

ABSTRACT
Cutaneous T-Cell Lymphomas (CTCL) are rare, but potentially devastating malignancies, whose pathogenesis remains poorly elucidated. Unfortunately, currently it is not possible to predict based on the available criteria in which patients the cancer will progress and which patients will experience an indolent disease course. Furthermore, at early stages this malignancy often masquerades as psoriasis, chronic eczema or other benign inflammatory dermatoses. As a result, it takes on average 6 y to diagnose this lymphoma since its initial presentation. In this study, we performed transcription expression profiling using TruSeq targeted RNA gene expression on 181 fresh and formalin-fixed and paraffin-embedded (FFPE) skin samples from CTCL patients and patients affected by benign inflammatory dermatoses that often mimic CTCL clinically and on histology (e.g., psoriasis, chronic eczema, etc.) We also analyzed multiple longitudinal biopsies that were obtained from the same patients over time. Our results underscore significant molecular heterogeneity with respect to gene expression between different patients and even within the same patients over time. Our study also confirmed TOX, FYB, LEF1, CCR4, ITK, EED, POU2AF, IL26, STAT5, BLK, GTSF1 and PSORS1C2 genes as being differentially expressed between CTCL and benign skin biopsies. In addition, we found that differential expression for a subset of these markers (e.g., TOX, FYB, GTSF1 and CCR4) may be useful in prognosticating this disease. This research, combined with other molecular analyses, prepares the foundation for the development of personalized molecular approach toward diagnosis and management of CTCL.
KEYWORDS: Cutaneous T-Cell Lymphoma (CTCL), diagnostic markers, expression profiling, Mycosis Fungoides (MF), prognostic markers, Sézary Syndrome (SS)
Abbreviations
- CLA
cutaneous lymphocyte-associated antigen
- CTCL
Cutaneous T-Cell Lymphoma
- ELM4
Extension Ligation Mix 4
- FFPE
fresh and formalin-fixed and paraffin-embedded
- MDACC
MD Anderson Cancer Center
- MUHC
McGill University/McGill University Health Center
- MF
Mycosis Fungoides
- cALCL
primary cutaneous anaplastic large cell lymphoma
- SS
Sézary Syndrome
- TRM
skin resident memory T
- TCM
cells, skin-tropic central memory T cells
- TPM
transcript per million
- UHR
Universal Human RNA
Introduction
Cutaneous T-Cell Lymphomas (CTCL) are rare, but potentially devastating malignancies. These cancers are characterized by localization of neoplastic T lymphocytes to the skin. Mycosis Fungoides (MF), its leukemic form, Sézary Syndrome (SS) and primary cutaneous anaplastic large cell lymphoma (cALCL) are the most common variants and account for ∼80% of all cutaneous lymphomas.1
At early stages this cancer typically presents with erythematous patches and plaques on the trunk following a bathing suit distribution. The diagnosis of CTCL is often challenging, since this cancer can masquerade clinically as other entities, such as chronic eczematous dermatitis, psoriasis, pityriasis rubra pilaris, drug eruptions or fungal infections.2 Even, histopathology and PCR studies for T cell receptor clonality are sometimes insufficient for a definitive diagnosis, particularly at early and erythrodermic stages of the disease.2 Based on available estimates in literature, it takes on average 6 y to diagnose this malignancy since its initial presentation.3 Recent discoveries describing the use of high-throughput T cell receptor sequencing instead of traditional PCR methods to detect clonality promise to significantly shorten the delay in the diagnosis of CTCL.3,4 This powerful method may also be suitable to help distinguish drug, viral or other reactive skin eruptions from MF in CTCL patients.3,4
In recent years, great strides were made to elucidate the cellular and molecular pathogenesis of this cancer. Specifically, the cellular origin of CTCL is believed to be very important.5 MF was hypothesized to arise from skin resident memory T (TRM) cells, whose normal function is to defend the body from external pathogens. These cells were shown to express cutaneous lymphocyte-associated antigen (CLA) and CCR4, but lack L-selectin and CCR7 expression, which would enable them to access lymph nodes and circulation.5 SS and leukemic-CTCL cancers on the other hand were proposed to arise from skin-tropic central memory T cells (TCM) that express L-Selectin and CCR7 as well as CLA and CCR4, which enables them to affect, skin, lymph nodes and blood resulting in greater morbidity and mortality to the host.5 Consequently, while skin-limited MF often exhibits indolent clinical behavior and does not impact survival, leukemic variants of CTCL and SS often have median disease survival of only 2–4 y.6-9
Recent research further aided our understanding of underlying molecular pathogenesis for CTCL. Specifically, large-scale mutational genome profiling analyses, including our own group's work, identified genomic alterations in several putative oncogenes and tumor suppressor genes, including CARD11, CCR4, TP53, NF-κB and Janus Kinase signaling members.10-14 Some of these results may have important clinical relevance, where for instance, mogamulizumab (anti-CCR4 antibody) recently showed promising clinical response in advanced CTCL patients.15,16 It was most striking, however, that there was very little overlap with respect to genomic alterations/mutations between these large-scale genome profiling studies, therefore, highlighting this cancer genetic heterogeneity.10-14 Similar, molecular heterogeneity was also seen previously with respect to chromosomal translocations, where only few chromosomal alterations were noted across multiple studies (e.g., loss of 10q24, 10p12.1-726.3 9p21, 17p13.2-p11.2 or gain of 17p11.2-q25.3 and 8q24.1-q24.3 etc.), while most chromosomal alteration were only reported to occur sporadically.10,17-21
In addition to the highlighted difficulties in diagnosing this cancer, it also remains very challenging to prognosticate this disease. Clinical disease stage at the time of diagnosis for early stage MF as well as clinical disease stage combined with age (≥60), presence of large cell transformation, elevated lactate dehydrogenase for advanced stages of CTCL were established as powerful prognostic markers and are very useful in a clinical setting.6-9,22 In addition to the aforementioned criteria, the presence of folliculotropic/syringotropic disease on histology and the ability to detect the same T cell receptor clone longitudinally also confers a grave cancer prognosis.23,24 Notably, it was found that Sézary cell count was a powerful predictor of disease-related mortality where patients with <1,000 Sézary cells/L had an overall survival of 7.6 y, while patients with >10,000 Sézary cells/L in blood survived only 2.1 y.9
Most patients (i.e., 70–80%) present with limited skin stage I patch/plaque disease and only ∼15–20% of stage I patients progress to higher stages. There remains a critical unanswered challenge to accurately predict which patients will progress compared with those, who will experience an indolent disease course. Multiple reports highlighted that high expression of TOX25,26 as well as miR-155, miR-21 and let-7i microRNAs12,27 may predict poor disease outcome.
To discover novel prognostic molecular markers and to gain additional insight in disease etiology, we previously conducted a series of gene expression analyses using RT-PCR on lesional MF/SS skin samples. We also compared gene expression between CTCL skin samples and skin affected by benign inflammatory dermatoses that often mimic MF/SS. As described in the protocol of our previous work, we reviewed over 400 different studies to select 284 genes that were reported to play an important role in CTCL pathogenesis and progression.28 This analysis demonstrated that CCL18, CCL26, CCR4, FYB, T3JAM, MMP12, LEF1, LCK, ITK, GNLY, IL2RA, IL-26, IL-22, IL1F, BLK, CDO1, GTSF1, EED, THAP11, SYCP1, cTAGE1, POU5F1, POU2AF, STAT4, STAT5 and TOX could jointly be used as diagnostic and poor prognostic markers in CTCL patients, while SERPINB13, PSORS1C2, WIF1 and BCL7A were preferentially upregulated in benign skin conditions and/or in indolent CTCL.29-31
To further validate and extend our findings, we have tested the expression of these 284 genes in a new cohort of American and Canadian patients with MF/SS, in patient skin samples affected by benign inflammatory dermatoses or in benign skin tag (i.e., acrochordon) samples using TruSeq targeted RNA gene expression analysis by Illumina.
Results
Study population
In this study, we analyzed a total of 181 samples from 157 patients. This included (A) 29 formalin-fixed and paraffin-embedded (FFPEs) tissues from benign inflammatory dermatoses and skin tag biopsies (1 sample per patient) (B) 134 FFPE samples of lesional CTCL skin from 110 patients (18 patients in this cohort had 2–5 skin longitudinal biopsies available for analysis) and (C) an additional 18 samples of freshly-obtained and liquid nitrogen snap-frozen skin samples from a different group of CTCL patients (Table S1). The latter enabled us to compare the overall gene expression signatures between the above 134 FFPE CTCL and 18 freshly-obtained CTCL samples.
We collected clinical data on the 110 MD Anderson Cancer Center (MDACC) CTCL patients. General patient characteristics for the MDACC cohort are presented in Table 1. Average age at the time of diagnosis was 56.9 ± 16.12 y. As expected and observed in the general population, our study consisted of a greater percentage of male patients (n = 66/110; 60%) than females and most were Caucasian (n = 75/110; 68.2%), while African–American and Hispanic patients were also proportionally represented in our cohort (20.9% and 9.1%, respectively). Only half of the patients in this cohort (n = 55/110; or 50%) at the time of the study biopsy were at the early stage of the disease (i.e., ≤IIA stage). This is not surprising considering that the MDACC is a highly specialized referral center for complex cases. Almost half of our patients (n = 53/110; 48.2%) progressed to higher stages during the 5 y in the study and, unfortunately, 30% of patients in our study succumbed to their disease.
