

ABSTRACT
Tumor microenvironment (TME) contains a variety of infiltrating immune cells. Among them, tumor-associated macrophages (TAMs) and their alternative activation contribute greatly to the progression of tumors. The mechanisms governing macrophage polarization in the TME are unclear. Here, we show that in TAMs or macrophages under tumor-conditioned medium treatment, the expression of transcription factor EB (TFEB) is reduced and more of the TFEB protein is in an inactive cytosolic form. Transforming growth factor (TGF)-β is identified as a main driving force for the reduced TFEB expression and activity in TAMs via activating ERK signaling. TFEB interference in macrophages significantly enhanced their alternative activation, with reduced expression of MHC-II and co-stimulatory molecule CD80, decreased ability to activate T cells, and increased ability to attract tumor cells. When co-inoculated with tumor cells, macrophages with TFEB knockdown significantly enhanced tumor growth with increased infiltration of M2-like macrophages, reduced infiltration of CD8+ T cells, and enhanced angiogenesis in the tumors. Mechanistic studies revealed that TFEB downregulation resulted in macrophage M2 polarization through reducing SOCS3 production and enhancing STAT3 activation. We further demonstrate that the activation of TFEB by hydroxypropyl-β-cyclodextrin in macrophages suppressed their M2 polarization and tumor-promoting capacity, and that macrophage-specific TFEB overexpression inhibited breast tumor growth in mice. Therefore, our data suggest that TFEB plays critical roles in macrophage polarization, and the downregulation of TFEB expression and activation is an integral part of tumor-induced immune editing in the TME. This study provides a rationale for a new cancer treatment strategy by modulating macrophage polarization through activating TFEB.
KEYWORDS: Breast cancer, macrophage, polarization, TFEB, tumor microenvironment
Abbreviations
- HPβCD
hydroxypropyl-β-cyclodextrin
- IFNγ
interferon-gamma
- IL
interleukin
- LLC
Lewis lung carcinoma
- LPS
lipopolysaccharides
- pMΦs
peritoneal macrophages
- SOCS3
suppressor of cytokine signaling 3
- STAT3
signal transducer and activator of transcription 3
- TAMs
tumor-associated macrophages
- TCM
tumor-conditioned medium
- TFEB
transcription factor EB
- TGF-β
transforming growth factor-β
- TME
tumor microenvironment
Introduction
`Tumor microenvironment (TME) comprises a variety of immune cells that modulate tumor initiation and progression.1 Among them, macrophages are a major component of the leukocyte infiltrate of tumors; they are educated in the TME and contribute to tumor growth, angiogenesis, metastasis, and treatment resistance through releasing various soluble factors including chemokines, cytokines, and growth factors.2,3
Macrophages display tremendous plasticity during their activation in response to varied environmental cues. At the two extreme ends of the spectrum of activation, are two polarization states: the classical activation state (M1) and the alternative activation state (M2). Classically activated M1 macrophages, generated upon the exposure to lipopolysaccharides (LPS) and interferon-gamma (IFNγ), produce proinflammatory cytokines, such as interleukin (IL)-12, and exert strong microbicidal and tumoricidal activities. Conversely, in response to Th2 cytokines such as IL-4 and IL-13, macrophages are polarized to an immunosuppressive M2 state and play roles in parasite containment, tissue remodeling, and tumor progression.4 In the TME, tumor cells strategically influence the activation and function of macrophages, educating them into immunosuppressive and tumor-promoting tumor-associated macrophages (TAMs).5 Previous studies suggested that TAMs mainly adopt a M2-like phenotype.6 TAMs execute immune suppressive and tumor-promoting functions by interacting with both tumor cells and other stromal cells, such as endothelial cells, T cells, and fibroblasts, and also represent a major obstacle for effective cancer therapy. Thus, a better understanding of the regulation of TAMs is important for developing more effective therapies for cancers.
The recently discovered transcription factor EB (TFEB) is known to control the expression of genes involved in autophagy and lysosomal biogenesis in response to various stimuli, such as growth factors and nutrients.7-9 TFEB positively regulates cellular degradative pathways and has been demonstrated to increase lysosomal lipid catabolism, lipolysis, and cellular fatty acid oxidation.9,10 Due to its role in stress responses, enhancement of TFEB activity has emerged as a potential therapeutic approach for multiple lysosomal and protein aggregation disorders.11-13 TFEB is also activated in macrophages during bacterial infection or stimulation with bacterial components, suggesting TFEB might perform evolutionarily conserved defense functions in the innate immune system.14-17 In addition, TFEB has been shown to act as a molecular switch that regulates dendritic cell antigen presentation pathways.18 Very recently, TFEB has been shown to be upregulated in cancer cells where it helps to increase tumor malignancy through enhancing cancer cell autophagy.19,20 However, the role and regulation of TFEB in immune cells in tumors, including TAMs, have not been investigated.
Here, we report that TFEB plays a major role in macrophage polarization and function in the context of cancer. The expression and activation of TFEB are suppressed in macrophages in response to cancer cell derived transforming growth factor-β (TGF-β) via elevated ERK signaling. Reduced TFEB expression and activation in macrophages promote their M2-like polarization via reduced SOCS3 expression and enhanced STAT3 signaling. Macrophage–cancer cell co-inoculation experiments demonstrated that knockdown of TFEB in macrophages promoted tumor growth in orthotopic mouse cancer models. Furthermore, chemical activation of TFEB in macrophages by HPβCD inhibited M2 macrophage polarization and suppressed tumor growth, and macrophage specific TFEB overexpression inhibited orthotopic breast tumor growth in mice. Taken together, our study has elucidated the regulation and function of TFEB on macrophage activation in cancer and suggests that TFEB could be a potential therapeutic target in cancer therapy.
Results
TFEB is involved in the regulation of macrophage M2 polarization
We first examined the expression of TFEB in macrophages freshly isolated from EO771 and 4T1 tumors in orthotopic mouse models. The results showed that TFEB mRNA expression in these macrophages was significantly lower than that in naive mouse peritoneal macrophages (pMΦs) (Fig. 1A). To mimic the TME, tumor-conditioned medium (TCM) was collected from LLC or EO771 cell culture and used to treat naive mouse pMΦs. Freshly isolated pMΦs were treated with TCM for 24 h. The results showed that both LLC TCM and EO771 TCM decreased TFEB mRNA and protein levels in pMΦs (Fig. 1B and C); however, EO771 TCM did not have an effect on the expression of two related proteins TFE3 and MITF (Fig. S1A).
Figure 1.
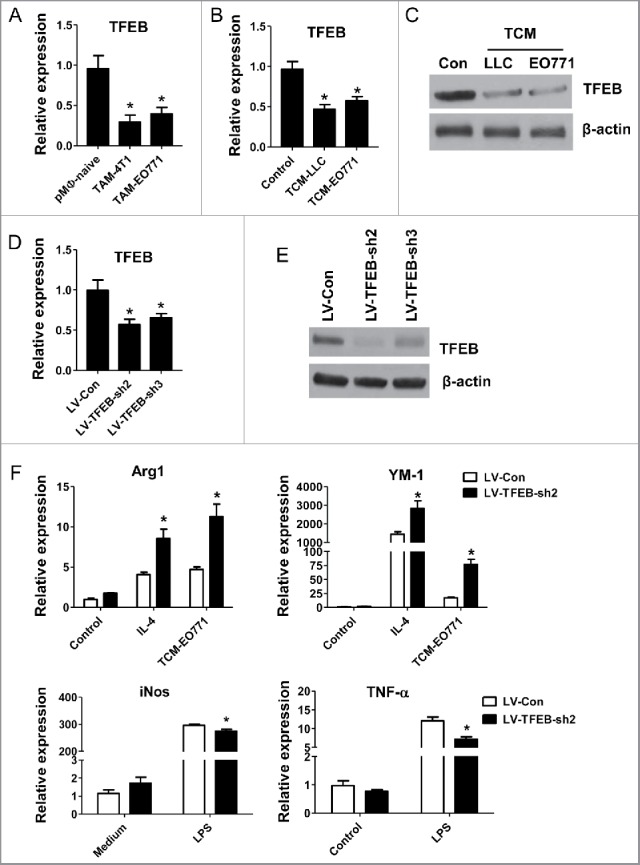
TFEB is involved in the regulation of macrophage M2 polarization. (A) MΦs were isolated from tumor tissues of mice bearing 4T1 (n = 8) or EO771 (n = 8) tumors. TFEB mRNA expression in macrophages was determined by qPCR. *p < 0.05 vs. pMΦ-naive, two-tailed Student's t-test. (B) Peritoneal macrophages were cultured with or without EO771 or LLC tumor-conditioned medium (TCM) for 24 h. *p < 0.05 vs. control; two-tailed Student's t-test. (C) Western blot analysis of TFEB protein in mouse peritoneal MΦs treated as in (B). (D) TFEB expression in mouse peritoneal MΦs transduced with control shRNA lentiviruses (LV-Con) or two different TFEB shRNA lentiviruses (LV-TFEB-sh2 or LV-TFEB-sh3). *p < 0.05 vs. LV-Con; two-tailed Student's t-test. (E) Western blot analysis of TFEB in peritoneal MΦs treated as in (D). (F) Effects of TFEB knockdown using LV-TFEB-sh2 in peritoneal MΦs on the expression of M2 markers Arg-1 and YM-1 and M1 markers iNOS and TNF-α. Peritoneal MΦs were treated with IL-4 (15 ng/mL) or TCM, or LPS (10 ng/mL) for 24 h. *p < 0.05 vs. LV-Con under the same treatment; one-way ANOVA followed by the post-hoc Dunnett's test.
