chenchik AA, Sen GC, Komarova EA, Gudkov AV. p53 cooperates with DNA methylation and a suicidal interferon response to maintain epigenetic silencing of repeats and noncoding RNAs. Proc Natl Acad Sci U S A 2013; 110:E89-98; PMID:23236145; https://doi.org/ 10.1073/pnas.1216922110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uze G, Schreiber G, Piehler J, Pellegrini S. The receptor of the type I interferon family. Curr Top Microbiol Immunol 2007; 316:71-95; PMID:17969444 [DOI] [PubMed] [Google Scholar]
- 43.Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol 2014; 14:36-49; PMID:24362405; https://doi.org/ 10.1038/nri3581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khoo JJ, Forster S, Mansell A. Toll-like receptors as interferon-regulated genes and their role in disease. J Interferon Cytokine Res 2011; 31:13-25; PMID:21198355; https://doi.org/ 10.1089/jir.2010.0095 [DOI] [PubMed] [Google Scholar]
- 45.Piganis RA, De Weerd NA, Gould JA, Schindler CW, Mansell A, Nicholson SE, Hertzog PJ. Suppressor of cytokine signaling (SOCS) 1 inhibits type I interferon (IFN) signaling via the interferon alpha receptor (IFNAR1)-associated tyrosine kinase Tyk2. J Biol Chem 2011; 286:33811-8; PMID:21757742; https://doi.org/ 10.1074/jbc.M111.270207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science 2001; 294:1540-3; PMID:11711679; https://doi.org/ 10.1126/science.1064890 [DOI] [PubMed] [Google Scholar]
- 47.Gresser I, Maury C, Bandu MT, Foiret D, Trojan J, Uriel J. Inhibitory effect of mouse interferon on the growth of an embryonal carcinoma in mice. J Interferon Res 1984; 4:375-81; PMID:6208297; https://doi.org/ 10.1089/jir.1984.4.375 [DOI] [PubMed] [Google Scholar]
- 48.Thyrell L, Erickson S, Zhivotovsky B, Pokrovskaja K, Sangfelt O, Castro J, Einhorn S, Grandér D. Mechanisms of Interferon-alpha induced apoptosis in malignant cells. Oncogene 2002; 21:1251-62; PMID:11850845; https://doi.org/ 10.1038/sj.onc.1205179 [DOI] [PubMed] [Google Scholar]
- 49.Jensen KE, Neal AL, Owens RE, Warren J. Interferon responses of chick embryo fibroblasts to nucleic acids and related compounds. Nature 1963; 200:433-4; PMID:14076723; https://doi.org/ 10.1038/200433a0 [DOI] [PubMed] [Google Scholar]
- 50.Lindahl P, Leary P, Gresser I. Enhancement by interferon of the expression of surface antigens on murine leukemia L 1210 cells. Proc Natl Acad Sci U S A 1973; 70:2785-8; PMID:4517933; https://doi.org/ 10.1073/pnas.70.10.2785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kudryavets YI, Bezdenezhnykh NO, Lykhova OO, Semesiuk NI, Vorontsova AL. The role of interferon as a modifier of epithelial-mesenchymal transition in tumor cells. Exp Oncol 2011; 33:178-81; PMID:21956475 [PubMed] [Google Scholar]
- 52.Balkwill F, Watling D, Taylor-Papadimitriou J. Inhibition by lymphoblastoid interferon of growth of cells derived from the human breast. Int J Cancer 1978; 22:258-65; PMID:700890; https://doi.org/ 10.1002/ijc.2910220307 [DOI] [PubMed] [Google Scholar]
- 53.Hobeika AC, Subramaniam PS, Johnson HM. IFNalpha induces the expression of the cyclin-dependent kinase inhibitor p21 in human prostate cancer cells. Oncogene 1997; 14:1165-70; PMID:9121765; https://doi.org/ 10.1038/sj.onc.1200939 [DOI] [PubMed] [Google Scholar]
- 54.Matsuoka M, Tani K, Asano S. Interferon-alpha-induced G1 phase arrest through up-regulated expression of CDK inhibitors, p19Ink4D and p21Cip1 in mouse macrophages. Oncogene 1998; 16:2075-86; PMID:9572488; https://doi.org/ 10.1038/sj.onc.1201745 [DOI] [PubMed] [Google Scholar]
- 55.Sangfelt O, Erickson S, Grander D. Mechanisms of interferon-induced cell cycle arrest. Front Biosci 2000; 5:D479-87; PMID:10762599; https://doi.org/ 10.2741/Sangfelt [DOI] [PubMed] [Google Scholar]