Table 1.
Clinical characteristics of the MD Anderson Cancer Center CTCL patients in the study.
| Patients in the study |
|||
|---|---|---|---|
| n | % | ||
| Number of patients | 110 | ||
| Age at diagnosis | 56.9 ± 16.12 | ||
| Sex | Female | 44 | 40 |
| Male | 66 | 60 | |
| Race | Caucasian | 75 | 68.2 |
| African–American | 23 | 20.9 | |
| Hispanic | 10 | 9.1 | |
| Other | 2 | 1.8 | |
| CTCL clinical stage | IA | 25 | 22.7 |
| IB | 23 | 20.9 | |
| IIA | 7 | 6.4 | |
| IIB | 25 | 22.7 | |
| III | 2 | 1.8 | |
| IVA | 18 | 16.4 | |
| IVB | 10 | 9.1 | |
| Disease progression | Stable disease | 57 | 51.8 |
| Progressive disease | 53 | 48.2 | |
| Disease mortality | Patients surviving | 77 | 70% |
| Patient deaths | 33 | 30% | |
We also performed logistic regression analyses to assess whether clinical features (sex, race, age) were associated with particular disease outcomes (Table S2A–F). As highlighted in our analyses, African–American patients had a significantly higher risk than Caucasians to present with their disease at an earlier age (OR = 5.84; 95% Confidence Intervals (CI): 1.31, 26.05). Also, as expected, advanced clinical stage at the time of biopsy (i.e., ≥IIB) conferred significantly higher risk of cancer progression (OR = 4.28; 95% CI: 1.89, 9.71) and was significantly associated with disease-related mortality (OR = 20.56; 95% CI: 5.58, 75.73). The demographic findings in the studied cohort of patients are consistent with previously reported trends for CTCL.28,32
From the review of pathological slides, we observed that the spectrum of disease observed our patients was representative of the overall spectrum of CTCL seen in our center. In particular, the majority of cases exhibited typical changes of patch/plaque CD4+ MF. Tumor lesions, folliculotropic MF, large cell transformation or younger patients with CD8+ disease did not appear to be either over- or under-represented in this population. Where appropriate patients from endemic areas were tested for HTLV1 serology and only one patient in this cohort was HTLV1+.
Gene expression analysis using TruSeq targeted RNA expression assay
Our previous work using RT-PCR on a smaller cohort of 60 patients, whose biopsies were obtained at the time of diagnosis and before any treatments, identified several important gene expression changes. We therefore, wanted to validate these identified putative diagnostic and prognostic markers in FFPE tissue samples. For this study, we used TruSeq targeted RNA expression assay.
Comparison of gene expression of all samples using unsupervised clustering analysis demonstrated that freshly-obtained and liquid nitrogen snap-frozen CTCL samples clustered together with other FFPE CTCL samples and exhibited a similar gene expression signature pattern, although clustering was more pronounced in fresh samples (Fig. 1). When performing unsupervised clustering analysis, we observed significant cross-classification between CTCL and benign samples. However, an overall trend was noted. The first cluster contained the majority (22/29 or 75.8%) of benign skin samples (spongiotic dermatitides, psoriasis, pityriasis rubra pilaris, lichen planus and fibroepithelial polyps), 29 out of 64 (45.3%) early stage (≤IIA) CTCL samples and 17 out of 70 (24.3%) of mid (stages IIB and III) and advanced (stage IV) cases. In contrast, the second cluster contained all fresh CTCL samples, 53 mid and advanced CTCL samples (75.7%), 35 (54.7%) of early stage CTCL and only 7 (24.2%) benign samples, which were mostly chronic eczemas (spongiotic dermatidities). This unsupervised clustering analysis highlights a significant degree of heterogeneity with respect to gene expression changes within different CTCL samples obtained from a diverse population of real-world patients some with new diagnosis, while others undergoing various medical and physical (e.g., phototherapy) treatments of CTCL.
Figure 1.

Unsupervised clustering analysis based on TruSeq targeted RNA gene expression of 284 select genes in skin tag lesions/benign inflammatory dermatoses (green), freshly-obtained and liquid nitrogen snap-frozen CTCL samples (gray) and FFPE CTCL tissues (yellow, light red and dark red).
In our previous studies, we have identified 26 genes (TOX, FYB, ITK, CCR4, POU2AF, LEF1, GNLY, EED, GTSF1, CCL26, IL2RA, STAT5, CCL18, IL-22, T3JAM, THAP11, STAT4, MMP12, LCK, IL-26, IL1F7, BLK, CDO1, SYCP1, cTAGE1 and PSORS1C2) whose differential expression was able to distinguish benign dermatoses from CTCL.29,31 In the current study, we used Welch's t-test to identify differentially expressed genes between benign skin samples (benign dermatoses and skin tags) and all CTCL samples or between benign skin samples and stage IV CTCL (Table 2). q values were also calculated for our data set to assess for false discovery rate (Table 2). These analyses revealed 75 genes whose expression was significantly different between these groups. As expected, the expression differences for the majority of cancer promoting genes were larger between benign skin samples and advanced CTCL than between benign skin samples and all CTLC (Table 2). For example, average expression of TOX oncogene in benign skin samples was 410.1 transcripts per million, whereas in all CTCL samples it was 2669.4 transcripts per million and in stage IV samples 3916.4 transcripts per million were detected. Notably, our previously identified putative diagnostic/prognostic markers TOX, FYB, LEF1, CCR4, ITK, EED, POU2AF, IL-26, STAT5, BLK, GTSF1 and PSORS1C2 were also highlighted in this cohort of patients (Table 2). Also, similar to the previous studies, we observed a significant upregulation of various inflammation mediating genes in CTCL, including CD70, STAT signaling genes, LTA, NFKB1, NFKB2, IL-15 and other inflammatory cytokines, when compared with benign samples (Table 2).