To test whether TFEB plays a role in macrophage polarization in the context of tumors, we transduced pMΦs with TFEB shRNA lentivirus. After transduction with LV-TFEB-Sh2 or LV-TFEB-Sh3 lentiviruses, we observed a 40–50% reduction of TFEB transcript (Fig. 1D), and an 85% (Sh2) or 70% (Sh3) reduction of total TFEB protein compared with control lentivirus (LV-Con) (Fig. 1E). The knockdown effects of LV-TFEB-sh2 lasted at least 7 d (Fig. S1B). We incubated the transduced cells with EO771 TCM, IL-4 or LPS for 24 h, and analyzed the expression of M2 or M1 macrophage markers via qPCR. We found that lentiviral knockdown of TFEB (LV-TFEB-Sh2) significantly enhanced Arg-1 and YM-1 expression in IL-4 or TCM treated macrophages, while it significantly suppressed the expression of iNOS and TNF-α in macrophages treated with LPS (Fig. 1F). To exclude the non-specific activity of lentiviral knockdown vectors, we compared the effects of LV-TFEB-sh2 and LV-TFEB-sh3 on TCM-induced macrophage M2 polarization, and similar results were obtained (Fig. S1C). These data suggest that decreased expression of TFEB in the TME renders macrophages prone to M2-like polarization.
Tumor cell derived TGF-β suppresses TFEB activation and expression in macrophages.
Emerging evidence has shown that cancer cells educate macrophages in the TME as an important component of immune editing.21 Tumor cell derived soluble factors, including IL-10, M-CSF (CSF1), IL-6, and TGF-β, induce macrophages to adopt an M2-like, tumor-promoting phenotype. To examine the regulator of TFEB in the TME, we stimulated pMΦs with several cytokines and growth factors for 24 h. As shown in Fig. 2A and B, only TGF-β strongly inhibited the TFEB production similar to what was observed after TCM treatment; IL-4 and IL-10 did not have an effect, while M-CSF and IL-6 slightly reduced TFEB expression but without statistical significance.
Figure 2.
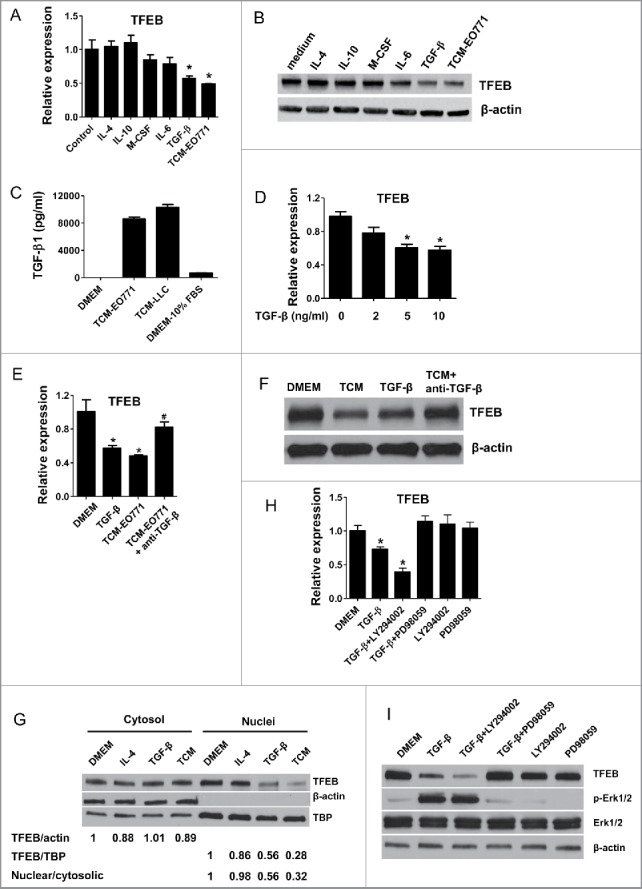
TGF-β decreases the expression of TFEB in macrophages. (A) Mouse peritoneal MΦs were cultured with serum-free DMEM alone (control) or with IL-4 (15 ng/mL), IL-10 (20 ng/mL), m-CSF (25 ng/mL), IL-6 (20 ng/mL), TGF-β (10 ng/mL), or EO771 tumor-conditioned medium for 24 h. TFEB expression was analyzed by qPCR. *p < 0.05 vs. control. (B) Western blot assay of TFEB in MΦs treated as in (A). (C) TGF-β concentrations in indicated media were measured by ELISA. (D) Mouse peritoneal MΦs were treated with TGF-β at various concentrations for 24 h. TFEB expression was analyzed by qPCR. *p < 0.05 vs. control (0 ng/mL TGF-β). (E) Mouse peritoneal MΦs were treated with TGF-β (10 ng/mL) or EO771 TCM in the presence or absence of TGF-β-neutralizing antibody (20 μg/mL) for 24 h, TFEB expression was analyzed by qPCR. *p < 0.05 vs. DMEM. (F) Western blot analysis of TFEB protein in MΦs treated as in (E). (G) Western blot analysis of TFEB protein levels in cytosolic or nuclear subcellular fractions of MΦs treated with IL-4 (15 ng/mL), TGF-β (10 ng/mL), or EO771 TCM. TATA-box-binding protein (TBP) and actin represent control proteins for the nuclear and cytosolic fraction, respectively. Quantification of relative intensity of the protein bands is shown under the lanes. (H) TFEB expression in mouse peritoneal MΦs treated with PD98059 (25 μM), LY294002 (40 μM), and EO771 TCM for 24 h. *p < 0.05 vs. DMEM. (I) Western blot analysis of cell lysates of peritoneal MΦs treated as in (H).
It has been well known that TGF-β is upregulated in tumors and plays a tumor-promoting role via various mechanisms.22,23 Indeed, abundant TGF-β was detected by ELISA in EO771 and LLC TCM (Fig. 2C); and TGF-β at comparable concentrations substantially reduced TFEB expression in MΦs (Fig. 2D). To confirm the role of TGF-β in the regulation of MΦ TFEB in the TME, we administered a TGF-β blocking antibody to examine if this would diminish the effects of TCM on macrophage TFEB expression. As can be seen in Fig. 2E and F, EO771 TCM significantly decreased TFEB expression in pMΦs, whereas the anti-TGF-β antibody reversed the effect.
Previous studies demonstrated that the transcription-regulating activity of TFEB depends on its dephosphorylation and nuclear translocation, rather than its overall cellular protein level.8,24 To test if TGF-β also reduces TFEB nuclear translocation in macrophages, we examined the subcellular localization of TFEB in TGF-β treated pMΦs. Indeed, the result showed that TGF-β and EO771 TCM treatment diminished TFEB nuclear localization (Fig. 2G). These results demonstrate that TGF-β could not only decrease the expression of TFEB, but also suppress its activation (nuclear translocation).
We next examined the mechanism by which TFEB is downregulated by TGF-β. It has been reported that TFEB is inactivated by phosphorylation via phosph-ERK1/2 in the MAPK signaling pathway8 and dephosphorylation and activation of TFEB positively feeds back to further upregulate its own expression.9 MAPK and PI3K-AKT pathways are known to be affected by TGF-β signaling. Thus, we treated pMΦs with TGF-β supplemented with LY294002 (a PI3K inhibitor) or PD980059 (a MEK inhibitor). As shown in Fig. 2H and I, PD980059 significantly prevented TGF-β-induced TFEB downregulation and Erk1/2 phosphorylation, while LY294002 did not have this effect. Taken together, these results indicated that ERK signaling plays a key role in TGF-β-induced downregulation of TFEB in TAMs.
