

Abstract
CIC-DUX4 gene fusion, resulting from either a t(4;19) or t(10;19) translocation, is the most common genetic abnormality detected in EWSR1-negative small blue round cell tumors (SBRCTs). Following their discovery it was debated if these tumors should be classified as variants of Ewing sarcoma (i.e. atypical Ewing sarcoma) or as a stand-alone pathologic entity. As such the WHO classification temporarily grouped the CIC-rearranged tumors under undifferentiated sarcomas with round cell phenotype, until further clinical evidence was available. However, most studies reported so far include small series with limited follow-up information which preclude a more definitive assessment. The present work investigates the clinicopathologic features of a large cohort of sarcomas with CIC gene rearrangement, in order to define their clinical presentation, morphologic spectrum, and outcome. Our study further examines the overall survival of the CIC-positive cohort compared to a control group of EWSR1-rearranged Ewing sarcoma matched for age and stage. The study cohort included 115 patients, with a mean age of 32 years and a slight male predominance. Most tumors occurred in the soft tissue (86%), predominantly deep-seated and equally divided among trunk and extremity, followed by visceral locations (12%) and rarely in the bone (3%). Microscopically, most tumors showed round to ovoid cytomorphology but half of the cases showed also focal areas of spindling and epithelioid/rhabdoid phenotype, with frequent myxoid stromal changes. Variable CD99 reactivity was seen in 84% cases, with a diffuse pattern only in 23% of cases, while nuclear WT1 was seen in 92%. A CIC-DUX4 fusion was detected in 57% of cases, with either DUX4 on 4q35 (35%) or on 10q26 in 25 (22%) cases. No FOXO4 gene rearrangements were present in 39 cases tested. Clinical follow-up was available in 57 patients, with a 5-year survival of 43%, which was significantly lower than the 77% 5-year survival in the control Ewing sarcoma group (p=0.002). Our findings show that CIC-DUX4 sarcomas occur most commonly in young adults within the somatic soft tissues, having a wide spectrum of morphology including round, epithelioid and spindle cells, and associated with an aggressive clinical course, with an inferior overall survival compared to Ewing sarcoma. The results support the classification of CIC-rearranged tumors as an independent molecular and clinical subset of SBRCTs distinct from Ewing sarcoma.
Keywords: CIC, DUX4, SBRCT, round cell sarcoma
INTRODUCTION
According to the current WHO classification1, undifferentiated round cell sarcomas are characterized by relatively monotonous round to ovoid cytomorphology, with a high nuclear to cytoplasmic ratio, and no distinct line of differentiation, lacking consistent genetic abnormalities. However, as they most often resemble Ewing sarcoma, for practical and treatment purposes, round cell undifferentiated sarcomas have been regarded as ‘Ewing sarcoma-like’ and are often managed similarly to the Ewing sarcoma family of tumors. In contrast to classic Ewing sarcoma, round cell undifferentiated sarcomas lack the pathognomonic translocations involving the EWSR1 gene on chromosome 22 fused to a member of the ETS transcription factor family, either FLI1,2 ERG,3,4 or other less common variants. Within this undifferentiated subgroup, the present WHO classification included round cell sarcomas with CIC-DUX4 fusions, as a temporary subset, until further evidence, both at the clinical and molecular level, could determine if they represent an independent pathologic entity.
CIC-rearranged sarcomas have been relatively recently described as aggressive tumors arising in soft tissues of children and young adults. Although they share partial morphologic overlap with Ewing sarcoma and variable CD99 expression, emerging molecular data suggest that CIC-DUX4 tumors have a distinct pathogenesis. The CIC-DUX4 fusion results from either a t(4;19)(q35;q13) or a t(10;19)(q26;q13) translocation5,6,7,8,9. The genes involved in the fusion are CIC, a transcriptional repressor on chromosome 19q13.1, and DUX4, a double homeobox transcription factor, located on either 4q35 or chromosome 10q26.3. Whether the group of CIC-DUX4-positive round cell sarcomas represents a stand-alone category of tumors or a subgroup of the Ewing sarcoma family has been a matter of debate. In the present study we attempt to elucidate this controversy by investigating a large cohort of 115 molecularly confirmed cases to provide a more definitive classification based on detailed clinical presentation, histologic spectrum, immunoprofile and outcome.