Table 2.
Genes with statistically significant differences (based on Welch's t-test) in expression between benign skin lesions vs. all CTCL samples (left panel) and benign skin vs. stage IV CTCL samples (right panel). Average expression is presented as transcripts per million. p or q values for all analyses are reported. The q values that failed to reach statistical significance are presented in light grey font.
| Benign FFPE sample vs. all CTCL samples |
Benign FFPE sample vs. Stage IV CTCL FFPE samples |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gene Name | Benign vs. CTCL t-test p value | Benign vs. CTCL q-value | Benign vs. CTCL FFPE log2-ratio (absolute value) | Benign Samples Average Expression | CTCL Samples Average Expression | Benign vs. stage IV CTCL t-test p value | Benign vs stage IV CTCL q-value | Benign vs. stage IV CTCL log2-ratio (absolute value) | Benign samples Average Expression | Stage IV CTCL Samples Average Expression |
| LTA | 2.08E-06 | 1.08E-05 | 2.8 | 189.2 | 1360.0 | 1.13E-03 | 4.61E-03 | 2.9 | 189.2 | 1450.0 |
| TOX | 4.00E-16 | 1.67E-14 | 2.7 | 410.1 | 2669.4 | 1.22E-06 | 5.98E-05 | 3.3 | 410.1 | 3916.4 |
| ITK | 3.67E-08 | 3.40E-07 | 2.6 | 140.1 | 841.3 | 3.41E-05 | 4.77E-04 | 2.7 | 140.1 | 916.4 |
| POU2AF1 | 3.90E-03 | 5.53E-03 | 2.4 | 151.4 | 800.2 | 2.96E-02 | 2.09E-01 | 2.6 | 151.4 | 948.5 |
| CD70 | 4.92E-06 | 2.40E-05 | 2.4 | 813.9 | 4375.0 | 8.24E-04 | 3.84E-03 | 2.5 | 813.9 | 4642.7 |
| SKAP1 | 2.24E-08 | 2.34E-07 | 2.4 | 373.9 | 1970.3 | 2.75E-05 | 4.49E-04 | 2.7 | 373.9 | 2402.8 |
| CCR4 | 3.14E-09 | 5.24E-08 | 2.4 | 1375.9 | 7192.2 | 4.10E-05 | 5.02E-04 | 2.6 | 1375.9 | 8592.0 |
| CCR7 | 8.16E-03 | 9.87E-03 | 2.4 | 91.6 | 492.5 | 3.91E-03 | 1.16E-02 | 2.1 | 91.6 | 396.3 |
| LEF1 | 9.34E-08 | 6.49E-07 | 2.3 | 161.0 | 783.8 | 1.48E-05 | 3.16E-04 | 2.4 | 161.0 | 878.7 |
| FYB | 8.08E-19 | 6.74E-17 | 2.3 | 1337.9 | 6636.5 | 1.72E-07 | 1.69E-05 | 2.9 | 1337.9 | 9750.4 |
| IL21R | 5.64E-05 | 1.57E-04 | 1.9 | 256.3 | 963.6 | 5.32E-04 | 3.66E-03 | 2.1 | 256.3 | 1136.5 |
| SH2D1A | 9.15E-06 | 3.47E-05 | 1.9 | 162.9 | 602.5 | 6.89E-03 | 1.69E-02 | 2.0 | 162.9 | 668.3 |
| MMP9 | 7.23E-03 | 8.87E-03 | 1.8 | 149.2 | 533.1 | 7.54E-03 | 1.69E-02 | 2.8 | 149.2 | 1038.3 |
| SERPINB4 | 1.23E-02 | 1.37E-02 | 1.8 | 37557.1 | 10458.7 | 2.54E-02 | 4.15E-02 | 1.6 | 37557.1 | 12550.8 |
| TRAF1 | 6.20E-06 | 2.64E-05 | 1.7 | 221.1 | 741.6 | 6.61E-04 | 3.81E-03 | 2.2 | 221.1 | 1048.3 |
| ZAP70 | 1.64E-05 | 5.07E-05 | 1.7 | 915.7 | 3006.2 | 3.40E-03 | 1.07E-02 | 1.8 | 915.7 | 3219.1 |
| EED | 6.07E-10 | 1.69E-08 | 1.6 | 1110.6 | 3364.1 | 5.60E-04 | 3.66E-03 | 1.5 | 1110.6 | 3047.6 |
| IL7R | 1.20E-05 | 4.01E-05 | 1.6 | 523.8 | 1567.4 | 3.80E-03 | 1.16E-02 | 1.7 | 523.8 | 1697.3 |
| PDCD1 | 5.51E-03 | 7.54E-03 | 1.6 | 199.9 | 621.3 | 3.74E-02 | 5.31E-02 | 2.1 | 199.9 | 860.0 |
| TBX3 | 2.70E-02 | 2.62E-02 | 1.6 | 164.6 | 497.9 | 3.47E-02 | 5.15E-02 | 1.7 | 164.6 | 549.4 |
| PSORS1C2 | 3.18E-03 | 4.74E-03 | 1.5 | 56092.9 | 19852.0 | 1.21E-02 | 2.42E-02 | 1.2 | 56092.9 | 24132.8 |
| CNOT3 | 6.32E-06 | 2.64E-05 | 1.5 | 268.2 | 754.1 | 2.99E-03 | 9.77E-03 | 1.5 | 268.2 | 762.8 |
| STAT1 | 1.46E-08 | 1.74E-07 | 1.5 | 1714.1 | 4796.4 | 9.97E-05 | 1.09E-03 | 1.6 | 1714.1 | 5279.1 |
| STAG3 | 5.47E-08 | 4.56E-07 | 1.5 | 1118.5 | 3149.1 | 8.25E-06 | 2.69E-04 | 1.8 | 1118.5 | 3848.8 |
| PILRB | 1.55E-09 | 3.23E-08 | 1.5 | 2562.3 | 7305.2 | 7.99E-04 | 3.84E-03 | 1.4 | 2562.3 | 6818.3 |
| NKG7 | 2.83E-03 | 4.29E-03 | 1.5 | 2964.4 | 8484.3 | 4.02E-02 | 5.62E-02 | 1.2 | 2964.4 | 6967.7 |
| PTPN6 | 6.59E-09 | 9.16E-08 | 1.4 | 746.3 | 2021.7 | 2.51E-04 | 2.24E-03 | 1.4 | 746.3 | 1981.0 |
| IRF4 | 8.00E-08 | 6.07E-07 | 1.4 | 192.3 | 505.3 | 6.27E-04 | 3.81E-03 | 1.3 | 192.3 | 477.6 |
| NFKB2 | 1.50E-05 | 4.81E-05 | 1.4 | 320.1 | 852.4 | 4.33E-03 | 1.25E-02 | 1.6 | 320.1 | 966.2 |
| IL32 | 1.09E-06 | 6.50E-06 | 1.4 | 2781.6 | 7520.3 | 1.48E-04 | 1.45E-03 | 1.5 | 2781.6 | 7979.7 |
| LCP2 | 1.00E-05 | 3.48E-05 | 1.4 | 536.0 | 1421.1 | 1.61E-05 | 3.16E-04 | 1.7 | 536.0 | 1696.3 |
| ZFX | 5.38E-07 | 3.45E-06 | 1.3 | 116.1 | 295.5 | 1.48E-02 | 2.84E-02 | 1.1 | 116.1 | 253.0 |
| STAT2 | 2.05E-05 | 6.11E-05 | 1.3 | 324.8 | 803.3 | 9.39E-03 | 1.96E-02 | 1.6 | 324.8 | 968.0 |
| IL15 | 1.91E-03 | 3.01E-03 | 1.3 | 274.7 | 694.3 | 4.08E-02 | 5.62E-02 | 1.6 | 274.7 | 843.3 |
| ZBTB16 | 1.24E-04 | 3.34E-04 | 1.2 | 1100.6 | 2606.4 | 7.02E-04 | 3.82E-03 | 1.5 | 1100.6 | 3168.9 |
| RAC2 | 1.31E-03 | 2.45E-03 | 1.2 | 4324.0 | 10279.5 | 1.22E-03 | 4.78E-03 | 1.4 | 4324.0 | 11662.1 |
| CXCL9 | 5.80E-03 | 7.81E-03 | 1.2 | 2507.9 | 5798.2 | 8.66E-03 | 1.89E-02 | 1.5 | 2507.9 | 6862.0 |
| ANKRD11 | 1.47E-06 | 8.18E-06 | 1.2 | 987.6 | 2236.8 | 1.46E-02 | 2.84E-02 | 1.0 | 987.6 | 2001.2 |
| EZH2 | 5.95E-04 | 1.31E-03 | 1.1 | 466.1 | 996.7 | 2.87E-02 | 4.50E-02 | 0.9 | 466.1 | 876.4 |
| CDKN2B | 1.31E-03 | 2.45E-03 | 1.1 | 476.2 | 1024.1 | 4.13E-02 | 5.62E-02 | 1.2 | 476.2 | 1065.9 |
| CCL5 | 1.33E-03 | 2.45E-03 | 1.1 | 18304.2 | 38736.5 | 3.18E-04 | 2.60E-03 | 1.3 | 18304.2 | 43788.2 |
| HDAC1 | 5.18E-06 | 2.40E-05 | 1.1 | 1225.0 | 2568.8 | 1.04E-03 | 4.43E-03 | 1.2 | 1225.0 | 2906.2 |
| NME1 | 1.50E-03 | 2.50E-03 | 1.1 | 1861.2 | 3878.3 | 3.73E-02 | 5.31E-02 | 1.1 | 1861.2 | 3929.5 |
| CD52 | 3.72E-04 | 8.87E-04 | 1.1 | 39313.9 | 81513.5 | 7.47E-04 | 3.84E-03 | 1.2 | 39313.9 | 90909.3 |
| MTF2 | 3.05E-04 | 7.49E-04 | 1.1 | 217.4 | 453.9 | 1.82E-03 | 6.37E-03 | 1.2 | 217.4 | 498.4 |
| ITGA3 | 6.71E-03 | 8.61E-03 | 1.0 | 182.7 | 369.1 | 1.02E-02 | 2.08E-02 | 1.9 | 182.7 | 665.6 |
| RASA1 | 9.05E-04 | 1.85E-03 | 1.0 | 338.1 | 677.7 | 9.12E-04 | 4.06E-03 | 1.1 | 338.1 | 745.1 |
| SUZ12 | 1.00E-05 | 3.48E-05 | 1.0 | 340.2 | 676.1 | 4.65E-04 | 3.51E-03 | 1.2 | 340.2 | 795.6 |