Knockdown of TFEB in macrophages reduces their capacity to activate T cells and increases their capacity to promote tumor cell migration and proliferation
T-cell activation and proliferation are essential to antitumor immune response. When TAMs adopt an M2-like phenotype, they exhibit poor antigen-presenting capacity and instead can suppress T-cell activation and proliferation by producing suppressive mediators.25 We tested whether TFEB knockdown may affect this suppressive function of macrophages on T-cell activation and proliferation. In this assay, macrophages were transduced with control or TFEB shRNA lentiviruses and treated with EO771 TCM; then, CFSE-labeled T cells were co-cultured with these macrophages and stimulated with CD3/CD28. T-cell division was then detected by flow cytometry as a decrease in the intensity of the CFSE signal. Titration of T-cell/macrophage ratios revealed that knockdown of TFEB in macrophages enhanced their ability to suppress T-cell proliferation. When TFEB-knockdown macrophages were over 5% of the total cell number (20:1), proliferation of T cells was markedly reduced compared with T cells co-cultured with control macrophages (Fig. 3A). Additionally, we found that TFEB-knockdown macrophages expressed lower level of CD80 and MHC-II compared with control macrophages (Fig. 3B).
Figure 3.
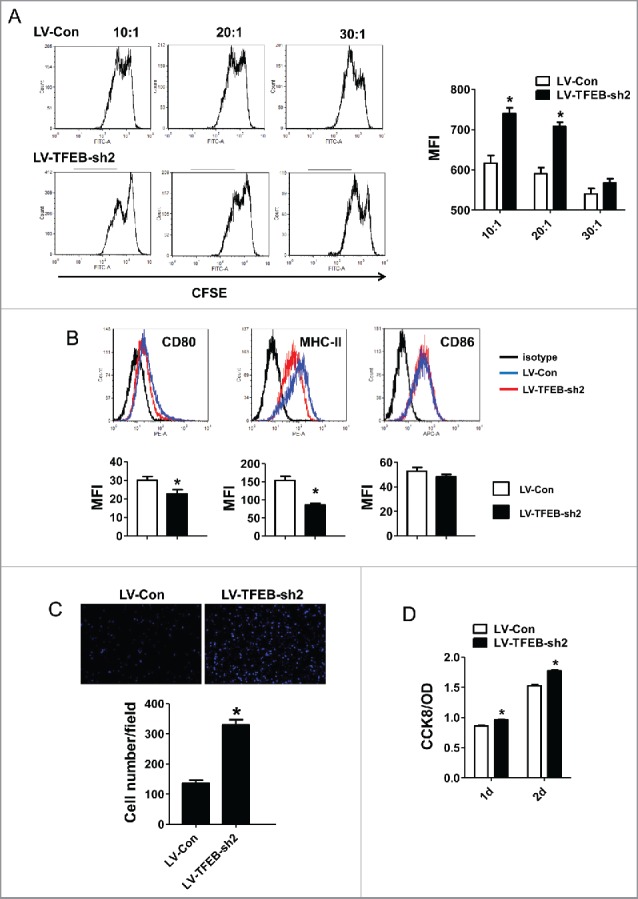
The effects of macrophage TFEB silencing on T-cell activation, proliferation, and tumor cell migration and proliferation. (A) Purified T-cells were labeled with CFSE, activated in vitro by CD3/CD28 and co-cultured with MΦs at the indicated T-cell:MΦ ratio for 60 h in the presence of EO771 TCM. Representative flow cytometry histograms (left) and quantitation (right) of T-cell proliferation as measured by dilution of CFSE are shown. *p < 0.05 vs. corresponding LV-Con group. (B) MΦs were transduced with control or TFEB shRNA lentiviruses and treated with LPS for 24 h. Cell surface CD86, CD80, and MHC II expression was measured by flow cytometry. Representative histograms (upper) and quantification of MFI (lower) were shown. *p < 0.05 vs. LV-Con. (C) TFEB knockdown enhanced the migration of EO771 cells. Transwell assays were performed as described in the Materials and methods section. (D) TFEB silencing in MΦs enhanced proliferation of tumor cells. EO771 cells were cultured in the presence of supernatant from LV-TFEB-sh2 or control virus transduced MΦs as described in the Methods.
M2-like TAMs promote cancer cell migration and proliferation, facilitating tumor development and metastasis.26-28 Transwell migration assays were performed to determine if TFEB knockdown in macrophages promotes tumor cell migration. As shown in Fig. 3C, migration of EO771 cells toward TCM-treated TFEB-knockdown macrophages was significantly enhanced compared with that toward TCM-treated control macrophages, indicating that interference with TFEB activity in alternatively activated macrophages greatly increased their chemoattractant ability for tumor cells. Furthermore, conditioned medium of macrophages with TFEB knockdown significantly increased EO771 cell proliferation compared with that of control macrophages (Fig. 3D).
TFEB knockdown in macrophages enhances tumor growth in co-inoculation mouse models
To examine the effects of TFEB manipulation in TAMs on tumor progression, macrophage-cancer cell co-inoculation mouse models were used. TFEB knockdown macrophages together with EO771 or LLC cancer cells were injected into C57BL/6 mice as described in the Materials and methods section. As shown in Fig. 4A and B, TFEB-knockdown macrophages significantly enhanced the growth of EO771 and LLC tumors, compared with control macrophages.
Figure 4.
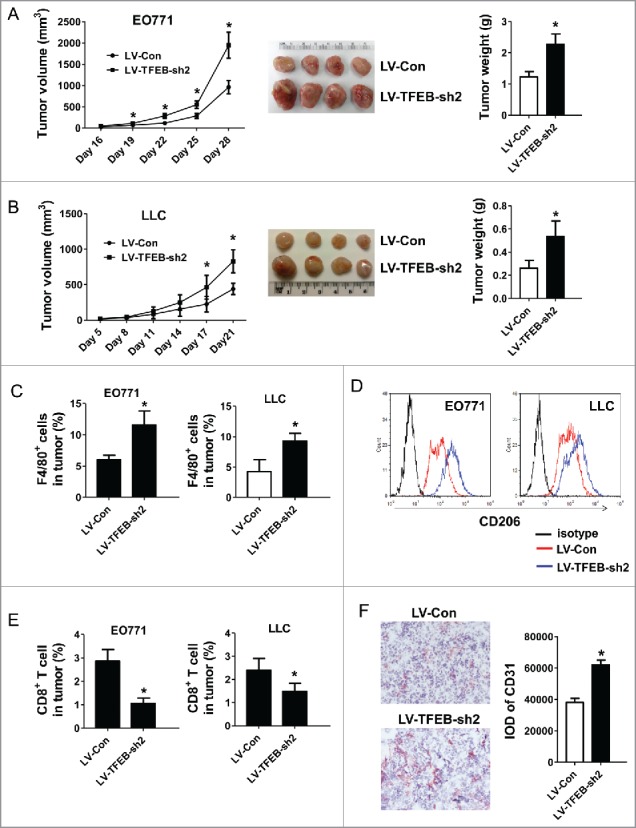
TFEB knockdown in macrophages promotes tumor growth in vivo. EO771 cells (A) or LLC cells (B) were injected into mice along with MΦs transduced with control or TFEB shRNA lentiviruses as described in the Materials and methods section. The tumor size was measured with a caliper at the indicated time points. Tumor volume is shown as mm3. The growth curve, representative images, and quantification of the primary tumors at the end-point are shown. Each data point represents the mean ± SD of eight mice (EO771) or five mice (LLC). *p < 0.05 vs. LV-Con; Student's t-test. (C) Percentage of F4/80+ MΦs in primary tumors at the end-point as determined by flow cytometry. *p < 0.05 vs. LV-Con; Student's t-test. Representative histograms are shown in Fig. S1A. (D) Flow cytometry analysis of CD206 expression in tumor infiltrating MΦs. (E) Percentage of CD8+ T cells in primary tumors at the end-point as determined by flow cytometry. Data are presented as the mean ± SD, *p < 0.05 vs. LV-Con; Student's t-test. Representative histograms are shown in Fig. S1B. (F) Representative immunohistochemical staining images and quantification of CD31 in EO771 tumor sections. *p < 0.05 vs. LV-Con; Student's t-test.