MATERIAL AND METHODS
Patients and Tumor Characteristics
The MSKCC files and personal consultation files of the senior authors (CRA, CDF) were searched for the diagnosis of small blue round cell tumors (SBRCTs) and Ewing sarcoma-like tumors with available tissue for molecular analysis. All cases included were positive for CIC gene rearrangements by FISH, being selected from SBRCTs lacking EWSR1 and FUS gene rearrangements (or other common sarcoma-associated translocations, e.g. SS18). A total of 115 SBRCTs with CIC-related fusions were identified during a two-decade period (1995–2016). Hematoxylin and eosin-stained slides and previously performed immunohistochemical stains were reviewed in all cases. All cases were handled in accordance with the ethical rules of the respective institutions.
For each case, the location of the tumor was recorded, along with the anatomic structures involved. The tumors were assessed for growth pattern, cytomorphology (round, oval, spindle, epithelioid, plasmacytoid/rhabdoid phenotype), cellular pleomorphism, nuclear features including nuclear contour, chromatin pattern and presence of nucleoli, mitotic activity, necrosis, type of stroma and myxoid change. The immunohistochemical stains were re-reviewed and a minimum panel was available for review in most cases, including CD99, WT1, cytokeratin and/or EMA, and desmin. However, most cases in fact had a much wider panel of stains performed, including neural/neuroendocrine markers, lymphoid markers which were negative and which excluded other diagnostic considerations.
As most of the cases tested were received in consultation, complete follow-up data was available in only 57 (50%) patients. The following clinical data were retrieved: tumor size, stage at diagnosis (primary versus distant metastasis at diagnosis), modality of initial therapy, recurrence, vital status and survival time. In addition, from a large cohort of 127 Ewing sarcoma patients with molecular confirmation treated at MSKCC, we selected a group of 57 Ewing sarcomas equivalent to the number of the CIC-positive cohort, closely matched for age and stage, for an accurate comparison for overall survival (OS). A further statistical analysis was performed focusing only on patients with localized disease at diagnosis, thus comparing 45 CIC-rearranged SBRCTs with 45 primary Ewing sarcoma patients for OS. The diagnosis of Ewing sarcoma was confirmed by either RT-PCR amplification of an EWSR1-FLI1/ERG transcript or an EWSR1 gene rearrangement by FISH.
Fluorescence in situ Hybridization (FISH)
FISH analysis was performed on interphase nuclei from paraffin-embedded 4 μm sections using bacterial artificial chromosomes (BAC clones), flanking CIC in 19q13 and DUX4 on 4q35 and 10q26.3 as previously described7. Two hundred tumor nuclei were evaluated using a Zeiss fluorescence microscope (Zeiss Axioplan, Oberkochen, Germany), controlled by Isis 5 software (Metasystems). Cases that were negative for DUX4 gene abnormalities were also investigated for the FOXO4 gene rearrangements, as previously described10,11 Supplementary Table 1). A cut-off of >20% nuclei showing a break-apart signal was considered to be positive for rearrangement. Nuclei with incomplete set of signals were omitted from the score.
Statistical analysis
Statistical analysis was performed on an SPSS platform (version 24.0; IBM Corp., Armonk, NY). The associations between the clinical variables and matched groups were evaluated by Fisher’s exact test. The OS time was measured in months from the date of diagnosis to the date of death. Kaplan-Meier estimate was used to calculate the OS. The statistical significance of different clinicopathologic variables (gender, age group [adult/pediatric], tumor size [≤5cm/>5cm], presence of metastasis at diagnosis, and recurrence status) in relation to survival was assessed by log-rank analysis. The OS between CIC fusion positive SBRCT and Ewing sarcoma patients were compared by log-rank analysis. A p<0.05 was considered as significant for all statistical analyses.