| IRF7 | 3.91E-03 | 5.53E-03 | 1.0 | 841.7 | 1658.5 | 1.82E-02 | 3.11E-02 | 1.0 | 841.7 | 1660.2 |
| PRDM2 | 1.83E-02 | 1.91E-02 | 1.0 | 37.1 | 72.2 | 4.49E-02 | 5.84E-02 | 1.4 | 37.1 | 95.6 |
| MAP2K1 | 1.38E-03 | 2.45E-03 | 0.9 | 412.6 | 794.8 | 8.94E-03 | 1.90E-02 | 1.0 | 412.6 | 803.8 |
| KLHL42 | 1.73E-03 | 2.78E-03 | 0.9 | 136.5 | 260.4 | 5.18E-03 | 1.45E-02 | 1.0 | 136.5 | 273.5 |
| EP400 | 1.40E-03 | 2.45E-03 | 0.9 | 1879.3 | 3608.5 | 1.46E-03 | 5.50E-03 | 1.1 | 1879.3 | 4107.7 |
| JARID2 | 5.21E-03 | 7.25E-03 | 0.9 | 698.2 | 1292.2 | 1.87E-02 | 3.11E-02 | 0.9 | 698.2 | 1326.6 |
| NFKB1 | 9.07E-04 | 1.85E-03 | 0.9 | 498.1 | 915.7 | 7.38E-03 | 1.69E-02 | 1.0 | 498.1 | 979.2 |
| WWOX | 8.98E-03 | 1.06E-02 | 0.9 | 953.4 | 1733.4 | 7.15E-03 | 1.69E-02 | 1.1 | 953.4 | 2071.6 |
| NUB1 | 8.89E-03 | 1.06E-02 | 0.8 | 5078.1 | 8783.1 | 1.82E-02 | 3.11E-02 | 0.9 | 5078.1 | 9771.0 |
| STAT5B | 4.80E-02 | 3.86E-02 | 0.8 | 210.0 | 369.8 | 7.01E-03 | 1.69E-02 | 1.2 | 210.0 | 498.2 |
| SMAD2 | 3.99E-02 | 3.43E-02 | 0.8 | 139.7 | 238.3 | 4.44E-02 | 5.84E-02 | 1.1 | 139.7 | 292.7 |
| IL17RA | 2.33E-03 | 3.60E-03 | 0.8 | 455.6 | 809.4 | 3.04E-02 | 4.65E-02 | 0.8 | 455.6 | 783.5 |
| MCL1 | 6.99E-06 | 2.78E-05 | 0.9 | 1349.3 | 2433.9 | 1.80E-03 | 6.37E-03 | 0.8 | 1349.3 | 2360.7 |
| TRAF2 | 2.37E-02 | 2.35E-02 | 0.8 | 2425.2 | 4113.7 | 1.56E-02 | 2.85E-02 | 0.9 | 2425.2 | 4613.1 |
| MYC | 1.41E-03 | 2.45E-03 | 0.8 | 380.8 | 663.6 | 2.35E-03 | 7.94E-03 | 1.2 | 380.8 | 873.1 |
| SOCS3 | 3.32E-02 | 3.04E-02 | 0.8 | 1008.8 | 1713.3 | 2.89E-02 | 4.50E-02 | 1.1 | 1008.8 | 2102.9 |
| E2F4 | 5.97E-03 | 7.91E-03 | 0.8 | 773.6 | 1314.5 | 1.87E-02 | 3.11E-02 | 1.0 | 773.6 | 1566.6 |
| TRRAP | 5.38E-04 | 1.25E-03 | 0.8 | 113.8 | 199.2 | 1.57E-02 | 2.85E-02 | 0.9 | 113.8 | 211.1 |
| NME4 | 1.38E-02 | 1.50E-02 | 0.8 | 3075.1 | 5452.4 | 4.53E-02 | 5.84E-02 | 0.9 | 3075.1 | 5723.2 |
| TGFB1 | 9.62E-03 | 1.11E-02 | 0.7 | 394.7 | 649.9 | 5.47E-03 | 1.49E-02 | 0.9 | 394.7 | 759.3 |
| ACVRL1 | 3.44E-02 | 3.08E-02 | 0.7 | 414.1 | 659.3 | 2.71E-02 | 4.35E-02 | 0.9 | 414.1 | 789.9 |
| ANPEP | 7.20E-03 | 8.87E-03 | 0.7 | 820.7 | 1308.1 | 7.58E-03 | 1.69E-02 | 0.9 | 820.7 | 1512.5 |
| NOTCH1 | 3.78E-02 | 3.29E-02 | 0.5 | 393.3 | 557.5 | 1.64E-02 | 2.92E-02 | 0.9 | 393.3 | 712.9 |
| GTSF1 | 3.35E-05 | 9.64E-05 | ∞ | 0 | 1852.2 | 1.73E-03 | 1.57E-02 | ∞ | 0 | 2573.3 |
| BLK | 1.96E-04 | 5.11E-04 | ∞ | 0 | 416.7 | 3.97E-02 | 6.21E-02 | ∞ | 0 | 283.5 |
| IL26 | 1.61E-03 | 2.63E-03 | ∞ | 0 | 1373.4 | 4.88E-01 | 2.16E-01 | ∞ | 0 | 144.6 |
| TNFSF11 | 4.76E-02 | 3.86E-02 | ∞ | 0 | 243.6 | 1.59E-01 | 1.42E-01 | ∞ | 0 | 280.3 |
Independent unsupervised clustering analysis of benign skin samples vs. stage IV CTCL (63 samples analyzed) using these genes produced two clusters, where cluster 2 had multiple sub-clusters (Fig. 2). Cluster 2C contained only CTCL samples (n = 13) and Clusters 1 and 2A contained 13 CTCL and only 3 benign samples (n = 16) (Fig. 2). On the other hand, Cluster 2B contained only benign samples (n = 19), while cluster 2D contained a mix of CTCL (eight samples) and benign (seven samples) cases.
Figure 2.

Unsupervised clustering analysis based on TruSeq targeted RNA gene expression of 284 select genes in benign (green) vs. stage IV (red) FFPE CTCL tissue samples.
We then wanted to compare different clinical stages of CTCL and analyze early (stage ≤IIA) vs. mid (stages IIB and III) vs. advanced (stage IV) disease. Interestingly, unsupervised clustering analysis of all FFPE CTCL samples failed to reveal three clusters that we earlier observed in the Boston cohort of patients33 and similarly underscored significant heterogeneity of gene expression as many early, mid stage and advanced disease samples were scattered across different clusters (Fig. 3). Even comparison of stage I vs. stage IV disease similarly failed to reveal clustering based on the TruSeq analysis of 284 selected genes (Fig. S1). However, statistical analyses of gene expression highlighted several genes that were differentially expressed between stage ≤IIA vs. stage≥IIB disease (Table 3A). We searched for genes that were differentially expressed in the analysis of stage ≤IIA vs. ≥IIB disease and in the analysis of stage I vs. stage IV disease. Four genes of interest fulfilled these criteria: TOX, FYB and GTSF1 were upregulated, while LTB4 was downregulated in advanced stages of CTCL (Table 3A). These genes were previously independently identified as putative disease progression molecular markers in other gene expression profiling studies.25,29,30,33,34
Figure 3.
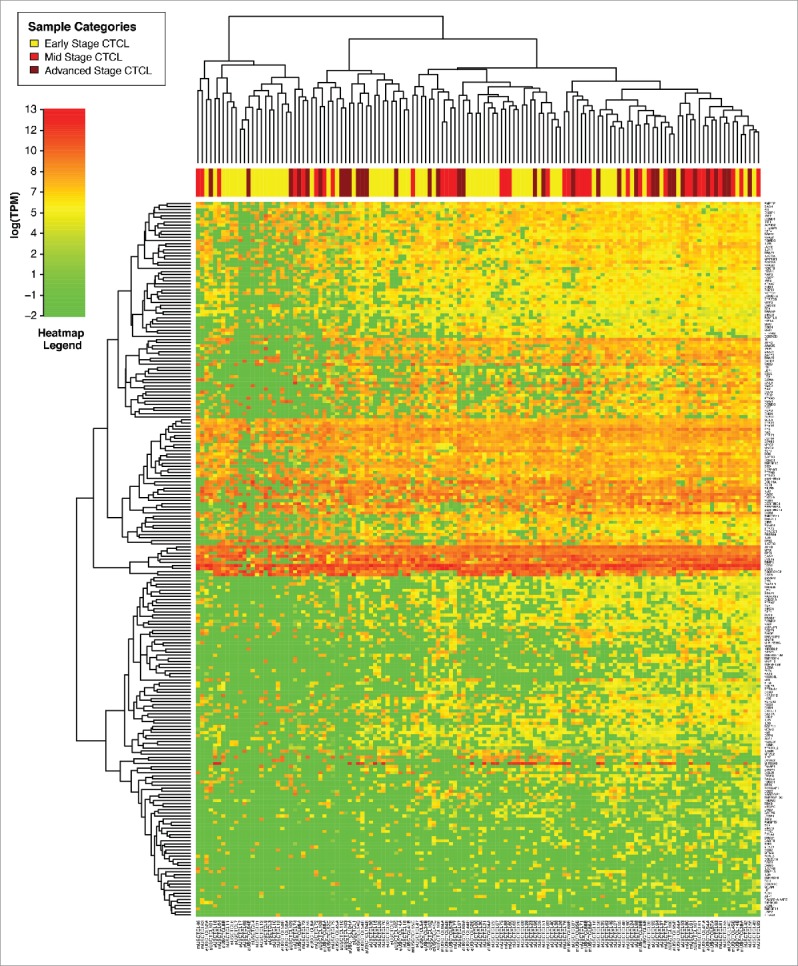
Unsupervised clustering analysis based on TruSeq targeted RNA gene expression of 284 select genes in early stage (≤IIA, yellow) vs. stage IIB and III (light red) vs. stage IV (dark red) FFPE CTCL tissue samples.
Table 3.
(A) Genes with statistically significant differences in expression between early (≤IIa) vs. mid (staged IIB and III) and advanced (stage IV) CTCL samples (left panel). Results for the same gene presented for analysis of stage I vs. stage IV CTCL samples (right panel). While trends in gene expression were confirmed in both analyses only expression differences for GTSF1, LTBP4, IL18, CCL5, LCP2, TOX and FYB were statistically significant in both data sets. Expression differences for other genes in the latter analysis did not reach statistical significance due to lower number of samples in that set. The data that failed to reach statistical significance are presented in light grey font and p values for all analyses are reported. Average expression is presented as transcripts per million. (B) Genes with statistically significant differences in expression between stage I stable/indolent disease vs. stage I progressive disease.