- 56.Katayama T, Nakanishi K, Nishihara H, Kamiyama N, Nakagawa T, Kamiyama T, Iseki K, Tanaka S, Todo S. Type I interferon prolongs cell cycle progression via p21WAF1/CIP1 induction in human colon cancer cells. Int J Oncol 2007; 31:613-20; PMID:17671689; https://doi.org/ 10.3892/ijo.31.3.613 [DOI] [PubMed] [Google Scholar]
- 57.Einat M, Resnitzky D, Kimchi A. Close link between reduction of c-myc expression by interferon and, G0/G1 arrest. Nature 1985; 313:597-600; PMID:3881681; https://doi.org/ 10.1038/313597a0 [DOI] [PubMed] [Google Scholar]
- 58.Lu M, Zhang W, Li Y, Berenzon D, Wang X, Wang J, Mascarenhas J, Xu M, Hoffman R. Interferon-alpha targets JAK2V617F-positive hematopoietic progenitor cells and acts through the p38 MAPK pathway. Exp Hematol 2010; 38:472-80; PMID:20303384; https://doi.org/ 10.1016/j.exphem.2010.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sangfelt Strander H. Apoptosis and cell growth inhibition as antitumor effector functions of interferons. Med Oncol 2001; 18:3-14; PMID:11778967; https://doi.org/ 10.1385/MO:18:1:3 [DOI] [PubMed] [Google Scholar]
- 60.Jewell AP, Worman CP, Lydyard PM, Yong KL, Giles FJ, Goldstone AH. Interferon-alpha up-regulates bcl-2 expression and protects B-CLL cells from apoptosis in vitro and in vivo. Br J Haematol 1994; 88:268-74; PMID:7803269; https://doi.org/ 10.1111/j.1365-2141.1994.tb05017.x [DOI] [PubMed] [Google Scholar]
- 61.Chawla-Sarkar M, Lindner DJ, Liu YF, Williams BR, Sen GC, Silverman RH, Borden EC. Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis 2003; 8:237-49; PMID:12766484; https://doi.org/ 10.1023/A:1023668705040 [DOI] [PubMed] [Google Scholar]
- 62.Lanza L, Peirano L, Bosco O, Contini P, Filaci G, Setti M, Puppo F, Indiveri F, Scudeletti M. Interferons up-regulate with different potency HLA class I antigen expression in M14 human melanoma cell line. Possible interaction with glucocorticoid hormones. Cancer Immunol Immunother 1995; 41:23-8; PMID:7543821; https://doi.org/ 10.1007/BF01788956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Giacomini P, Fraioli R, Nistico P, Tecce R, Nicotra MR, Di Filippo F, Fisher PB, Natali PG. Modulation of the antigenic phenotype of early-passage human melanoma cells derived from multiple autologous metastases by recombinant human leukocyte, fibroblast and immune interferon. Int J Cancer 1990; 46:539-45; PMID:2118485; https://doi.org/ 10.1002/ijc.2910460334 [DOI] [PubMed] [Google Scholar]
- 64.Dunn IS, Haggerty TJ, Kono M, Durda PJ, Butera D, Macdonald DB, Benson EM, Rose LB, Kurnick JT. Enhancement of human melanoma antigen expression by IFN-beta. J Immunol 2007; 179:2134-42; PMID:17675472; https://doi.org/ 10.4049/jimmunol.179.4.2134 [DOI] [PubMed] [Google Scholar]
- 65.Nieto MA, Huang RY, Jackson RA, Thiery JP. Emt: 2016. Cell 2016; 166:21-45; PMID:27368099; https://doi.org/ 10.1016/j.cell.2016.06.028 [DOI] [PubMed] [Google Scholar]
- 66.Li S, Xie Y, Zhang W, Gao J, Wang M, Zheng G, Yin X, Xia H, Tao X. Interferon alpha-inducible protein 27 promotes epithelial-mesenchymal transition and induces ovarian tumorigenicity and stemness. J Surg Res 2015; 193:255-64; PMID:25103640; https://doi.org/ 10.1016/j.jss.2014.06.055 [DOI] [PubMed] [Google Scholar]
- 67.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer 2005; 5:275-84; PMID:15803154; https://doi.org/ 10.1038/nrc1590 [DOI] [PubMed] [Google Scholar]
- 68.Manic G, Signore M, Sistigu A, Russo G, Corradi F, Siteni S et al.. CHK1-targeted therapy to deplete DNA replication-stressed, p53-deficient, hyperdiploid colorectal cancer stem cells. Gut 2017; PMID:28389531; https://doi.org/ 10.1136/gutjnl-2016-312623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhu Y, Karakhanova S, Huang X, Deng SP, Werner J, Bazhin AV. Influence of interferon-alpha on the expression of the cancer stem cell markers in pancreatic carcinoma cells. Exp Cell Res 2014; 324:146-56; PMID:24726912; https://doi.org/ 10.1016/j.yexcr.2014.03.020 [DOI] [PubMed] [Google Scholar]