RESULTS
Patients Demographics and Clinical Presentation
Among the 115 patients included, there was a slight male dominance, with 63 (55%) males and 52 (45%) females. The age at diagnosis ranged from 6–81 years, with a mean of 32 years. Twenty-five patients (22%) were in the pediatric age group (<18 years of age). The anatomic location of the primary tumor was known in 111 cases (Table 1). Two additional patients presented with metastases to brain and lung (unknown primary tumors). Ninety-five cases (86%) arose within soft-tissue, 3 (3%) cases in bone (all involving pelvic bones) and 13 (12%) cases were visceral (mostly involving GI and GU organs, see Table 1). Among the soft tissue tumors, most were located deep within the muscle, with only 7 (7%) cases being superficial. Soft tissue tumors were equally distributed between trunk (n=39) and extremity (n=38), with less common presentation in the head and neck (n=12) and retroperitoneum/intra-abdominal/pelvic location (n=6). Within the limbs, 31 tumors occurred in the lower and 7 in the upper extremity.
Table 1.
Anatomic location of CIC-rearranged sarcomas
| Location of the tumor | Number of cases (n=111) |
|---|---|
| Soft tissue | 95 (86%) |
| Trunk | 39 |
| Lower extremity | 31 |
| Upper extremity | 7 |
| Head/neck | 12 |
| Retroperitoneum/perineum/pelvis | 6 |
|
| |
| Viscera | 13 (12%) |
| Stomach | 1 |
| Small/large intestine | 5 |
| Kidney/prostate | 4 |
| Tonsils/parapharyngeal | 3 |
|
| |
| Bone | 3 (3%) |
| Pelvic bones | 3 |
Microscopic Findings
All cases showed solid proliferation of small to medium-sized neoplastic cells. One-third of the cases showed a nodular or vaguely nodular appearance with thick collagenous septa separating the tumor into compartments (Fig. 1A). Less common growth patterns included intersecting short fascicles in 2 cases (Fig. 1B) and a reticular architecture pattern (Fig. 1C) in one case. Thirty-five cases showed variably prominent myxoid stromal change, which was moderate to diffuse in 12 cases (Fig. 1D). No prominent collagenous stroma was identified in these tumors.
Figure 1. Morphologic spectrum of CIC-rearranged sarcomas.
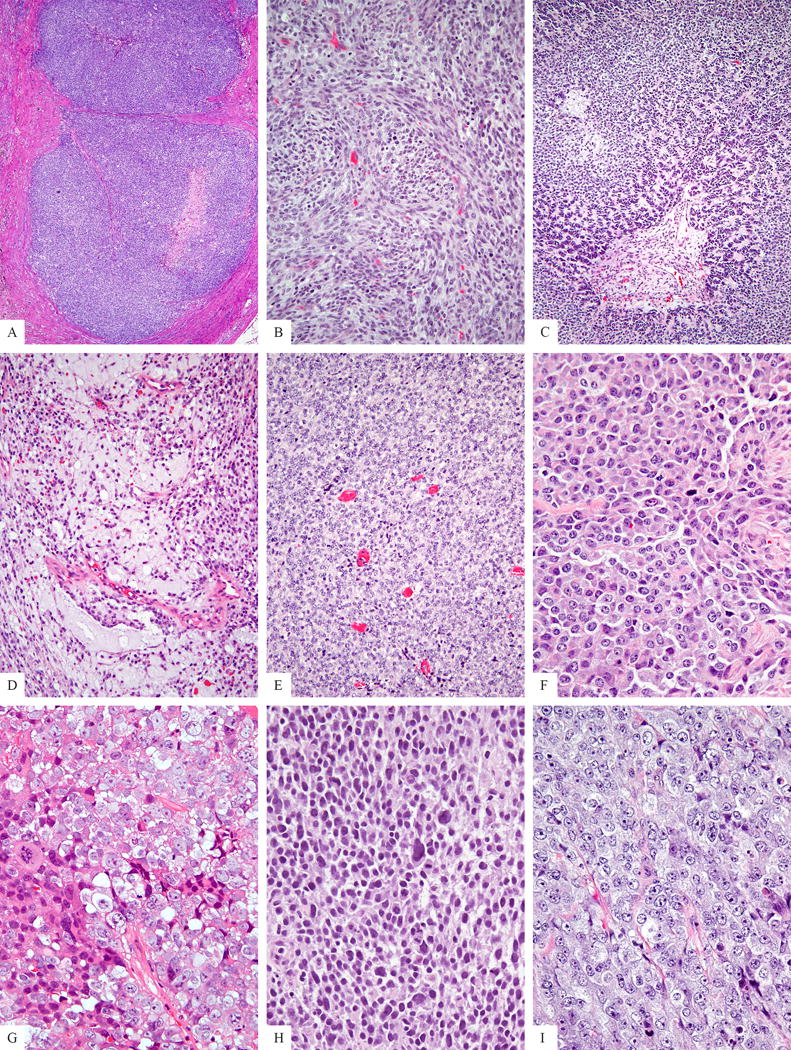
Tumors showed a solid and often nodular growth pattern (A). Less common architectural patterns included intersecting short fascicles in tumors with a spindle cell component (B), a rare reticular growth (C). Myxoid stromal component was seen in a third of cases (D). The predominant phenotype was that of solid sheets of round to ovoid cells (E), however, focal areas of more epithelioid or plasmacytoid appearance was also noted with light eosinophilic cytoplasm and eccentric nuclei (F). The nuclear features showed variable chromatin patterns, with either fine (G), dark, hyperchromatic (H) or vesicular (I). Most cases showed minor variability in nuclear size and shape, except for a small subset revealing moderate degree of nuclear pleomorphism (H). Small to medium sized nucleoli were a common finding (G,I).