| A. | ||||||||
|---|---|---|---|---|---|---|---|---|
| Early (≤IIA) vs. Advanced (≥IIB) CTCL cases |
Stage I vs. Stage IV CTCL cases |
|||||||
| Gene names | Early vs. mid and advanced stages t- test | Early vs. mid and advanced log2-ratio (absolute value) | Stages ≤IIA average expression | Stages ≥IIB average expression | Stage I vs. Stage IV CTCL t-test | Stage I vs. Stage IV CTCL log2-ratio (absolute value) | Stage I CTCL samples average expression | Stage IV CTCL samples average expression |
| GTSF1 | 2.41E-03 | 2.0 | 739.6 | 2981.9 | 5.73E-03 | 2.6 | 434.1 | 2573.3 |
| TRIP13 | 2.94E-02 | 1.8 | 316.0 | 1088.7 | 3.55E-01 | 1.7 | 316.0 | 1013.0 |
| GNLY | 1.39E-02 | 1.8 | 3168.5 | 10687.9 | 3.88E-01 | 0.5 | 3168.5 | 4505.9 |
| LTBP4 | 2.20E-02 | 1.7 | 395.1 | 123.7 | 2.31E-02 | 1.7 | 348.6 | 103.9 |
| CD70 | 3.79E-03 | 1.2 | 2852.7 | 6416.3 | 8.05E-02 | 0.8 | 2725.1 | 4642.7 |
| SH2D1A | 5.80E-03 | 1.1 | 395.5 | 860.5 | 1.57E-01 | 0.7 | 411.0 | 668.3 |
| GZMA | 2.29E-02 | 1.0 | 2202.7 | 4393.7 | 7.48E-01 | 0.1 | 2288.1 | 2513.3 |
| IL18 | 2.02E-02 | 1.1 | 2984.0 | 1436.6 | 3.80E-02 | 1.0 | 3089.1 | 1590.5 |
| PTPN7 | 2.10E-03 | 0.9 | 364.1 | 682.7 | 5.02E-02 | 0.9 | 350.7 | 647.8 |
| TP63 | 2.30E-02 | 0.8 | 8855.4 | 5243.4 | 2.90E-01 | 0.4 | 8781.1 | 6581.0 |
| CCL5 | 1.32E-02 | 0.7 | 31726.6 | 52914.6 | 3.70E-02 | 0.5 | 30877.9 | 43788.2 |
| LCP2 | 1.64E-04 | 0.7 | 1109.7 | 1850.7 | 2.78E-02 | 0.5 | 1172.5 | 1696.3 |
| PSORS1C2 | 4.74E-02 | 0.7 | 28499.0 | 17235.7 | 9.46E-01 | 0.0 | 24564.2 | 24132.8 |
| IL21R | 1.88E-02 | 0.7 | 825.6 | 1307.7 | 2.34E-01 | 0.4 | 851.8 | 1136.5 |
| NKG7 | 4.91E-02 | 0.6 | 6593.1 | 10265.8 | 7.65E-01 | 0.1 | 6472.6 | 6967.7 |
| TIMP1 | 9.17E-03 | 0.6 | 11146.9 | 16890.6 | 2.84E-01 | 0.3 | 11764.0 | 14446.1 |
| TOX | 1.95E-02 | 0.6 | 2329.9 | 3485.6 | 3.67E-02 | 0.7 | 2461.2 | 3916.4 |
| SERPINB5 | 4.08E-02 | 0.6 | 5283.5 | 3558.5 | 1.01E-01 | 0.5 | 5378.8 | 3717.3 |
| FYB | 7.41E-03 | 0.5 | 5987.4 | 8708.4 | 1.16E-02 | 0.7 | 6032.9 | 9750.4 |
| B. | ||||
|---|---|---|---|---|
| Stage I indolent disease vs. Stage I progressive disease | ||||
| Gene name | Indolent vs. progressive Stage I CTCL t-test | Indolent vs. progressive Stage I CTCL log2-ratio | Indolent Stage I CTCL average | Progressive Stage I CTCL average |
| CCR4 | 4.14E-02 | 1.3 | 4039.5 | 10134.0 |
| TOX | 1.34E-02 | 1.1 | 1695.3 | 3695.3 |
| SERPINB3 | 2.90E-02 | 1.0 | 10073.5 | 5063.5 |
| FYB | 3.52E-02 | 0.7 | 4988.2 | 8018.0 |
| PTPN6 | 4.93E-02 | 0.7 | 1767.8 | 2793.4 |
Our population included 58 stage I patient samples (collected from 48 patients), who were followed clinically for up to 5 y (depending on the time of biopsy) and, hence, we wanted to compare stage I patients with stable disease (38 samples) vs. stage I patients who have progressed to higher clinical stages (20 skin biopsy samples). While the independent clustering analysis demonstrated significant heterogeneity with respect to gene expression (Fig. S2), Welch's t-test highlighted TOX, CCR4, FYB, SERPINB3 and PTPN6 genes as being differentially expressed (Table 3B).
Analysis of patients with longitudinal skin biopsies
In our study, 18 patients had 2–5 biopsies (i.e., 42 total skin samples) that were obtained over several years (individual years of skin biopsy specimens are indicated in Fig. 4). Each biopsy from these patients was reviewed, and in each case, the diagnosis of MF or SS was confirmed. Five of these patients did not progress, while 12 did progress to higher stages and 50% (or six patients) died from their disease. This higher rate of progression and disease-related mortality, when compared with the overall cohort, was not surprising: the need to perform multiple biopsies suggests a clinical suspicion for CTCL progression. As demonstrated in Fig. 4, unsupervised clustering analysis demonstrated that 6 out of 18 multiple biopsy specimen sets clustered together according to their gene expression profiles (cases USCTCL 8 A and B; 39 A and B; 50 A and B; 84 A and B; 86 A and B; and 54 A, B and C, but not 54 D and E samples). In all of these cases, samples had been collected in different years. Also, the ability to cluster samples together was noted for patch/plaque (two cases), tumor (three cases) and stage IV (one case) disease. Inability to cluster together multiple biopsies from the remaining 12 patients highlights disease heterogeneity over time with respect to gene expression within the same individual. Also, interestingly, when we analyzed several select genes (GNLY, CD30/TNFRSF8, CD70, GTSF1, BCL7A and IL-9) within thesemultiple biopsy specimens with respect to their clinical stage at the time of biopsy, similar patterns emerged where poor prognosis/pro-inflammatory genes (CD30, GNLY, CD70 and GTSF1) were upregulated in higher disease stage patients, while BCL7A, a favorable prognosis gene,35 was preferentially expressed in early disease stages (Fig. 5A–E). Interestingly, IL-9, which was previously shown to be expressed in a subset of MF cases,36 was consistently expressed across multiple biopsies in only one patient (Fig. 5E).
Figure 4.
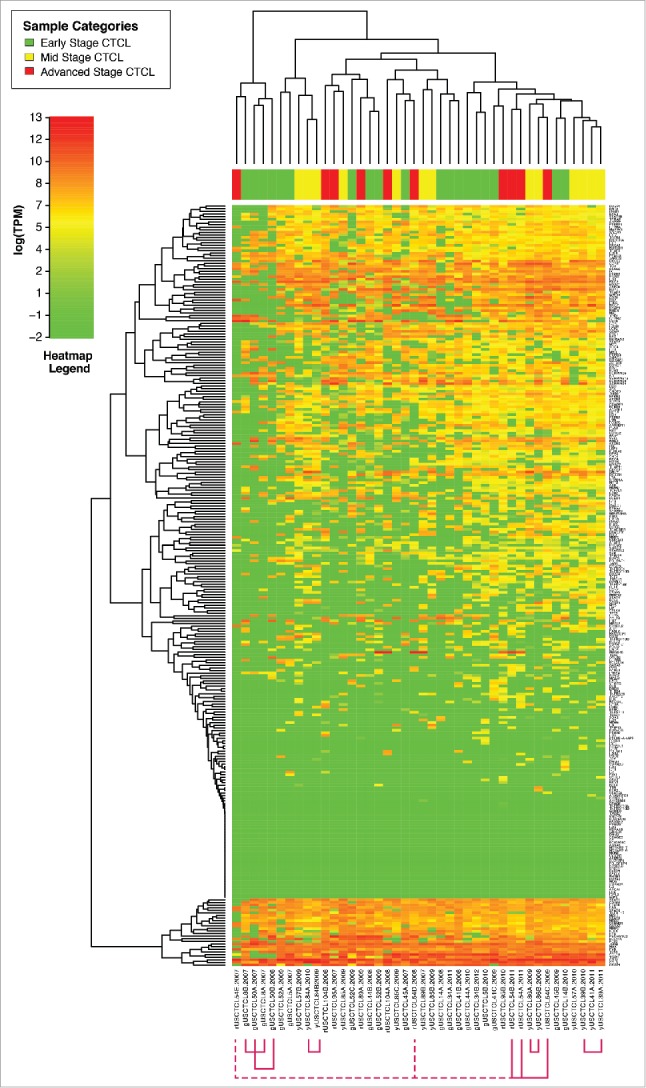
Unsupervised clustering analysis based on TruSeq targeted RNA gene expression of 284 select genes in CTCL FFPE tissue samples. Multiple (2–5) skin samples were obtained in different years (as indicated in the figure) form patients.
Figure 5.
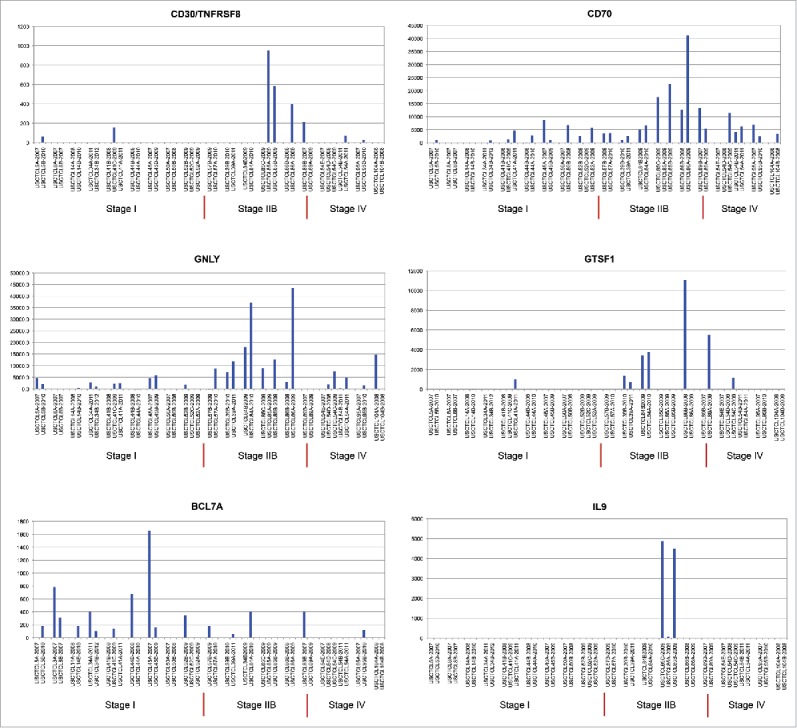
Individual gene expression findings in different patients, who had multiple skin biopsies performed in different years of the study, as indicated.