TAMs are a major constituent of the infiltrating inflammatory cells in the TME. It has been reported that increased TAMs are associated with tumor vascularization, metastases, and poor prognosis. Interestingly, although it is unlikely that the initially injected macrophages could survive until the end point of the experiment, initial co-inoculation of TFEB-knockdown macrophages significantly increased the infiltration of total macrophages (F4/80+) at the end point (Figs. 4C and S1A); and these macrophage expressed M2 marker CD206 at a higher level (Fig. 4D). We believe the injected macrophages may have affected the outcome of the endogenous macrophages, thus leading to a long-lasting effect on the TME. In addition, there were fewer CD8+ T cells in the tumors with initial TFEB-knockdown macrophage co-inoculation (Figs. 4E and S1B). Furthermore, we conducted immunohistochemistry to detect the expression of mouse endothelial cell marker CD31, a specific marker of angiogenesis. Co-inoculation with TFEB-knockdown macrophages increased CD31+ cells in the tumors, implying more angiogenesis occurred (Fig. 4F). These results demonstrate that TFEB-knockdown macrophages enhance the progression of tumors.
STAT3 is involved in TFEB-knockdown mediated promotion of macrophage M2 polarization
We next sought to examine the molecular mechanisms by which TFEB-knockdown increased macrophage M2 polarization. It has recently been shown that suppressor of cytokine signaling 3 (SOCS3) is induced by TFEB during infection, and SOCS3 then inhibits the action of signal transducer and activator of transcription 3 (STAT3).4 We found that IL-4 and TCM-treated TFEB-knockdown macrophages exhibited a significantly decreased level of SOCS3 compared with control macrophages (Fig. 5A). As shown in Fig 5B, TFEB knockdown in macrophages enhanced the activation of STAT3 post IL-4 and TCM stimulation, and a reverse relationship was observed with SOCS3, while phosphorylation of STAT6 did not show any difference. The STAT3 signaling pathway plays critical roles in the macrophage M2 polarization.4 To directly investigate the contribution of STAT3 activation to the enhanced M2 polarization of TFEB-knockdown macrophages, we used the STAT3 inhibitor stattic, which inhibited phosphorylation of STAT3.29 We found that stattic significantly diminished STAT3 phosphorylation in IL-4 or TCM treated macrophages (Fig. 5C). Importantly, as shown in Fig. 5D, stattic treatment completely blocked TFEB-knockdown enhanced M2 polarization in macrophages induced by IL-4 or TCM, but had much less profound effects on control macrophages. These data suggested that TFEB-knockdown mediated promotion of macrophage M2 polarization is mainly due to increased activation of the STAT3 signaling pathway.
Figure 5.
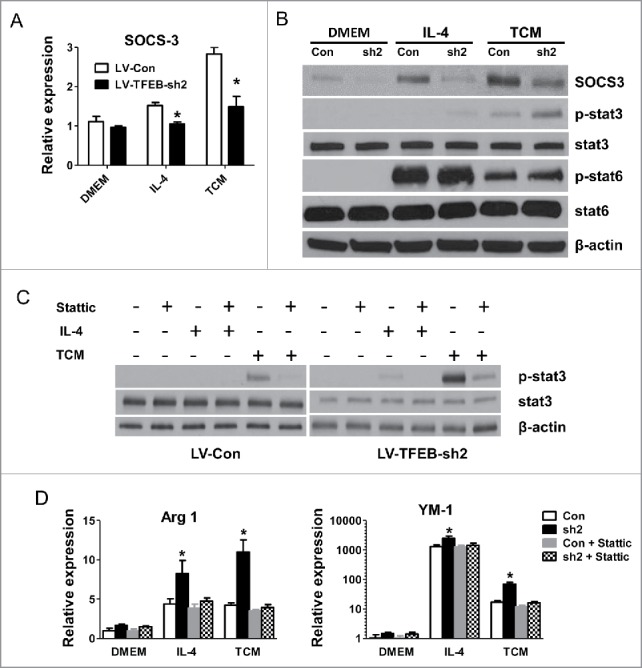
SOCS3-STAT3 pathway plays a key role in TFEB silencing-enhanced macrophage M2 polarization. (A) MΦs were transduced with control or TFEB shRNA lentiviruses and treated with IL-4 (15 ng/mL) or EO771 TCM for 24 h. The expression of SOCS3 was analyzed by qPCR. *p < 0.05 vs. corresponding LV-Con group. (B) Cellular proteins were analyzed by Western blot analysis after the cells were treated as in (A). (C) Cellular proteins were analyzed by Western blot analysis after MΦs were treated as in (A), with or without pretreatment with stattic (5 μM) for 2 h. (D) MΦs were treated as in (C). The expression of Arg1 and YM-1 mRNA was analyzed by qPCR. *p < 0.05 vs. corresponding control.
Activation of TFEB by HPβCD attenuates macrophage M2 polarization and their tumor-promoting properties
To explore the translatability of the above findings, we tested if pharmacological activation of TFEB can alter macrophage phenotype and function in the context of tumors. A chemical named 2-hydroxypropyl-β-cyclodextrin (HPβCD) was recently reported to function as a direct chemical activator of TFEB.30,31 We found that HPβCD treatment significantly increased nuclear translocation of TFEB in pMΦs (Fig. 6A), indicating TFEB was activated and adopted a dephosphorylated form. HPβCD treatment under these conditions was also confirmed not to induce apoptosis of macrophages (data not shown).
Figure 6.
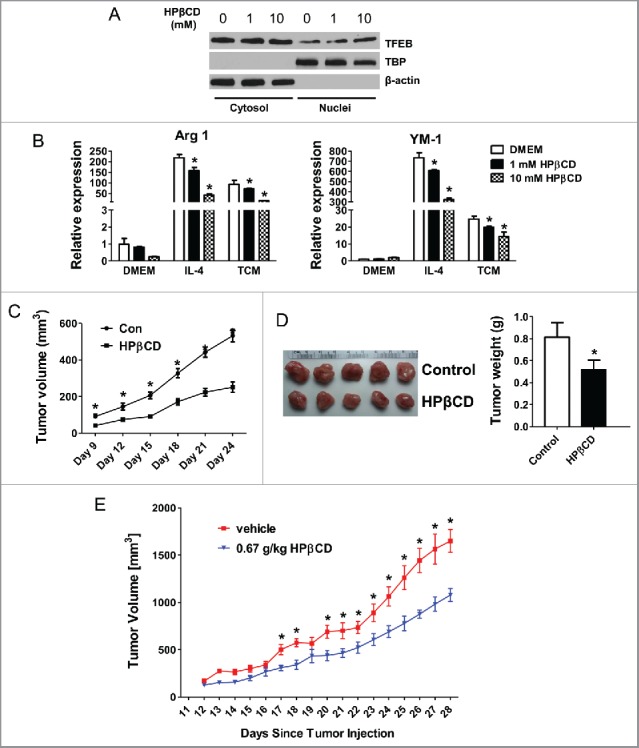
Activation of TFEB suppresses tumor growth in mice. (A) Western blot analysis of cytosolic and nuclear fractions obtained from MΦs treated with HPβCD for 24 h. (B) Expression of Arg1 and YM-1 in MΦs treated with IL-4 (15 ng/mL) or EO771 TCM with or without HPβCD for 24 h. (C) 4T1 cells and HPβCD-treated macrophages were co-injected into BALB/c mice. The tumor size was measured with a caliper at the indicated time points. Tumor volume is shown as mm3. Primary tumor growth curve is shown. Each data point represents the mean ± SD of 10 mice. *p < 0.05 vs. Con. (D) Representative images of primary tumors in mice at the end-point are shown (left); and tumor weight was measured and analyzed (right). (E) Effects of intraperitoneal administration of HPβCD on the tumor growth in Balb/c mice inoculated with 4T1 breast cancer cells (n = 10). Data are presented as the mean ± SD, *p < 0.05 vs. control; Student's t-test.
To evaluate the impact of HPβCD-induced TFEB activation on macrophage polarization, we treated pMΦs with HPβCD alone, or with IL4 or EO771 TCM. We found HPβCD dose-dependently reduced Arg-1 and YM-1 expression in IL-4 or TCM treated pMΦs after 24 h treatment, indicating an attenuation of M2 polarization by HPβCD (Fig. 6B). The impact of HPβCD on TCM-induced macrophage M2 polarization maintained after 48 h (Fig. S3A). Consistently, LPS-stimulated macrophages displayed enhanced expression of M1 markers iNOS and IL-12 with HPβCD treatment (Fig. S3B).
We then tested the effects of TFEB activation in macrophages on tumor growth. TCM-treated macrophages with or without HPβCD treatment were mixed with 4T1 cells and were co-inoculated in BALB/c mice. Tumor growth was monitored for 24 d. The results showed that co-inoculation of HPβCD-treated macrophages significantly halted tumor growth in mice, compared with co-inoculation of control macrophages (Fig. 6C and D). Taken together, our data suggested that activation of TFEB in macrophages by HPβCD attenuates the M2-like phenotype and pro-tumor function of TAMs.