Most of the cases displayed round to ovoid cytomorphology, with 57 (50%) cases being composed purely of round/ovoid cells (Fig 1E). Twelve cases (10%) showed a mixture of round/ovoid and spindle cells, with one of the cases displaying a predominant (>50%) spindle cell morphology (Fig 1B). Twenty-seven (23%) cases had a mixture of round/ovoid and plasmacytoid/rhabdoid cells, with 12 (11%) showing predominantly (>50%) a plasmacytoid/rhabdoid phenotype (Fig. 1F). Nineteen (17%) cases showed a mixture of round/ovoid, spindle and plasmacytoid/rhabdoid, however, the predominant (>50%) component was composed of round/ovoid cells.
Most tumors showed cells with scant lightly eosinophilic to clear cytoplasm, which was more abundant in the epithelioid/rhabdoid component. The nuclear features displayed various chromatin patterns: including 33 cases with fine (Fig 1G), 22 cases with hyperchromatic (Fig. 1H), 60 with a vesicular chromatin pattern (Fig. 1I). Multifocal prominent nucleoli were observed in 38 (33%) of the tumors (Fig. 1G, I). Most tumors had relatively monomorphic cytomorphology, with only mild nuclear pleomorphism. However, in 15 (13%) cases there was some degree of moderate nuclear pleomorphism (Fig. 1H), in 6 being more diffuse and in 9 cases only focal. The mitotic count varied from 6–99 per 10 HPFs, with a mean of 32 MF/10 HPFs. Most (84%) cases showed geographic necrosis.
Immunohistochemical Findings
Immunohistochemical stains including CD99, WT1, AE1/AE3, and desmin were evaluated. One hundred and ten cases were evaluated for CD99: 25 (23%) showed diffuse positivity (Fig. 2A), 67 (36 multifocal, 31 focal)(61%) cases multifocal/focal positivity (Fig. 2B), while 18 (16%) were negative. Forty-eight (74%) of 65 cases evaluated showed diffuse WT1 nuclear expression (Fig. 2C), while 12 (18%) cases were multifocally/focally positive. No staining for WT1 was noted in 4 cases. Focal staining for AE1:AE3 and desmin was present in 16 and 4 cases, respectively, of the 110 cases evaluated.
Figure 2. Immunohistochemical and FISH ancillary methods in diagnosis of CIC fusion positive sarcomas.