- 70.Lee J, Sayed N, Hunter A, Au KF, Wong WH, Mocarski ES, Pera RR, Yakubov E, Cooke JP. Activation of innate immunity is required for efficient nuclear reprogramming. Cell 2012; 151:547-58; PMID:23101625; https://doi.org/ 10.1016/j.cell.2012.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jia D, Yang W, Li L, Liu H, Tan Y, Ooi S, Chi L, Filion LG, Figeys D, Wang L. beta-Catenin and NF-kappaB co-activation triggered by TLR3 stimulation facilitates stem cell-like phenotypes in breast cancer. Cell Death Differ 2015; 22:298-310; PMID:25257174; https://doi.org/ 10.1038/cdd.2014.145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, Trumpp A. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature 2009; 458:904-8; PMID:19212321; https://doi.org/ 10.1038/nature07815 [DOI] [PubMed] [Google Scholar]
- 73.Tasdogan A, Kumar S, Allies G, Bausinger J, Beckel F, Hofemeister H, Mulaw M, Madan V, Scharfetter-Kochanek K, Feuring-Buske M et al.. DNA damage-induced HSPC malfunction depends on ROS accumulation downstream of IFN-1 signaling and bid mobilization. Cell Stem Cell 2016; 19:752-67; PMID:27641306; https://doi.org/ 10.1016/j.stem.2016.08.007 [DOI] [PubMed] [Google Scholar]
- 74.Yang X, Zhang X, Fu ML, Weichselbaum RR, Gajewski TF, Guo Y, Fu YX. Targeting the tumor microenvironment with interferon-beta bridges innate and adaptive immune responses. Cancer Cell 2014; 25:37-48; PMID:24434209; https://doi.org/ 10.1016/j.ccr.2013.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Blank C, Mackensen A. Contribution of the PD-L1/PD-1 pathway to T-cell exhaustion: an update on implications for chronic infections and tumor evasion. Cancer Immunol Immunother 2007; 56:739-45; PMID:17195077; https://doi.org/ 10.1007/s00262-006-0272-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shen W, Patnaik MM, Ruiz A, Russell SJ, Peng KW. Immunovirotherapy with vesicular stomatitis virus and PD-L1 blockade enhances therapeutic outcome in murine acute myeloid leukemia. Blood 2016; 127:1449-58; PMID:26712908; https://doi.org/ 10.1182/blood-2015-06-652503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer 2016; 16:131-44; PMID:26911188; https://doi.org/ 10.1038/nrc.2016.14 [DOI] [PubMed] [Google Scholar]
- 78.Indraccolo S. Interferon-alpha as angiogenesis inhibitor: learning from tumor models. Autoimmunity 2010; 43:244-7; PMID:20166871; https://doi.org/ 10.3109/08916930903510963 [DOI] [PubMed] [Google Scholar]
- 79.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science 2011; 331:1565-70; PMID:21436444; https://doi.org/ 10.1126/science.1203486 [DOI] [PubMed] [Google Scholar]
- 80.Dunn GP, Bruce AT, Sheehan KC, Shankaran V, Uppaluri R, Bui JD, Diamond MS, Koebel CM, Arthur C, White JM et al.. A critical function for type I interferons in cancer immunoediting. Nat Immunol 2005; 6:722-9; PMID:15951814; https://doi.org/ 10.1038/ni1213 [DOI] [PubMed] [Google Scholar]
- 81.Gresser I, Maury C, Brouty-Boye D. Mechanism of the antitumour effect of interferon in mice. Nature 1972; 239:167-8; PMID:4561966; https://doi.org/ 10.1038/239167a0 [DOI] [PubMed] [Google Scholar]
- 82.Ferrantini M, Proietti E, Santodonato L, Gabriele L, Peretti M, Plavec I, Meyer F, Kaido T, Gresser I, Belardelli F. Alpha 1-interferon gene transfer into metastatic Friend leukemia cells abrogated tumorigenicity in immunocompetent mice: antitumor therapy by means of interferon-producing cells. Cancer Res 1993; 53:1107-12; PMID:8439955 [PubMed] [Google Scholar]