We further explored the possibility to use HPβCD as a therapy for breast cancer. Ten-week-old female Balb/c mice were inoculated 2 × 105 4T1 cells in the fourth mammary fat pad. After 3 d, mice began daily intraperitoneal injections with 0.67 g/kg HPβCD in PBS, or same volume of PBS as control. These injections continued for 4 weeks during which tumor size were measured daily. Fig. 6E shows that HPβCD injection significantly halted tumor growth in mice.
Overexpression of TFEB in macrophages suppresses breast tumor growth
To further examine the impact of macrophage TFEB on breast tumor growth, we generated chimeric mice to introduce TFEB overexpression specifically in macrophages. Mice with macrophage specific overexpression of TFEB (MΦ-TFEBtg) were generated and characterized as described previously in Dr. Babak Razani's laboratory at Washington University, St. Louis. 12 We transplanted bone marrow cells from female MΦ-TFEBtg mice (TFEBtg bone marrow) or control LysM-Cre mice (WT bone marrow) to lethally irradiated female WT C57Bl/6 mice. Four weeks after the bone marrow transplantation, the recipients were inoculated with EO771 cells, and the tumor growth was monitored for 34 d. The mice were then killed and leukocytes in the tumors were analyzed by flow cytometry. As shown in Fig. 7A, the tumor growth was significantly suppressed in the mice received bone marrow cells with macrophage specific TFEB overexpression. At the end point, the tumors were significantly smaller in these mice (Fig. 7B). Flow cytometry analysis showed that although the total leukocytes (CD45+ cells) in the tumors, the percentage of macrophages (F4/80+ cells) in tumor-infiltrating leukocytes, and the percentage of M2 macrophages (CD206+ cells) in tumor-infiltrating macrophages were not different in the tumors of the two groups of mice (Fig. 7C), CD206 intensity in tumor-infiltrating macrophages in the mice with macrophage TFEB overexpression was significantly lower (Fig. 7D). Moreover, CD3+CD8+ T cells were significantly increased in the tumors of the mice with macrophage TFEB overexpression (Fig. 7E).
Figure 7.
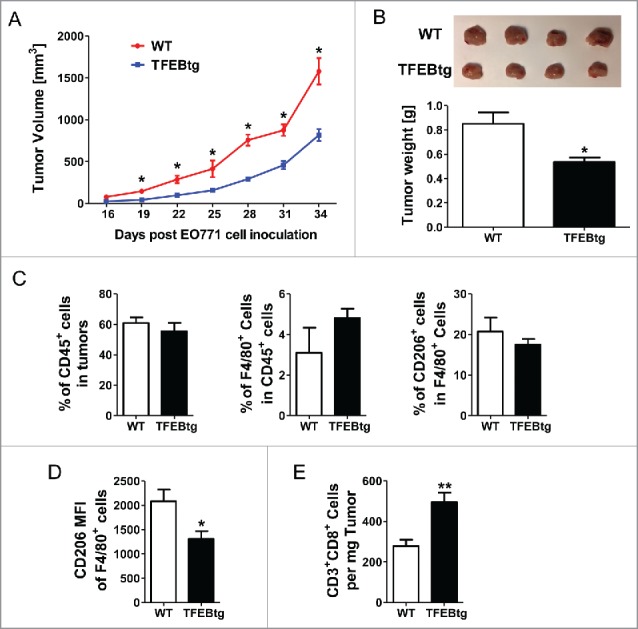
Macrophage specific TFEB overexpression suppressed breast tumor growth. Female C57BL/6 mice were transplanted with bone marrow cells from either control LysM-Cre mice (WT bone marrow) or macrophage specific TFEB transgenic mice (TFEBtg bone marrow) and inoculated with EO771 cells in mammary fat pads. Tumor growth curve (A) and representative tumor images and tumor weight quantification at the end point (B) are shown. Tumor tissue leukocytes were analyzed by flow cytometry. Percentage of CD45+ leukocytes in tumor cells, percentage of F4/80+ macrophages in total leukocytes, and percentage of CD206+ M2 macrophages in total macrophages (C). Mean fluorescent intensity (MFI) of CD206 in all F4/80+ cells (D), and the number of CD3+CD8+ T cells in tumor tissue (E) are shown. *p < 0.05; **p < 0.01; Student's t-test; n = 4–5.
Discussion
TFEB has been recognized as an important regulator of autophagy and lysosomal biogenesis and function, implying that it may play a role in many autophagy defect-related conditions and lysosomal and protein aggregation disorders.8,11-13 It has recently become appreciated that TFEB is linked to innate and adaptive immunity. For instance, TFEB is activated and required for a proper host response in macrophages infected by Staphylococcus aureus15 and is involved in IFN-independent activation of antiviral genes.32 It has also been reported that TFEB is critical for T-cell function and humoral immunity through its direct control of CD40L expression.33 Therefore, TFEB has been proposed as a key regulator of the immune system. However, the roles of TFEB in cancer immunity have not been studied. Here, for the first time, we demonstrated that the decreased TFEB expression in TAMs may be an intrinsic feature of the TME, and reduction of TFEB expression and activation confers TAMs an M2-like phenotype and renders them more tumor-promoting. We provide evidence that the macrophage polarization modulating effects of TFEB are controlled through a TFEB-SOCS3-STAT3 signaling pathway. We further showed that knockdown of TFEB in macrophages promoted tumor growth, and chemical activation of TFEB in macrophages or macrophage specific TFEB overexpression inhibited tumor growth in mice. This study suggests that macrophage TFEB may be a potential target for cancer therapy.
TAMs have been shown to promote tumor growth, angiogenesis, invasion, and metastasis in many cancer types. TAMs are generally thought to more closely resemble the M2-polarized phenotype.6 The TME includes several soluble factors, such as IL-4, M-CSF, TGF-β, and IL-10, all of which lead to polarization of macrophages toward an M2 phenotype.5,34 Our data suggested that the factors in the TME, particularly TGF-β, decreased TFEB expression in macrophages. TGF-β has been reported to be a multifunctional cytokine in cell proliferation, differentiation, and apoptosis.22 It has been found that there is a marked increase in the expression of TGF-β mRNA and protein in cancers. In many of these cancers, high expression of TGF-β correlates with more advanced stages of malignancy and decreased survival.23 Our study provides a novel mechanism by which TGF-β executes its tumor-promoting function. We also noticed that M-CSF and IL-6 also slightly reduced TFEB expression although without statistical significance; we believe it is possible that some other tumor-derived factors in addition to TGF-β in the TME may also impact TFEB activation in TAMs to various extents. This warrants further investigation.
It has been reported that TFEB activity is modulated by phosphorylation and the positive feedback of TFEB on its own expression.9 Upstream negative regulation by the kinases mTORC1 and ERK2 maintains TFEB in an inactive state.8,24,35 TGF-β utilizes a multitude of intracellular signaling pathways including various branches of the MAP kinase pathways and phosphatidylinositol-3-kinase/AKT/mTOR pathways.22,36 Therefore, we hypothesized that TGF-β might use one of these signaling axes to regulate TFEB expression and activity. In support of this hypothesis, we observed that a MEK inhibitor, PD980059, significantly reversed TGF-β-induced downregulation of TFEB, indicating that TGF-β inhibits TFEB transcription by activating ERK signaling.
It has recently been shown that suppressor of cytokine signaling 3 (SOCS3) might be induced by TFEB during infection.15 In accordance with this, our data demonstrated TFEB knockdown indeed decreased SOCS3 expression in IL-4 or TCM-treated macrophages. STATs are well-known transcription factors with important roles in macrophage polarization.4,37 STAT-mediated activation of macrophages is regulated by members of the SOCS family including SOCS3.38 STAT3 and STAT6 activation results in M2 macrophage polarization and associated immune suppression and tumor progression.37 Our results showed IL-4 and TCM induced a dramatic elevation of STAT3 phosphorylation in TFEB knockdown macrophages, while TFEB knockdown did not have an effect on STAT6 phosphorylation. Furthermore, the inhibition of STAT3 activation could sufficiently block TFEB knockdown-enhanced M2 macrophage polarization. Collectively, our data suggested that TFEB regulates macrophage M2 polarization through the SOCS3-STAT3 signaling axis.
While TFEB is well known as a master regulator of autophagy and lysosomal biogenesis,7,8 interestingly, not much is known regarding the role of autophagy and lysosome function in macrophage polarization, reports have suggested that autophagy actually promotes macrophage M2 polarization,39 and lysosomal lipolysis is essential for alternative activation of macrophages;40 however, our results showed that TFEB knockdown promoted macrophage M2 polarization, suggesting that the M2 polarization regulating effects of TFEB in the TME may be independent of its roles in autophagy and lysosome biogenesis. This warrants further investigation.