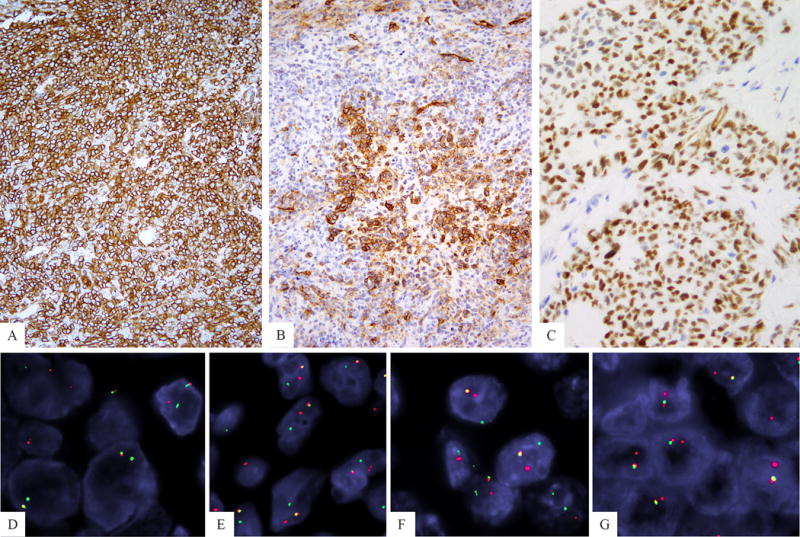
CD99 reactivity a hallmark for Ewing sarcoma was present with a diffuse membranous pattern in a minority of cases (A), while most showed patchy, focal staining (B). WT1 nuclear staining was a consistent finding in overwhelming majority of cases (C). 3-color fusion FISH assay showing CIC (green, telomeric) fused to DUX4 (red, centromeric) gene located on 4q35 (D) or 10q26 (E). FISH break-apart showing split red centromeric and green telomeric CIC signals, in an SBRCT lacking DUX4 or FOXO4 gene abnormalities.
Fluorescence in situ Hybridization (FISH)
All cases showed a CIC gene break-apart signal. Cases with other CIC gene abnormalities, such as small, constant gaps suspicious for inversion, were not considered as a positive result and were not included in the study cohort. As the DUX4 gene is located in the subtelomeric region of 4q35 or 10q26, a FISH split assay could not be designed due its proximity to the telomeric area. Thus, the alternative method was to interrogate the CIC gene partner by using a 3-color FISH assay for telomeric CIC and centromeric DUX4, as previously described.7 The results showed that 40 (35%) cases were positive for fusion to DUX4 gene on 4q35 (Fig. 2D), while 25 (22%) were fused to DUX4 on 10q26 (Fig. 2E). In the remaining 43% of the cases a fusion partner could not be demonstrated (Fig. 2 F); however, in about one-third of these cases (n=14) the fusion assay was not informative due to a CIC telomeric deletion, which precluded evaluation of its fusion partner. From the 50 cases lacking a DUX4 gene partner, 39 cases had available material for further FISH FOXO4 break-apart assay, as previously described in 2 cases,10,11 however, no FOXO4 gene abnormalities were identified.
Follow-up data
Clinical follow-up information was available in 57 patients (50%), ranging from 1 to 269 months, median of 14 months. This cohort was representative of the entire group, with 33 (58%) males and 24 (42%) females, with an age range of 10–81 years (median of 31 years). Forty-eight (84%) patients were of adult age (>18 years). The tumor size ranged from 0.7–23 cm, with 32 (64%) patients presenting with large sized tumors (>5 cm). Nine (16%) patients presented with distant metastasis at initial diagnosis to the lung (n=9), liver, brain or supraclavicular lymph nodes. Among the patients who presented with primary disease, 25 developed recurrence following treatment: 4 patients had local recurrence (LR) alone, 17 patients developed distant recurrence (DR) and 4 patients developed both LR and DR (lungs and pelvis). Overall the metastatic rate was 53% (30/57) and 44% (21/48) in the localized group. The lungs were the most common site for distant recurrence (16/21, 76%). Twenty-three (40%) patients died of disease (DOD) at last follow up. Twenty-two of 23 (96%) patients who remained free of disease are alive, while only 5/21 (24%) patients who recurred distantly are still alive.
CIC-positive tumors treated with neoadjuvant chemotherapy showed an inferior pathologic response and poor outcome
52 patients had detailed therapy information, with 22 receiving neoadjuvant chemotherapy after the diagnosis was established on a core biopsy, while 29 patients had an initial surgical resection with curative intent, followed by adjuvant chemotherapy in 22 patients and radiation in 2. The overwhelming number of patients (>90%) were treated following a Ewing sarcoma chemotherapy regimen. Five patients were treated with surgery alone. One patient received only radiotherapy, due to advanced age and anatomic location of the tumor (80/M, paraspinal). Ten patients presenting with localized disease and treated with neoadjuvant chemotherapy could be evaluated for treatment response in the resection specimen showing that only 3 had >90% therapy-related changes (fibrosis), consistent with a grade III chemotherapy response. The remaining 7 (70%) patients had an inferior degree of pathologic response, either <50% necrosis (n=4, grade I response) or >50% but <90% (n=3, grade II), using the 4-tier grading system applied for Ewing sarcoma. However there was no correlation between the degree of pathologic response and survival in these 10 patients analyzed.