- 83.Ferrantini M, Giovarelli M, Modesti A, Musiani P, Modica A, Venditti M, Peretti E, Lollini PL, Nanni P, Forni G et al.. IFN-alpha 1 gene expression into a metastatic murine adenocarcinoma (TS/A) results in CD8+ T cell-mediated tumor rejection and development of antitumor immunity. Comparative studies with IFN-gamma-producing TS/A cells. J Immunol 1994; 153:4604-15; PMID:7963533 [PubMed] [Google Scholar]
- 84.Le Bon A, Schiavoni G, D'Agostino G, Gresser I, Belardelli F, Tough DF. Type i interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity 2001; 14:461-70; PMID:11336691; https://doi.org/ 10.1016/S1074-7613(01)00126-1 [DOI] [PubMed] [Google Scholar]
- 85.Santini SM, Lapenta C, Logozzi M, Parlato S, Spada M, Di Pucchio T, Belardelli F. Type I interferon as a powerful adjuvant for monocyte-derived dendritic cell development and activity in vitro and in Hu-PBL-SCID mice. J Exp Med 2000; 191:1777-88; PMID:10811870; https://doi.org/ 10.1084/jem.191.10.1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mattner J, Wandersee-Steinhauser A, Pahl A, Rollinghoff M, Majeau GR, Hochman PS, Bogdan C. Protection against progressive leishmaniasis by IFN-beta. J Immunol 2004; 172:7574-82; PMID:15187137; https://doi.org/ 10.4049/jimmunol.172.12.7574 [DOI] [PubMed] [Google Scholar]
- 87.Lorenzi S, Mattei F, Sistigu A, Bracci L, Spadaro F, Sanchez M, Spada M, Belardelli F, Gabriele L, Schiavoni G. Type I IFNs control antigen retention and survival of CD8alpha(+) dendritic cells after uptake of tumor apoptotic cells leading to cross-priming. J Immunol 2011; 186:5142-50; PMID:21441457; https://doi.org/ 10.4049/jimmunol.1004163 [DOI] [PubMed] [Google Scholar]
- 88.Eisenthal A, Cameron RB, Rosenberg SA. Induction of antibody-dependent cellular cytotoxicity in vivo by IFN-alpha and its antitumor efficacy against established B16 melanoma liver metastases when combined with specific anti-B16 monoclonal antibody. J Immunol 1990; 144:4463-71; PMID:2111349 [PubMed] [Google Scholar]
- 89.Tough DF. Modulation of T-cell function by type I interferon. Immunol Cell Biol 2012; 90:492-7; PMID:22391814; https://doi.org/ 10.1038/icb.2012.7 [DOI] [PubMed] [Google Scholar]
- 90.Boudreau JE, Stephenson KB, Wang F, Ashkar AA, Mossman KL, Lenz LL, Rosenthal KL, Bramson JL, Lichty BD, Wan Y. IL-15 and type I interferon are required for activation of tumoricidal NK cells by virus-infected dendritic cells. Cancer Res 2011; 71:2497-506; PMID:21307131; https://doi.org/ 10.1158/0008-5472.CAN-10-3025 [DOI] [PubMed] [Google Scholar]
- 91.O'Neill LA, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med 2016; 213:15-23; PMID:26694970; https://doi.org/ 10.1084/jem.20151570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.York AG, Williams KJ, Argus JP, Zhou QD, Brar G, Vergnes L, Gray EE, Zhen A, Wu NC, Yamada DH et al.. Limiting Cholesterol Biosynthetic Flux Spontaneously Engages Type I IFN Signaling. Cell 2015; 163:1716-29; PMID:26686653; https://doi.org/ 10.1016/j.cell.2015.11.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wu D, Sanin DE, Everts B, Chen Q, Qiu J, Buck MD, Patterson A, Smith AM, Chang CH, Liu Z et al.. Type 1 interferons induce changes in core metabolism that are critical for immune function. Immunity 2016; 44:1325-36; PMID:27332732; https://doi.org/ 10.1016/j.immuni.2016.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pietrocola F, Pol J, Vacchelli E, Rao S, Enot DP, Baracco EE, Levesque S, Castoldi F, Jacquelot N, Yamazaki T et al.. Caloric restriction mimetics enhance anticancer immunosurveillance. Cancer Cell 2016; 30:147-60; PMID:27411589; https://doi.org/ 10.1016/j.ccell.2016.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Quesada JR, Hersh EM, Manning J, Reuben J, Keating M, Schnipper E, Itri L, Gutterman JU. Treatment of hairy cell leukemia with recombinant alpha-interferon. Blood 1986; 68:493-7; PMID:3730612 [PubMed] [Google Scholar]