Discussion
In the current work, we for the first time applied the TruSeq targeted RNA gene expression analysis to study FFPE skin samples from a large cohort of diverse, unselected CTCL patients encountered in a tertiary referral setting. Some of these patients presented with a new diagnosis of MF/SS, while others have been receiving various treatments of their lymphoma. This study depicts real-world scenario of patients seen in a tertiary care center and highlights significant disease heterogeneity with respect to gene expression changes among different patients and even within the same patients (based on the analysis of 42 longitudinal biopsies). This gene expression profiling presents a comprehensive analysis of over 280 highly-studied MF/SS biomarkers and candidate genes for CTCL pathogenesis. Our study describes an important technique that enables the analysis of FFPE tissue samples that are readily available in multiple institutions. Our findings complement recent large-scale whole genome sequencing studies10-14 that similarly revealed significant disease heterogeneity with respect to gene mutations identified in MF/SS patients.
Our analysis was able to confirm several important gene expression changes that might be useful in combination with other techniques to diagnose and/or prognosticate CTCL patients. Specifically, our study highlighted upregulation of TOX, FYB, LEF1, CCR4, ITK, EED, POU2AF, IL-26, STAT5, BLK, GTSF1 and downregulation of PSORS1C2 in CTCL patients when compared with benign inflammatory dermatoses. The biologic significance of these independently validated in this study genes in relation to CTCL tumorigenesis has been described previously in detail in supplementary tables of our earlier publications.29,31 Specifically, TOX gene was time and again shown to be a bona fide oncogene in CTCL.25 It is important to note that TOX expression is not specific to CTCL tumors. This marker was shown to be expressed in primary cutaneous B-cell lymphomas,37,38 and at low levels in inflammatory T cells in benign inflammatory dermatoses (e.g., atopic dermatitis and drug eruptions).38 Our presented findings confirm these results and demonstrate that TOX is heterogeneously expressed at low levels in benign inflammatory dermatoses (410.1 transcripts per million). However, TOX expression was several folds higher in CTCL (2669.4 transcripts per million) and was found to be most elevated in advanced stage IV cancers (3916.4 transcripts per million).
Similarly, our findings highlight the importance of STAT5 expression in CTCL, which was documented to drive the expression of oncogenic miR-155.39 Furthermore, in CTCL40 and ectopic expression of B-lymphoid kinase (BLK) was previously reported was further confirmed by our findings. TOX, FYB and GTSF1 were significantly upregulated in advanced CTCL stages when compared with stage ≤IIA disease. Furthermore, TOX, FYB and CCR4 were upregulated in stage I patients that were at risk of cancer progression. Notably, these gene expression changes have been described in similar clinical scenarios and in our previously studied cohorts.25,29,30,33-35
Our findings did not confirm the three different transcriptional profile clusters that were reported in the historic Boston cohort of patients.29,33,41 This could be due to multiple reasons. In the analysis of samples from the Boston cohort of patients, we used freshly-obtained skin tissues and analyzed them using the “gold standard” method of RT-PCR to study gene expression changes. Furthermore, in the Boston cohort the patient biopsy sites were treatment naïve, since biopsies were performed at the time of the diagnosis, whereas in the current study many patients were already on one or more stage-appropriate treatments. Also, while the Boston cohort was mostly comprised of classic patch/plaque MF and erythrodermic SS samples from Caucasian patients, in this study there was a significantly greater patient and disease diversity. Despite these differences several important markers (e.g., TOX, CCR4, FYB, GTSF1, LEF1, ITK, BLK, PSORS1C2, etc) were cross-validated in both cohorts of patients. Also, from these combined studies an important theme emerged showing that CTCL expresses higher levels of various inflammatory markers when compared with benign dermatoses, such as eczema, psoriasis and lichen planus. This correlates clinically with the observation that while it is often relatively easy to achieve control of benign inflammation with topical steroids, phototherapy and/or other treatment modalities, the same treatments often fail in controlling malignant inflammation in CTCL.
In conclusion, this work together with other studies highlights several molecular markers that may be useful to diagnose and prognosticate CTCL. It would probably be necessary to use these expression changes in conjunction with other molecular and clinical diagnostic/prognostic indicators. Most importantly this works highlights a significant degree of CTCL molecular heterogeneity between different patients and even within the same patients over time.
Patients and methods
Patients and samples
All patients were enrolled in the study in accordance with the IRB-approved protocols: PA12–0267, PA12–0497 and Lab97–256 at the MDACC and A09-M106–13A and 13–201-GEN at McGill University/McGill University Health Center (MUHC). These protocols enabled us to identify patients with MF/SS cancers based on the existing research study database at MDACC (IRB approved studies PA12–0497 and Lab97–256). We were then able to request tissue blocks from skin biopsies performed during January 2007–2012 at the MDACC and obtain two 20 micron sections from these tissue blocks for subsequent TruSeq targeted RNA expression analysis (IRB approval PA12–0267). The diagnosis was established pathologically for all cases and available pathological slides were retrieved and reviewed by at least two pathologists to confirm the diagnosis and identify important pathological features (i.e., large cell transformation, folliculotropism, CD8+ positivity, CD30 positivity, granulomatous changes, etc.) We were also able to analyze demographic information and disease outcomes using the MDACC electronic medical records for these patients (IRB approvals PA12–0267 and PA12–0497).
Similarly, patients were enrolled in the study with informed consent in accordance with the Declaration of Helsinki and provided a skin sample for research at the McGill University/MUHC in accordance with the IRB approved protocol A09-M106–13A CTCL. The biopsies were performed and processed as described previously.29,33,41 In accordance to the MUHC IRB approved protocol 13–201-GEN, we were also able to request tissue blocks from skin biopsies performed during 2009–2014 for patients with skin tags or benign inflammatory dermatoses (e.g., psoriasis, eczema, pityriasis rubra pilaris and lichen planus) and to obtain two 20 micron sections from these tissue blocks for subsequent TruSeq targeted RNA expression analysis.
For all patients, where normal skin (e.g., fibroepithelial polyp) or lesional skin affected by a benign inflammatory dermatosis was collected only the diagnosis was reported and no clinical follow up was performed, as per the approved IRB protocol. Similarly, for all patients with freshly-collected and liquid nitrogen snap-frozen lesional CTCL samples only the diagnosis of MF and essential disease information was recorded in the study. For all patients for whom FFPE CTCL samples were obtained from the MDACC, the diagnosis, relevant pathology reports and clinical disease history were recorded. For all CTCL cases the diagnosis and clinical staging were followed in accordance to the established diagnostic criteria.1
RNA preparation and quality check
For this analysis, 181 samples were used for total RNA extraction. Total RNA extractions were performed on FFPE skin samples using RNeasy FFPE kit (Qiagen, Valencia, CA) according to the manufacturer's protocol. RNA was quantified using nanodrop ND-8000 spectrophotometer (Thermo Scientific, Wilmington, DE). RNA integrity was measured using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). RNA Integrity numbers were used to assess RNA quality. In this study, 181 samples were used to extract total RNA. Expression of 284 genes was analyzed using TruSeq targeted RNA expression.
TruSeq targeted RNA expression analysis
For this study, we used TruSeq targeted RNA expression assay by Illumina, which offers customizable gene expression profiling using as little as 50 ng of RNA in accordance to the published protocol (http://support.illumina.com/downloads/truseq-targeted-rna-expression-guide-15034665.html). Custom probe oligonucleotides targeting the 284 genes of interest were designed Design using Illumina Studio (Table S3). RNA samples (50–75 ng depending on sample availability) were diluted in nuclease free water and together with HeLa cells RNA and company provided two Universal Human RNA (UHR) control samples (Illumina) were subjected to reverse transcription using ProtoScript II reverse transcriptase (Illumina). Subsequently targeted oligonucleotide probes were hybridized to cDNA and were washed using the paramagnetic streptavidin beads in the presence of TE buffer (Illumina) starting at 80°C and subsequently gradually decreasing temperature down to 25°C.
cDNAs with annealed probes were then subjected to nucleotide chain extension and ligation reactions using Illumina Extension and Ligation Mix 4 (ELM4) reagent. Subsequently, obtained hybridized products were amplified 29 times by PCR using Illumina PCR Master Mix 2, A50X/R7XX primers and TruSeq DNA Polymerase 1 reagents. Reaction products were purified using AMPure XP beads (Beckman-Coulter, Brea, CA). Produced libraries were pooled into six groups according to their category and/or year of sample acquisition (e.g., benign samples, freshly obtained CTCL lesional skin samples, FFPE CTCL samples, etc.) and sequenced using MiSeq sequencing system.