We evaluated the effects of macrophage TFEB knockdown and activation on tumor growth using orthotopic tumor models. Our data show that knockdown of TFEB in macrophages significantly promoted tumor cell proliferation, migration, and T-cell immunosuppression in vitro, and accelerated tumor growth in vivo by increasing M2 macrophages, reducing CD8+ T cells, and enhancing angiogenesis in tumors. Conversely, chemical activation of TFEB in macrophages or macrophage-specific TFEB overexpression suppressed their M2 polarization and significantly attenuated tumor growth in mice.
On the basis of our data, we propose a model to describe the regulation and function of TFEB in TAMs as follows. Tumor cells produce and secrete TGF-β, which acts on the TAMs in the TME and increases the phosphorylation of Erk. Phosphorylated Erk increases phosphorylation of TFEB and thus reduces TFEB nuclear translocation. Reduced nuclear translocation of TFEB downregulates its own expression as well as the expression of SOCS3. Reduced SOCS3 leads to de-repression of STAT3 phosphorylation and thus its activation. Activation of STAT3 promotes M2-like polarization of TAMs. In these M2-like TAMs, the expression of Arg 1 and YM-1 is increased while the expression of CD80 and MHC-II is decreased. These TAMs acquire tumor-promoting functions, including promoting angiogenesis, tumor cell proliferation and migration, and inhibiting T-cell activation (Fig. 8). In conclusion, this study suggests that modulation of macrophage TFEB expression and activation may be of therapeutic value for cancers.
Figure 8.
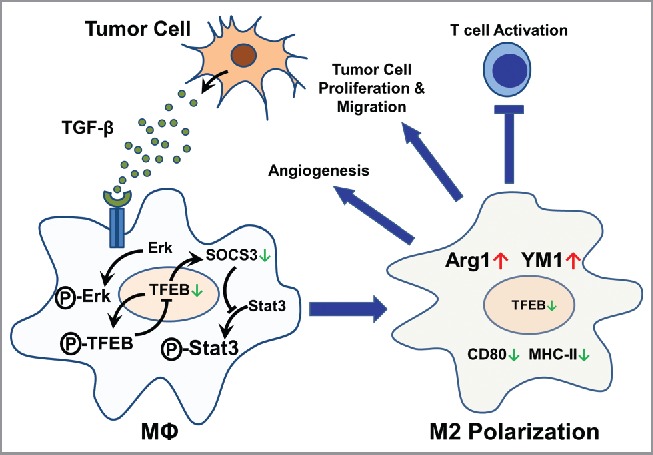
A model depicting the regulation and the role of TFEB in macrophage polarization in tumors.
Materials and methods
Animals
BALB/c and C57BL/6 mice (all female, 6–8 weeks old) were purchased from Jackson Laboratories (Bar Harbor, Maine) and housed in the University of South Carolina Animal Research Facility. Animal care procedures and experimental methods were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of South Carolina according to National Institutes of Health guidelines.
Cell culture
Lewis lung carcinoma (LLC) and 4T1 mouse mammary tumor cell lines were obtained from the American Type Culture Collection (ATCC). EO771 cells, developed from an ER+ spontaneous mammary adenocarcinoma,41,42 were maintained in culture as described previously.43 LLC cells were used within 6 mo after being obtained from ATCC. EO771 and 4T1 cell lines were authenticated in 2016 by IDEXX Laboratories (IDEXX BioResearch Case #7479-2016). The samples were confirmed to be of mouse origin and no mammalian interspecies contamination was detected. A genetic profile was generated for each sample using a panel of microsatellite markers for genotyping. The 4T1 cells were confirmed to match identically to the genetic profile established for this cell line. The genetic profile for the EO771 cell line is more consistent with having been derived from a mouse with a mixed/stock genetic background. A very similar profile has been seen in other sources of EO771 cell lines.44 The cells were cultured in high glucose Dulbecco's modified Eagle medium (DMEM; Invitrogen Life Technologies) with 10% FBS (Invitrogen) and penicillin/streptomycin at 37 °C in a humidified 5% CO2 atmosphere.
Tumor cell injection
For tumor growth experiment, 2 × 106 LLC cells (in 200 μL PBS) were subcutaneously injected into the back of each mouse. 4T1 cells (2 × 105) or EO771 cells (2 × 105) in 20 μL PBS were injected into both sides of the 4th pair of mammary fat pads of BALB/c or C57BL/6 mice, respectively. Tumor cells were mixed with mouse peritoneal macrophages (MΦs) (3:1) transduced with control lentivirus (LV-Con) or TFEB shRNA lentivirus (LV-TFEB-sh2) and pretreated with corresponding TCM for 24 h. After tumor cell inoculation, the tumors were measured using calipers every 3 d. The tumor volume was determined by the formula: (length × width2)/2.
Tumor conditioned medium
To obtain TCM, LLC or EO771 cells were seeded at 5 × 106 cells per 75-cm2 dish and cultured to 90% confluence. The media were then replaced with serum-free DMEM. After 24 h, the supernatant was collected and filtered through a 0.45-μm filter.
Cell isolation
Splenocytes were isolated by mechanical disruption. Tumors were weighed, cut into small fragments (< 3 mm) and digested in 5 mL of dissociation solution (RPMI 1640 medium supplemented with 10% FBS, Collagenase type I (200 U/mL) and DNase I (100 μg/mL)) for 60 min at 37 °C. Erythrocytes were lysed with red blood cell lysing buffer (Sigma, St. Louis, MO). Cell suspensions were passed through 70-μm cell strainers, then washed and resuspended in staining buffer. TAMs (F4/80+) were isolated from tumor suspensions by positive selection using the magnetic beads.
In vitro macrophage treatment
BALB/c or C57BL/6 mice were injected with 3 mL of 3% thioglycollate in sterile PBS intraperitoneally. Three days later, mice were killed and peritoneal macrophages (pMΦs) were harvested by lavaging the peritoneal cavity with 2 × 10 mL PBS. Macrophages were resuspended with DMEM containing 10% FBS and seeded in 6-well or 12-well plates. The non-adherent cells were removed by washing with PBS 2 h later, and the adhered macrophages were further cultured in serum-free DMEM overnight. Macrophages were then treated with serum-free DMEM, TCM or IL-4 (15 ng/mL, AASN BioAbChem Inc., Ladson, SC) for the indicated period of time.
Transwell migration assay
Cell migration assays were performed in 24-well plates with 8-μm polyethylene terephthalate membrane filters (Corning, USA) separating the lower and upper culture chambers. MΦs transduced with LV-TFEB-sh2 lentiviruses (LV-Con as control) were cultured in the lower chambers. TCM was added to the lower chambers. EO771 cells (1 × 104 cells) suspended in 100 μL serum-free DMEM were added to the upper chamber. After 2 h, tumor cells that migrated across the insert were stained with DAPI and counted under an inverted wide field fluorescence microscope at 400× magnification (20 fields per well, triplicate for each experimental group).
T-cell–macrophage co-culture proliferation assay
Resident peritoneal MΦs were isolated from mice as described above. Splenocytes were isolated from another C57BL/6 mouse. The spleen was removed and gently squeezed between frosted glass slides, and red blood cells were lysed with a hypotonic solution. T cells were purified using the CD4+ T Cell Isolation kit from STEMCELL Technologies by negative selection. The resulting purified T-cell suspension, lacking platelets and RBCs, was labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE, BioLegend). CFSE-labeled T cells (1 × 105) were plated in 24-well CD3+ antibody-coated (5 μg/mL) plates with CD28 soluble antibody (1 μg/mL) added to the medium to induce T-cell proliferation either with or without MΦs at an indicated ratio. MΦs were added 3 h after T-cell activation. Cells were then incubated for 60 h and then analyzed by flow cytometry.
Flow cytometry
Cells were stained with anti-CD80 PE mAb, anti-CD86 APC mAb, anti-CD3 PE mAb, anti-MHC-II PE mAb, anti-F4/80 FITC mAb, or anti-CD206 PE mAb (all from eBioscience, San Diego, CA) in staining buffer (PBS containing 2% FBS) for 30 min on ice in the dark. Samples were washed twice with staining buffer, and then analyzed by flow cytometry using a FACSAria flow cytometer.
Immunohistochemistry
Primary tumor tissues were fixed in 4% paraformaldehyde at 4 °C for 12 h, dehydrated in 30% sucrose overnight, and embedded in OCT. Serial sections (8 µm thick) were cut throughout the entire tumor tissue. For CD31 staining, the tumor tissue sections were fixed in 4% paraformaldehyde, rinsed with PBS, and incubated with 0.3% H2O2 in methanol for 10 min. After washing with PBS, anti-mouse CD31 antibody (1:200; Abcam) was applied. Immunocomplexes were detected with biotin-conjugated secondary antibodies and AEC chromogen/HRP substrate kit (GeneTex). The sections were counterstained with hematoxylin and mounted with a permanent mounting medium. Microvessel density was assessed with CD31 staining and counted on 200× magnification fields. CD31-positive endothelial cells or cell clusters clearly separate from adjacent structures were considered single vessels.