- 96.Quesada JR, Alexanian R, Hawkins M, Barlogie B, Borden E, Itri L, Gutterman JU. Treatment of multiple myeloma with recombinant alpha-interferon. Blood 1986; 67:275-8; PMID:3942826 [PubMed] [Google Scholar]
- 97.Goldman JM, Melo JV. Targeting the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med 2001; 344:1084-6; PMID:11287980; https://doi.org/ 10.1056/NEJM200104053441409 [DOI] [PubMed] [Google Scholar]
- 98.Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, Stark GR. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov 2007; 6:975-90; PMID:18049472; https://doi.org/ 10.1038/nrd2422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cohen HT, McGovern FJ. Renal-cell carcinoma. N Engl J Med 2005; 353:2477-90; PMID:16339096; https://doi.org/ 10.1056/NEJMra043172 [DOI] [PubMed] [Google Scholar]
- 100.Tsao H, Atkins MB, Sober AJ. Management of cutaneous melanoma. N Engl J Med 2004; 351:998-1012; PMID:15342808; https://doi.org/ 10.1056/NEJMra041245 [DOI] [PubMed] [Google Scholar]
- 101.Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol 2015; 15:405-14; PMID:26027717; https://doi.org/ 10.1038/nri3845 [DOI] [PubMed] [Google Scholar]
- 102.Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell 2015; 28:690-714; PMID:26678337; https://doi.org/ 10.1016/j.ccell.2015.10.012 [DOI] [PubMed] [Google Scholar]
- 103.Vacchelli E, Ma Y, Baracco EE, Sistigu A, Enot DP, Pietrocola F, Yang H, Adjemian S, Chaba K, Semeraro M et al.. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science 2015; 350:972-8; PMID:26516201; https://doi.org/ 10.1126/science.aad0779 [DOI] [PubMed] [Google Scholar]
- 104.Schiavoni G, Sistigu A, Valentini M, Mattei F, Sestili P, Spadaro F, Sanchez M, Lorenzi S, D'Urso MT, Belardelli F et al.. Cyclophosphamide synergizes with type I interferons through systemic dendritic cell reactivation and induction of immunogenic tumor apoptosis. Cancer Res 2011; 71:768-78; PMID:21156650; https://doi.org/ 10.1158/0008-5472.CAN-10-2788 [DOI] [PubMed] [Google Scholar]
- 105.Schiavoni G, Mattei F, Di Pucchio T, Santini SM, Bracci L, Belardelli F, Proietti E. Cyclophosphamide induces type I interferon and augments the number of CD44(hi) T lymphocytes in mice: implications for strategies of chemoimmunotherapy of cancer. Blood 2000; 95:2024-30; PMID:10706870 [PubMed] [Google Scholar]
- 106.Moschella F, Torelli GF, Valentini M, Urbani F, Buccione C, Petrucci MT, Natalino F, Belardelli F, Foà R, Proietti E et al.. Cyclophosphamide induces a type I interferon-associated sterile inflammatory response signature in cancer patients' blood cells: implications for cancer chemoimmunotherapy. Clin Cancer Res 2013; 19:4249-61; PMID:23759676; https://doi.org/ 10.1158/1078-0432.CCR-12-3666 [DOI] [PubMed] [Google Scholar]
- 107.Lim JY, Gerber SA, Murphy SP, Lord EM. Type I interferons induced by radiation therapy mediate recruitment and effector function of CD8(+) T cells. Cancer Immunol Immunother 2014; 63:259-71; PMID:24357146; https://doi.org/ 10.1007/s00262-013-1506-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li XD, Mauceri H, Beckett M, Darga T et al.. STING-dependent cytosolic DNA sensing promotes radiation-induced Type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 2014; 41:843-52; PMID:25517616; https://doi.org/ 10.1016/j.immuni.2014.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Twyman-Saint\sVictor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, Benci JL, Xu B, Dada H, Odorizzi PM et al.. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015; 520:373-7; PMID:25754329; https://doi.org/ 10.1038/nature14292 [DOI] [PMC free article] [PubMed] [Google Scholar]