For MiSeq sequencing qPCR was used to quantify the samples and 7pM was loaded on the MiSeq sequencer and 50 base reads were obtained using v2 chemistry42 and MiSeq Control Software 2.4 version (Illumina) in accordance with the manufacturer's instructions. Bcl2fastq Conversion Software v1.8.4 was used to demultiplex data and identify bases.
Data analysis and statistical analyses
Sequence read data core quality was assessed using FastQC software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Gene expression was quantified using the RSEM or RNA-Seq by Expectation Maximization algorithm,43 which estimates the gene abundance ratio against the RNA-Seq read set using a probabilistic model by expectation maximization. RSEM models the uncertainty occurring when a sequenced read aligns at multiple loci. The quantified values reported for each sample are presented as transcript per million (TPM). This measure is independent of the mean expressed transcript length and is, thus, robustly comparable across samples. Reads were aligned against a human transcriptome using Bowtie 44
Genes were selected as differentially expressed by applying a Welch's t-test against the null hypothesis that the expression is the same across the conditions (p value <0.05). In this testing algorithm each gene was independently observed (tested). We tested double with a base 2 log-ratio above 1 (i.e., 2-fold) on the expression average. Absolute log value was reported along with average expression for each category. q values were also calculated for our data set to assess for false discovery rate, as described previously.45
Demographic characteristics for our MDACC patient population, including race (Caucasian, African-American, Hispanic and other) and age groups (<40, 40–59 and ≥60 y old) were assessed and reported for this cohort of patients. To examine whether differences exist between men and women or different races logistic regression models were conducted adjusting for age, sex, race and stage of the disease at the time of diagnosis. Standard model selection procedures were used to select the final models.46
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the new investigator funding program from the Ottawa Hospital Research Institute to Dr. Litvinov and the Canadian Dermatology Foundation research grants to Dr. Sasseville and Dr. Litvinov, Joan Sealy Trust Cancer Research Fund (Ottawa Hospital Research Institute) grant to Dr. Litvinov and the Department of Medicine, The Ottawa Hospital research grant to Dr. Litvinov and the Fonds de la recherche en santé du Québec (FRSQ) research grant to Dr. Sasseville (FRQS# 22648). Dr Duvic is a Blanche Bender Professor of Cancer Research and received funding from the Dorothy and Martin Spatz Foundation.
References
- 1.Olsen E, Vonderheid E, Pimpinelli N, Willemze R, Kim Y, Knobler R, Zackheim H, Duvic M, Estrach T, Lamberg S et al.. Revisions to the staging and classification of Mycosis Fungoides and Sezary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood 2007; 110:1713-22; PMID:17540844; https://doi.org/ 10.1182/blood-2007-03-055749 [DOI] [PubMed] [Google Scholar]
- 2.Jawed SI, Myskowski PL, Horwitz S, Moskowitz A, Querfeld C. Primary cutaneous T-cell lymphoma (Mycosis Fungoides and Sezary syndrome): part I. Diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol 2014; 70:205 e1–16; quiz 21–2; PMID:24438969 [DOI] [PubMed] [Google Scholar]
- 3.Kirsch IR, Watanabe R, O'Malley JT, Williamson DW, Scott LL, Elco CP, Teague JE, Gehad A, Lowry EL, LeBoeuf NR et al.. TCR sequencing facilitates diagnosis and identifies mature T cells as the cell of origin in CTCL. Sci Transl Med 2015; 7:308ra158; PMID:26446955; https://doi.org/ 10.1126/scitranslmed.aaa9122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weed J, Girardi M. The difficult–and often delayed–diagnosis of CTCL. Sci Transl Med 2015; 7:308fs41; PMID:26446952; https://doi.org/ 10.1126/scitranslmed.aad2518 [DOI] [PubMed] [Google Scholar]
- 5.Clark RA. Resident memory T cells in human health and disease. Sci Transl Med 2015; 7:269rv1; PMID:25568072; https://doi.org/ 10.1126/scitranslmed.3010641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scarisbrick JJ, Prince HM, Vermeer MH, Quaglino P, Horwitz S, Porcu P, Stadler R, Wood GS, Beylot-Barry M, Pham-Ledard A et al.. Cutaneous Lymphoma International Consortium Study of outcome in advanced stages of Mycosis Fungoides and Sezary syndrome: effect of specific prognostic markers on survival and development of a prognostic model. J Clin Oncol 2015; 33:3766-73; PMID:26438120; https://doi.org/ 10.1200/JCO.2015.61.7142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alberti-Violetti S, Talpur R, Schlichte M, Sui D, Duvic M. Advanced-stage Mycosis Fungoides and Sezary syndrome: survival and response to treatment. Clin Lymphoma, Myeloma leuk 2015; 15:e105-12; PMID:25817937; https://doi.org/ 10.1016/j.clml.2015.02.027 [DOI] [PubMed] [Google Scholar]
- 8.Talpur R, Singh L, Daulat S, Liu P, Seyfer S, Trynosky T, Wei W, Duvic M. Long-term outcomes of 1,263 patients with Mycosis Fungoides and Sezary syndrome from 1982 to 2009. Clin Cancer Res 2012; 18:5051-60; PMID:22850569; https://doi.org/ 10.1158/1078-0432.CCR-12-0604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vidulich KA, Talpur R, Bassett RL, Duvic M. Overall survival in erythrodermic cutaneous T-cell lymphoma: an analysis of prognostic factors in a cohort of patients with erythrodermic cutaneous T-cell lymphoma. Int J Dermatol 2009; 48:243-52; PMID:19261011; https://doi.org/ 10.1111/j.1365-4632.2009.03771.x [DOI] [PubMed] [Google Scholar]
- 10.Wang L, Ni X, Covington KR, Yang BY, Shiu J, Zhang X, Xi L, Meng Q, Langridge T, Drummond J et al.. Genomic profiling of Sezary syndrome identifies alterations of key T cell signaling and differentiation genes. Nat Genet 2015; 47:1426-34; PMID:26551670; https://doi.org/ 10.1038/ng.3444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ungewickell A, Bhaduri A, Rios E, Reuter J, Lee CS, Mah A, Zehnder A, Ohgami R, Kulkarni S, Armstrong R et al.. Genomic analysis of Mycosis Fungoides and Sezary syndrome identifies recurrent alterations in TNFR2. Nat Geneti 2015; 47:1056-60; PMID:26258847; https://doi.org/ 10.1038/ng.3370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sandoval J, Diaz-Lagares A, Salgado R, Servitje O, Climent F, Ortiz-Romero PL, Pérez-Ferriols A, Garcia-Muret MP, Estrach T, Garcia M et al.. MicroRNA expression profiling and DNA methylation signature for deregulated microRNA in cutaneous T-cell lymphoma. J Invest Dermatol 2015; 135:1128-37; PMID:25405321; https://doi.org/ 10.1038/jid.2014.487 [DOI] [PubMed] [Google Scholar]
- 13.McGirt LY, Jia P, Baerenwald DA, Duszynski RJ, Dahlman KB, Zic JA, Zwerner JP, Hucks D, Dave U, Zhao Z et al.. Whole-genome sequencing reveals oncogenic mutations in Mycosis Fungoides. Blood 2015; 126:508-19; PMID:26082451; https://doi.org/ 10.1182/blood-2014-11-611194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.da Silva Almeida AC, Abate F, Khiabanian H, Martinez-Escala E, Guitart J, Tensen CP, Vermeer MH, Rabadan R, Ferrando A, Palomero T. The mutational landscape of cutaneous T cell lymphoma and Sezary syndrome. Nat Genet 2015; 47:1465-70; PMID:26551667; https://doi.org/ 10.1038/ng.3442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scarisbrick JJ. New drugs in cutaneous T-cell lymphomas. Curr Opin Oncol 2016; 28:384-9; PMID:27390044; https://doi.org/ 10.1097/CCO.0000000000000311 [DOI] [PubMed] [Google Scholar]
- 16.Duvic M, Pinter-Brown LC, Foss FM, Sokol L, Jorgensen JL, Challagundla P, Dwyer KM, Zhang X, Kurman MR, Ballerini R et al.. Phase 1/2 study of mogamulizumab, a defucosylated anti-CCR4 antibody, in previously treated patients with cutaneous T-cell lymphoma. Blood 2015; 125:1883-9; PMID:25605368; https://doi.org/ 10.1182/blood-2014-09-600924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wain EM, Mitchell TJ, Russell-Jones R, Whittaker SJ. Fine mapping of chromosome 10q deletions in Mycosis Fungoides and sezary syndrome: identification of two discrete regions of deletion at 10q23.33-24.1 and 10q24.33-25.1. Genes Chromosomes Cancer 2005; 42:184-92; PMID:15540164; https://doi.org/ 10.1002/gcc.20115 [DOI] [PubMed] [Google Scholar]