Interestingly, patients treated with neoadjuvant chemotherapy (n=22) showed an inferior survival compared to patients managed by surgery first (n=29) (p=0.025), although the latter group was heterogeneous and included 22 patients with adjuvant chemotherapy, 2 with adjuvant radiation, and 5 with surgery alone. This survival advantage was retained when comparing only patients with localized disease who received neoadjuvant chemotherapy (n=17, of whom 13 underwent surgical resection) versus those managed by surgery first (n=25, of whom 18 were treated with adjuvant chemo)(p=0.041). However, patients selected for neoadjuvant therapy had a larger tumor size (p<0.0001) compared to patients who were managed by surgery first, with no difference in age or anatomic location. Patients who were treated for localized disease with surgery and adjuvant chemotherapy (n=18) had only a trend for improved survival compared to patients treated with neoadjuvant chemotherapy (n=17)(p=0.084). Among the 5 patients who had surgery alone (tumor size range 3.5–5, mean 4.1 cm), none developed distant recurrences or died of disease. The 2 patients who were treated with surgery followed by radiation alone are both alive without distant recurrence.
Survival analysis of CIC-rearranged sarcoma patients
On survival analysis, the CIC-rearranged sarcoma patients showed a 53% 2-year OS and a 43% 5-year OS rates (Fig. 3A). When limited to the 45 CIC-rearranged sarcoma patients presenting with localized disease, the 2-year and 5-year OS rates were 59% and 49%. There was no significant effect on OS related to gender (male vs. female; p=0.178), age (adult vs. pediatric; p=0.269) and tumor size (≤5 cm vs. >5 cm; p=0.378). Patients who presented with metastatic disease at diagnosis had a poor prognosis compared to patients with primary disease (p<0.0001)(Fig. 3B). Also, presence of recurrence (NR vs. LR vs. DR) was statistically significant on OS (p<0.0001)(Fig. 3C). The survival analysis of CIC-rearranged sarcoma patients based on clinical variables is presented in Supplementary Table 2.
Figure 3. Survival analysis of CIC-rearranged sarcoma patients.
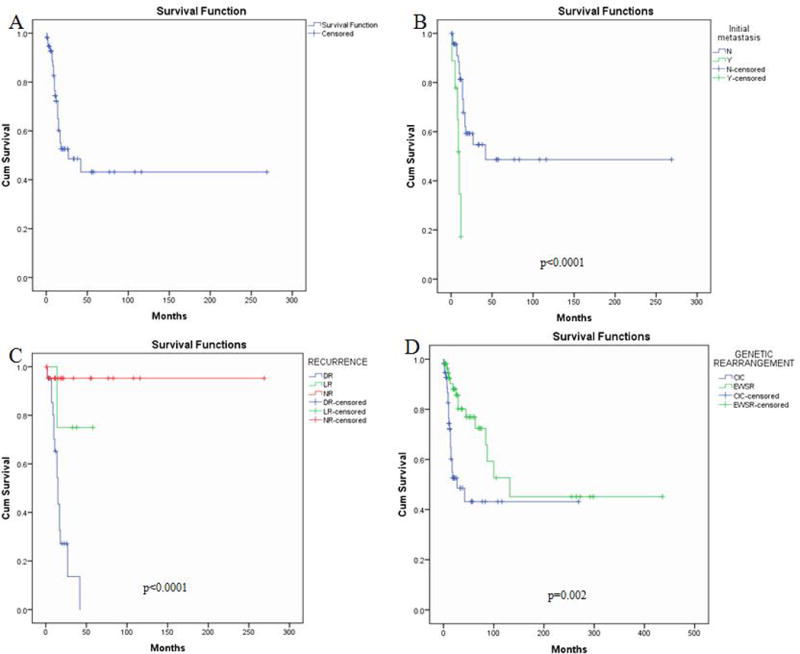
Overall survival of the 57 patients with follow-up information showed a 2-year OS: 53% and a 5-year OS: 43% (A). Patients who presented with metastatic disease at diagnosis followed an unfavorable clinical course compared to patients with localized disease (p<0.0001) (B). Distant recurrence was associated with an inferior survival (p<0.0001) (C). Patients with CIC-rearranged sarcoma have a significantly worse outcome compared to patients with Ewing sarcoma (matched for age/stage) (p=0.002) (D).