Quantitative real-time PCR (qPCR)
Total RNA was extracted using the TRIzol reagent (Invitrogen). RNA (1 μg) was reverse-transcribed using iScript cDNA Synthesis Kit (Bio-Rad, Life Science). qPCR was conducted on a CFX96 system (Bio-Rad) using iQ SYBR Green Supermix (Bio-Rad). All primers used for qPCR analysis were synthesized by Integrated DNA Technologies. All assays were conducted following the manufacturer's instructions. The relative amount of target mRNA was determined using the comparative threshold (Ct) method by normalizing target mRNA Ct values to those of 18S RNA. PCR thermal cycling conditions were 3 min at 95°C, and 40 cycles of 15 s at 95°C and 58 s at 60°C. Samples were run in triplicate. The primer sequences are listed in Table S1.
Western blot analysis
Cells were lysed in RIPA buffer (Pierce) supplemented with protease inhibitor cocktail and phosphatase inhibitor cocktail (Sigma). Total cellular extract (20 μg) was separated in 10% SDS-PAGE precast gels (Bio-Rad) and transferred onto nitrocellulose membranes (Millipore Corp.). Membranes were first probed with TFEB (1:2,000; Bethyl Laboratories, Inc.), TBP (1:1,000; Abcam), ERK1/2, phosphor-ERK1/2, STAT6, phosphor-STAT6 (1:1000, Cell Signaling), STAT3, phosphor-STAT3 (1:1,000 Santa cruz), SOCS3 (1:500 Proteintech), or β-actin (1:1,000; Sigma) antibodies, followed by goat anti-rabbit secondary antibody conjugated with horseradish peroxidase (HRP; Millipore). Protein detection was conducted using Pierce ECL Western Blotting Substrate (Pierce). Image-Pro Plus 6.0 analysis software was used to quantify signal intensities.
Lentiviral vector generation and lentiviral transduction
General procedures for lentivirus preparation and macrophage transduction were described previously.45-47
Bone marrow transplantation
Bone marrow cells from macrophage-specific TFEB overexpression mice12 were obtained from Washington University at St. Louis. Bone marrow transplantation was performed as described previously.48,49
Statistical analysis
Data were presented as mean ± SD or SEM as indicated. Statistical significance was calculated using the Student's t-test (two group comparison) or one-way ANOVA followed by post-hoc Dunnett's test (multi-group comparison) using the GraphPad Prism statistical program (GraphPad Prism; GraphPad Software, Inc.). p < 0.05 was considered significant.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgment
We would like to acknowledge Dr. Adam Hartstone-Rose for his assistance in the preparation of the figures.
Funding
This work was funded by NIH grants HL116626 and AT003961-8455 to DF, and HL125838 to BR.
Author contributions
L.F. and D.F. designed the experiments and wrote the manuscript. L.F. performed most of the study and performed the statistical analyses. J.W., S.I., F.S., Y.W. J.H., and T.D.E. assisted in sacrificing mice and collecting samples for the animal studies. Y.W. performed immunohistochemistry analysis. Y.H. assisted in lentivirus preparation. B.R. helped in interpreting the data, and in writing the manuscript. All authors have agreed to the submission of this manuscript for publication.
References
- 1.de Visser KE, Coussens LM. The inflammatory tumor microenvironment and its impact on cancer development. Contrib Microbiol 2006; 13:118-37; PMID:16627962; https://doi.org/ 10.1159/000092969 [DOI] [PubMed] [Google Scholar]
- 2.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell 2010; 141:39-51; PMID:20371344; https://doi.org/ 10.1016/j.cell.2010.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Doedens AL, Stockmann C, Rubinstein MP, Liao D, Zhang N, DeNardo DG, Coussens LM, Karin M, Goldrath AW, Johnson RS. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res 2010; 70:7465-75; PMID:20841473; https://doi.org/ 10.1158/0008-5472.CAN-10-1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 2012; 122:787-95; PMID:22378047; https://doi.org/ 10.1172/JCI59643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ostuni R, Kratochvill F, Murray PJ, Natoli G. Macrophages and cancer: from mechanisms to therapeutic implications. Trends Immunol 2015; 36:229-39; PMID:25770924; https://doi.org/ 10.1016/j.it.2015.02.004 [DOI] [PubMed] [Google Scholar]
- 6.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 2002; 23:549-55; PMID:12401408; https://doi.org/ 10.1016/S1471-4906(02)02302-5 [DOI] [PubMed] [Google Scholar]
- 7.Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, et al.. A gene network regulating lysosomal biogenesis and function. Science 2009; 325:473-7; PMID:19556463; https://doi.org/ 10.1126/science.1174447 [DOI] [PubMed] [Google Scholar]
- 8.Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P et al.. TFEB links autophagy to lysosomal biogenesis. Science 2011; 332:1429-33; PMID:21617040; https://doi.org/ 10.1126/science.1204592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Klisch TJ et al.. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol 2013; 15:647-58; PMID:23604321; https://doi.org/ 10.1038/ncb2718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schilling JD, Machkovech HM, He L, Diwan A, Schaffer JE. TLR4 activation under lipotoxic conditions leads to synergistic macrophage cell death through a TRIF-dependent pathway. J Immunol 2013; 190:1285-96; PMID:23275600; https://doi.org/ 10.4049/jimmunol.1202208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Decressac M, Bjorklund A. TFEB: Pathogenic role and therapeutic target in Parkinson disease. Autophagy 2013; 9:1244-6; PMID:23715007; https://doi.org/ 10.4161/auto.25044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Emanuel R, Sergin I, Bhattacharya S, Turner JN, Epelman S, Settembre C, Diwan A, Ballabio A, Razani B. Induction of lysosomal biogenesis in atherosclerotic macrophages can rescue lipid-induced lysosomal dysfunction and downstream sequelae. Arterioscler Thromb Vasc Biol 2014; 34:1942-52; PMID:25060788; https://doi.org/ 10.1161/ATVBAHA.114.303342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Bjorklund A. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc Natl Acad Sci USA 2013; 110:E1817-26; PMID:23610405; https://doi.org/ 10.1073/pnas.1305623110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tiller GR, Garsin DA. Of worms and men: HLH-30 and TFEB regulate tolerance to infection. Immunity 2014; 40:857-8; PMID:24950206; https://doi.org/ 10.1016/j.immuni.2014.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Visvikis O, Ihuegbu N, Labed SA, Luhachack LG, Alves AM, Wollenberg AC, Stuart LM, Stormo GD, Irazoqui JE. Innate host defense requires TFEB-mediated transcription of cytoprotective and antimicrobial genes. Immunity 2014; 40:896-909; PMID:24882217; https://doi.org/ 10.1016/j.immuni.2014.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gray MA, Choy CH, Dayam RM, Ospina-Escobar E, Somerville A, Xiao X, Ferguson SM, Botelho RJ. Phagocytosis enhances lysosomal and bactericidal properties by activating the transcription factor TFEB. Curr Biol 2016; 26:1955-64; PMID:27397893; https://doi.org/ 10.1016/j.cub.2016.05.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pastore N, Brady OA, Diab HI, Martina JA, Sun L, Huynh T, Lim JA, Zare H, Raben N, Ballabio A, Puertollano R. TFEB and TFE3 cooperate in the regulation of the innate immune response in activated macrophages. Autophagy 2016; 12:1240-1258; PMID:27171064; https://doi.org/ 10.1080/15548627.2016.1179405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Samie M, Cresswell P. The transcription factor TFEB acts as a molecular switch that regulates exogenous antigen-presentation pathways. Nat Immunol 2015; 16:729-736; PMID:26030023; https://doi.org/ 10.1038/ni.3196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, Lengrand J, Deshpande V, Selig MK, Ferrone CR et al.. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 2015; 524:361-5; PMID:26168401; https://doi.org/ 10.1038/nature14587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giatromanolaki A, Kalamida D, Sivridis E, Karagounis IV, Gatter KC, Harris AL, Koukourakis MI. Increased expression of transcription factor EB (TFEB) is associated with autophagy, migratory phenotype and poor prognosis in non-small cell lung cancer. Lung Cancer (Amsterdam, Netherlands) 2015; 90:98-105; PMID:26264650; https://doi.org/ 10.1016/j.lungcan.2015.07.008 [DOI] [PubMed] [Google Scholar]