- 18.van Doorn R, van Kester MS, Dijkman R, Vermeer MH, Mulder AA, Szuhai K, Knijnenburg J, Boer JM, Willemze R, Tensen CP. Oncogenomic analysis of Mycosis Fungoides reveals major differences with Sezary syndrome. Blood 2009; 113:127-36; PMID:18832135; https://doi.org/ 10.1182/blood-2008-04-153031 [DOI] [PubMed] [Google Scholar]
- 19.Laharanne E, Oumouhou N, Bonnet F, Carlotti M, Gentil C, Chevret E, Jouary T, Longy M, Vergier B, Beylot-Barry M et al.. Genome-wide analysis of cutaneous T-cell lymphomas identifies three clinically relevant classes. J Invest Dermatol 2010; 130:1707-18; PMID:20130593; https://doi.org/ 10.1038/jid.2010.8 [DOI] [PubMed] [Google Scholar]
- 20.Vermeer MH, van Doorn R, Dijkman R, Mao X, Whittaker S, van Voorst Vader PC, Gerritsen MJ, Geerts ML, Gellrich S, Söderberg O et al.. Novel and highly recurrent chromosomal alterations in Sezary syndrome. Cancer Res 2008; 68:2689-98; PMID:18413736; https://doi.org/ 10.1158/0008-5472.CAN-07-6398 [DOI] [PubMed] [Google Scholar]
- 21.Caprini E, Cristofoletti C, Arcelli D, Fadda P, Citterich MH, Sampogna F, Magrelli A, Censi F, Torreri P, Frontani M et al.. Identification of key regions and genes important in the pathogenesis of sezary syndrome by combining genomic and expression microarrays. Cancer Res 2009; 69:8438-46; PMID:19843862; https://doi.org/ 10.1158/0008-5472.CAN-09-2367 [DOI] [PubMed] [Google Scholar]
- 22.Talpur R, Sui D, Gangar P, Dabaja BS, Duvic M. Retrospective analysis of prognostic factors in 187 cases of transformed Mycosis Fungoides. Clin Lymphoma, Myeloma Leuk 2016; 16:49-56; PMID:26702474; https://doi.org/ 10.1016/j.clml.2015.11.010 [DOI] [PubMed] [Google Scholar]
- 23.Danish HH, Liu S, Jhaveri J, Flowers CR, Lechowicz MJ, Esiashvili N, Khan MK. Validation of cutaneous lymphoma international prognostic index (CLIPI) for Mycosis Fungoides and Sezary syndrome. Leuk Lymphoma 2016:57(12):2813-2819; PMID:27104864 [DOI] [PubMed] [Google Scholar]
- 24.Vega F, Luthra R, Medeiros LJ, Dunmire V, Lee SJ, Duvic M, Jones D. Clonal heterogeneity in Mycosis Fungoides and its relationship to clinical course. Blood 2002; 100:3369-73; PMID:12384439; https://doi.org/ 10.1182/blood.V100.9.3369 [DOI] [PubMed] [Google Scholar]
- 25.Huang Y, Litvinov IV, Wang Y, Su MW, Tu P, Jiang X, Kupper TS, Dutz JP, Sasseville D, Zhou Y. Thymocyte selection-associated high mobility group box gene (TOX) is aberrantly over-expressed in Mycosis Fungoides and correlates with poor prognosis. Oncotarget 2014; 5:4418-25; PMID:24947046; https://doi.org/ 10.18632/oncotarget.2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dulmage BO, Geskin LJ. Lessons learned from gene expression profiling of cutaneous T-cell lymphoma. Br J Dermatol 2013; 169:1188-97; PMID:23937674; https://doi.org/ 10.1111/bjd.12578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ralfkiaer U, Hagedorn PH, Bangsgaard N, Lovendorf MB, Ahler CB, Svensson L, Kopp KL, Vennegaard MT, Lauenborg B, Zibert JR et al.. Diagnostic microRNA profiling in cutaneous T-cell lymphoma (CTCL). Blood 2011; 118:5891-900; PMID:21865341; https://doi.org/ 10.1182/blood-2011-06-358382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun G, Berthelot C, Li Y, Glass DA 2nd, George D, Pandya A, Kurzrock R, Duvic M. Poor prognosis in non-Caucasian patients with early-onset Mycosis Fungoides. J Am Acad Dermatol 2009; 60:231-5; PMID:19026464; https://doi.org/ 10.1016/j.jaad.2008.09.063 [DOI] [PubMed] [Google Scholar]
- 29.Litvinov IV, Netchiporouk E, Cordeiro B, Dore MA, Moreau L, Pehr K, Gilbert M, Zhou Y, Sasseville D, Kupper TS. The use of transcriptional profiling to improve personalized diagnosis and management of cutaneous T-cell lymphoma (CTCL). Clin Cancer Res 2015; 21:2820-9; PMID:25779945; https://doi.org/ 10.1158/1078-0432.CCR-14-3322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Litvinov IV, Cordeiro B, Huang Y, Zargham H, Pehr K, Dore MA, Gilbert M, Zhou Y, Kupper TS, Sasseville D. Ectopic expression of cancer testis antigens in Cutaneous T-Cell Lymphoma (CTCL) patients. Clin Cancer Res 2014; 20(14):3799-808; PMID:24850846; https://doi.org/ 10.1158/1078-0432.CCR-14-0307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Litvinov IV, Netchiporouk E, Cordeiro B, Zargham H, Pehr K, Gilbert M, Zhou Y, Moreau L, Woetmann A, Ødum N et al.. Ectopic expression of embryonic stem cell and other developmental genes in cutaneous T-cell lymphoma. Oncoimmunology 2014; 3:e970025; PMID:25941598; https://doi.org/ 10.4161/21624011.2014.970025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Litvinov IV, Tetzlaff MT, Rahme E, Habel Y, Risser DR, Gangar P, Jennings MA, Pehr K, Prieto VG, Sasseville D et al.. Identification of geographic clustering and regions spared by cutaneous T-cell lymphoma in Texas using 2 distinct cancer registries. Cancer 2015; 121:1993-2003; PMID:25728286; https://doi.org/ 10.1002/cncr.29301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Litvinov IV, Jones DA, Sasseville D, Kupper TS. Transcriptional profiles predict disease outcome in patients with cutaneous T-cell lymphoma. Clin Cancer Res 2010; 16:2106-14; PMID:20233883; https://doi.org/ 10.1158/1078-0432.CCR-09-2879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Kester MS, Borg MK, Zoutman WH, Out-Luiting JJ, Jansen PM, Dreef EJ, Vermeer MH, van Doorn R, Willemze R, Tensen CP. A meta-analysis of gene expression data identifies a molecular signature characteristic for tumor-stage Mycosis Fungoides. J Inves Dermatol 2012; 132:2050-9; PMID:22513784; https://doi.org/ 10.1038/jid.2012.117 [DOI] [PubMed] [Google Scholar]
- 35.Litvinov IV, Zhou Y, Kupper TS, Sasseville D. Loss of BCL7A expression correlates with poor disease prognosis in patients with early-stage cutaneous T-cell lymphoma. Leuk Lymphoma 2012; 54(3):653-4; PMID:22856870 [DOI] [PubMed] [Google Scholar]
- 36.Vieyra-Garcia PA, Wei T, Naym DG, Fredholm S, Fink-Puches R, Cerroni L, Odum N, O'Malley JT, Gniadecki R, Wolf P. STAT3/5-dependent IL9 overexpression contributes to neoplastic cell survival in Mycosis Fungoides. Clin Cancer Res 2016; 22:3328-39; PMID:26851186; https://doi.org/ 10.1158/1078-0432.CCR-15-1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schrader AM, Jansen PM, Willemze R. TOX expression in cutaneous B-cell lymphomas. Arch Dermatol Res 2016; 308:423-7; PMID:27180090; https://doi.org/ 10.1007/s00403-016-1654-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schrader AM, Jansen PM, Willemze R. TOX expression in cutaneous T-cell lymphomas: an adjunctive diagnostic marker that is not tumour specific and not restricted to the CD4(+) CD8(−) phenotype. Br J Dermatol 2016; 175:382-6; PMID:26931394; https://doi.org/ 10.1111/bjd.14508 [DOI] [PubMed] [Google Scholar]
- 39.Kopp KL, Ralfkiaer U, Gjerdrum LM, Helvad R, Pedersen IH, Litman T, Jønson L, Hagedorn PH, Krejsgaard T, Gniadecki R et al.. STAT5-mediated expression of oncogenic miR-155 in cutaneous T-cell lymphoma. Cell Cycle 2013; 12:1939-47; PMID:23676217; https://doi.org/ 10.4161/cc.24987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krejsgaard T, Vetter-Kauczok CS, Woetmann A, Kneitz H, Eriksen KW, Lovato P, Zhang Q, Wasik MA, Geisler C, Ralfkiaer E, et al.. Ectopic expression of B-lymphoid kinase in cutaneous T-cell lymphoma. Blood 2009; 113:5896-904; PMID:19351960; https://doi.org/ 10.1182/blood-2008-09-181024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shin J, Monti S, Aires DJ, Duvic M, Golub T, Jones DA, Kupper TS. Lesional gene expression profiling in cutaneous T-cell lymphoma reveals natural clusters associated with disease outcome. Blood 2007; 110:3015-27; PMID:17638852; https://doi.org/ 10.1182/blood-2006-12-061507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR et al.. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008; 456:53-9; PMID:18987734; https://doi.org/ 10.1038/nature07517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform 2011; 12:323; PMID:21816040; https://doi.org/ 10.1186/1471-2105-12-323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009; 10:R25; PMID:19261174; https://doi.org/ 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA 2003; 100:9440-5; PMID:12883005; https://doi.org/ 10.1073/pnas.1530509100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hosmer DWL. Applied Logistic Regression. New York, NY: John Wiley & Sons Inc., 2000; 1-397. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.