CIC-fusion positive SBRCTs have a significantly unfavorable outcome compared to Ewing sarcoma
The survival comparison between CIC-rearranged SBRCTs and Ewing sarcoma patients is presented in Supplementary Table 3. The 57 Ewing sarcoma cohort (matched for stage and age) had a significantly longer OS compared to CIC-rearranged sarcoma patients (p=0.002), with a 2-year and 5-year survival of 87% and 77%, respectively (Fig. 3D). This survival difference was retained when comparing only patients with localized disease for both cohorts (n=45), with the 5-year survival for CIC-positive cases at 49% compared to 76% for Ewing sarcoma patients (p=0.035). In fact, the 5 year-survival for the whole cohort of localized Ewing sarcoma patients, not skewed by age (n=106, out of 127 cases total available from our database), was even more favorable at 82% (p=0.005).
DISCUSSION
This study investigates a large cohort of 115 CIC-rearranged sarcomas providing further and more definitive evidence on its distinctive clinical and pathologic characteristics, in favor of a stand-alone molecularly defined entity. Initially classified under the umbrella of the Ewing sarcoma family, our findings reinforce important differences between these two round cell sarcomas. First, the anatomic distribution is different, with the overwhelming majority of CIC-DUX4-sarcomas, 86% in this series, occurring in the somatic soft tissues, equally distributed between extremities and trunk, followed by visceral location and infrequently in the bone. In contrast, most Ewing sarcomas occur in skeletal locations, either in the flat bones of the pelvis or chest wall or the diaphysis of long bones.1 Second, CIC-DUX4-positive tumors preferentially affect young adults with a peak incidence in the fourth decade, with slight male predominance. In fact only 22% of the patients included in this study were of pediatric age group. In contrast, patients with Ewing sarcoma have a mean age at diagnosis of 15 years,12 also with a slight male predominance. Although both tumor entities share an undifferentiated and mostly monotonous proliferation of round to ovoid cells, there are certain distinctive features associated with CIC-DUX4-positive tumors, most importantly a wider spectrum of cytomorphologies, with mixture of round, spindle and epithelioid cells. Although half of the cases showed mainly a round to ovoid phenotype, the remaining cases revealed, in addition, focal areas of spindle or epithelioid cells. Furthermore, the tumor cells showed increased nuclear size and shape variability, vesicular chromatin with focally prominent nucleoli, in addition to more abundant, typically light eosinophilic cytoplasm. Stromal myxoid change was a common finding in CIC-positive tumors, which is typically absent in Ewing sarcoma.13,14
Immunophenotypically, as previously reported, most CIC-rearranged tumors (84%) showed variable expression of CD99, but only 23% with a diffuse pattern and 16% were completely negative. This is in contrast with classic Ewing sarcomas which typically show diffuse and strong membranous reactivity with CD99 in most if not all cases. Furthermore, nuclear WT1 reactivity was also a consistent finding in CIC-positive sarcomas, in contrast with Ewing sarcoma, as previously noted, most likely related to its transcriptional up-regulation.15
A CIC-DUX4 fusion was detected in 57% of cases with either DUX4 on 4q35 (35%) or DUX4 on 10q26 in 25 (22%) cases. CIC-DUX4 fusion appears functionally unrelated to EWSR1-ETS, with the transcriptional profile of CIC-DUX4-positive sarcomas being distinct from that of Ewing sarcoma or other sarcoma subtypes, in keeping with separate tumor entities.15 Prior evidence has demonstrated that CIC-DUX4 sarcomas overexpress the PEA3 (polyoma enhancer activator 3) subfamily of transcription factors, including ETV1, ETV4, and ETV5, both at mRNA and protein levels.5,15–17 CIC is the human homologue of Drosophila capicua, a gene identified in a screen for mutations affecting the anterior-posterior pattern of Drosophila embryos.18 CIC gene abnormalities have been implicated in various neoplastic conditions, such as CIC loss of function mutations being identified in 