- 21.Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity 1951; 41:49-61; PMID:25035953; https://doi.org/ 10.1016/j.immuni.2014.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kubiczkova L, Sedlarikova L, Hajek R, Sevcikova S. TGF-beta – an excellent servant but a bad master. J Transl Med 2012; 10:183; PMID:22943793; https://doi.org/ 10.1186/1479-5876-10-183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neuzillet C, Tijeras-Raballand A, Cohen R, Cros J, Faivre S, Raymond E, de Gramont A. Targeting the TGFbeta pathway for cancer therapy. Pharmacol Ther 2015; 147:22-31; PMID:25444759; https://doi.org/ 10.1016/j.pharmthera.2014.11.001 [DOI] [PubMed] [Google Scholar]
- 24.Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, Ferguson SM. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal 2012; 5:ra42; PMID:22692423; https://doi.org/ 10.1126/scisignal.2002790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang Y, Li Y, Zhu B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis 2015; 6:e1792; PMID:26086965; https://doi.org/ 10.1038/cddis.2015.162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER, Graf T, Pollard JW, Segall J, Condeelis J. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res 2004; 64:7022-9; PMID:15466195; https://doi.org/ 10.1158/0008-5472.CAN-04-1449 [DOI] [PubMed] [Google Scholar]
- 27.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006; 124:263-6; PMID:16439202; https://doi.org/ 10.1016/j.cell.2006.01.007 [DOI] [PubMed] [Google Scholar]
- 28.Zhang B, Zhang Y, Yao G, Gao J, Yang B, Zhao Y, Rao Z, Gao J. M2-polarized macrophages promote metastatic behavior of Lewis lung carcinoma cells by inducing vascular endothelial growth factor-C expression. Clinics (Sao Paulo, Brazil) 2012; 67:901-6; PMID:22948457; https://doi.org/ 10.6061/clinics/2012(08)08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol 2006; 13:1235-42; PMID:17114005; https://doi.org/ 10.1016/j.chembiol.2006.09.018 [DOI] [PubMed] [Google Scholar]
- 30.Song W, Wang F, Lotfi P, Sardiello M, Segatori L. 2-Hydroxypropyl-beta-cyclodextrin promotes transcription factor EB-mediated activation of autophagy: implications for therapy. J Biol Chem 2014; 289:10211-22; PMID:24558044; https://doi.org/ 10.1074/jbc.M113.506246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kilpatrick K, Zeng Y, Hancock T, Segatori L. Genetic and chemical activation of TFEB mediates clearance of aggregated alpha-synuclein. PLoS One 2015; 10:e0120819; PMID:25790376; https://doi.org/ 10.1371/journal.pone.0120819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hasan M, Koch J, Rakheja D, Pattnaik AK, Brugarolas J, Dozmorov I, Levine B, Wakeland EK, Lee-Kirsch MA, Yan N. Trex1 regulates lysosomal biogenesis and interferon-independent activation of antiviral genes. Nat Immunol 2013; 14:61-71; PMID:23160154; https://doi.org/ 10.1038/ni.2475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huan C, Kelly ML, Steele R, Shapira I, Gottesman SR, Roman CA. Transcription factors TFE3 and TFEB are critical for CD40 ligand expression and thymus-dependent humoral immunity. Nat Immunol 2006; 7:1082-91; PMID:16936731; https://doi.org/ 10.1038/ni1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mia S, Warnecke A, Zhang XM, Malmstrom V, Harris RA. An optimized protocol for human M2 macrophages using M-CSF and IL-4/IL-10/TGF-beta yields a dominant immunosuppressive phenotype. Scand J Immunol 2014; 79:305-14; PMID:24521472; https://doi.org/ 10.1111/sji.12162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pena-Llopis S, Vega-Rubin-de-Celis S, Schwartz JC, Wolff NC, Tran TA, Zou L, Xie XJ, Corey DR, Brugarolas J. Regulation of TFEB and V-ATPases by mTORC1. EMBO J 2011; 30:3242-58; PMID:21804531; https://doi.org/ 10.1038/emboj.2011.257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiao YQ, Malcolm K, Worthen GS, Gardai S, Schiemann WP, Fadok VA, Bratton DL, Henson PM. Cross-talk between ERK and p38 MAPK mediates selective suppression of pro-inflammatory cytokines by transforming growth factor-beta. J Biol Chem 2002; 277:14884-93; PMID:11842088; https://doi.org/ 10.1074/jbc.M111718200 [DOI] [PubMed] [Google Scholar]
- 37.Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, Rimoldi M, Biswas SK, Allavena P, Mantovani A. Macrophage polarization in tumour progression. Semin Cancer Biol 2008; 18:349-55; PMID:18467122; https://doi.org/ 10.1016/j.semcancer.2008.03.004 [DOI] [PubMed] [Google Scholar]
- 38.Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol 2014; 5:614; PMID:25506346; https://doi.org/ 10.3389/fimmu.2014.00614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu K, Zhao E, Ilyas G, Lalazar G, Lin Y, Haseeb M, Tanaka KE, Czaja MJ. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy 2015; 11:271-284; PMID:25650776; https://doi.org/ 10.1080/15548627.2015.1009787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang SC, Everts B, Ivanova Y, O'Sullivan D, Nascimento M, Smith AM, Beatty W, Love-Gregory L, Lam WY, O'Neill CM et al.. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol 2014; 15:846-855; PMID:25086775; https://doi.org/ 10.1038/ni.2956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Casey AE, Laster WR Jr, Ross GL. Sustained enhanced growth of carcinoma EO771 in C57 black mice. Proc Soc Exp Biol Med 1951; 77:358-62; PMID:14854049; https://doi.org/ 10.3181/00379727-77-18779 [DOI] [PubMed] [Google Scholar]
- 42.Nachat-Kappes R, Pinel A, Combe K, Lamas B, Farges MC, Rossary A, Goncalves-Mendes N, Caldefie-Chezet F, Vasson MP, Basu S. Effects of enriched environment on COX-2, leptin and eicosanoids in a mouse model of breast cancer. PLoS One 2012; 7:e51525; PMID:23272114; https://doi.org/ 10.1371/journal.pone.0051525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jia X, Yu F, Wang J, Iwanowycz S, Saaoud F, Wang Y, Hu J, Wang Q, Fan D. Emodin suppresses pulmonary metastasis of breast cancer accompanied with decreased macrophage recruitment and M2 polarization in the lungs. Breast Cancer Res Treat 2014; 148:291-302; PMID:25311112; https://doi.org/ 10.1007/s10549-014-3164-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Incio J, Tam J, Rahbari NN, Suboj P, McManus DT, Chin SM, Vardam T, Batista A, Babykutty S, Jung K et al.. PlGF/VEGFR-1 signaling promotes macrophage polarization and accelerated tumor progression in obesity. Clin Cancer Res 2016; 22:2293-3004; https://doi.org/ 10.1158/1078-0432.CCR-15-1839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fan D, Qiu S, Overton CD, Yancey PG, Swift LL, Jerome WG, Linton MF, Fazio S. Impaired secretion of apolipoprotein E2 from macrophages. J Biol Chem 2007; 282:13746-53; PMID:17341585; https://doi.org/ 10.1074/jbc.M611754200 [DOI] [PubMed] [Google Scholar]
- 46.Fan D, Yancey PG, Qiu S, Ding L, Weeber EJ, Linton MF, Fazio S. Self-association of human PCSK9 correlates with its LDLR-degrading activity. Biochemistry 2008; 47:1631-9; PMID:18197702; https://doi.org/ 10.1021/bi7016359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Su YR, Blakemore JL, Zhang Y, Linton MF, Fazio S. Lentiviral transduction of apoAI into hematopoietic progenitor cells and macrophages: applications to cell therapy of atherosclerosis. Arterioscler Thromb Vasc Biol 2008; 28:1439-46; PMID:18497309; https://doi.org/ 10.1161/ATVBAHA.107.160093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang J, Iwanowycz S, Yu F, Jia X, Leng S, Wang Y, Li W, Huang S, Ai W, Fan D. microRNA-155 deficiency impairs dendritic cell function in breast cancer. Oncoimmunology 2016; 5:e1232223; PMID:27999745; https://doi.org/ 10.1080/2162402X.2016.1232223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang J, Yu F, Jia X, Iwanowycz S, Wang Y, Huang S, Ai W, Fan D. MicroRNA-155 deficiency enhances the recruitment and functions of myeloid-derived suppressor cells in tumor microenvironment and promotes solid tumor growth. Int J Cancer 2015; 136:E602-613; PMID:25143000; https://doi.org/ 10.1002/ijc.29151 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.