83% of oligodendrogliomas.19,20 CIC encodes a transcriptional repressor with a high-mobility group (HMG)-box containing DNA binding domain that normally inhibits ETV1/4/5 expression and regulates receptor tyrosine kinase signaling pathways.21,22 Using an experimental cell line model system, Kawamura-Saito et al. demonstrated binding of this CIC HMG box to the promoter of PEA genes ETV1 and ETV5. Their results further revealed that fusion of DUX4 to CIC sequence provides strong transcriptional activity, resulting in mostly upregulated gene expression, with minimal down-regulated genes.5 Similar ETV1/4/5 upregulation is also present in other CIC related fusions, such as CIC-LEUTX positive angiosarcoma23 CIC-NUTM1-positive central nervous system primitive neuroectodermal tumor (PNET).23,24
In contrast, DUX4 gene is normally expressed in germ cells and is epigenetically silenced in somatic differentiated tissues through CpG methylation.25 Aberrant expression of DUX4 has been implicated in the development of facioscapulohumeral muscular dystrophy.26 The exact role in tumorigenesis of DUX4 dysregulation in the setting of CIC-DUX4 fusion remains poorly defined. Recently, CIC-DUX4 sarcoma has been shown to exhibit strong DUX4 immunoexpression, while other translocation-positive round cell sarcomas (i.e. Ewing sarcoma, alveolar rhabdomyosarcoma, synovial sarcoma, desmoplastic small round cell tumor) did not.27 DUX4 gene is located within the D4Z4 macrosatellite repeat region (11–100 copies of the repeat units) of the chromosomes 4 and 10 subtelomeric regions.28
Two SBRCT cases were reported quite recently to have CIC alternative fusions involving FOXO4 gene on Xq13.10,11 Of interest both cases occurred in the head and neck soft tissue (neck and scalp) in a 65 year-old male and 13 year-old male, respectively. No FOXO4 gene abnormalities were detected in any of the 39 cases tested lacking DUX4 abnormalities in the present series, including head and neck lesions, suggesting that CIC-FOXO4 fusion is a rare genetic event in the pathogenesis of SBRCTs. Also of interest, a small subset of high grade angiosarcomas occurring in the soft tissue of young adults have been recently shown to harbor CIC–related fusions, including one case fused to the LEUTX gene, which, similar to DUX4, belongs to the paired (PRD) homeobox genes.29,30
Finally patients with CIC-rearranged tumors followed an aggressive clinical course with a high metastatic rate, mainly to the lung. The 5-year overall survival was 43% for the entire group and 49% for the patients who presented with localized disease at diagnosis. The overall survival was significantly lower compared to the localized Ewing sarcoma cohort, matched for stage and age, which showed a 76% 5 year-survival. These findings confirm the results of Yoshida et al who found a statistically significant inferior overall survival in a smaller cohort of 20 CIC-rearranged sarcomas compared to a group of 53 Ewing sarcoma patients (unmatched for age or stage).14
In summary, this is the largest cohort to date of molecularly confirmed CIC-fusion positive sarcomas showing a relatively uniform clinical presentation, distinct from Ewing sarcoma. Most study group patients were young adults in their 4th decade (mean 32 years of age), presenting with a deep soft tissue tumor, either in the extremity or trunk. Microscopically, tumors had a more heterogeneous cytology, with a mixture of round, spindle and epithelioid cell types and often myxoid stroma. Immunohistochemically, the variable CD99 reactivity and strong WT1 are helpful ancillary techniques, diagnosis can be further confirmed by FISH for CIC gene rearrangements. A CIC-DUX4 fusion was documented in only half of the cases, while no FOXO4 gene abnormality was identified. Most importantly, the follow-up information revealed highly aggressive behavior, with a high metastatic rate of 53%, mainly to the lung and a 5 year-survival of 44%, which was significantly inferior to comparable Ewing sarcoma patients. Furthermore the patients who were treated with neoadjuvant chemotherapy using a similar regimen as for Ewing sarcoma showed a poor pathologic response in 70% of cases. All these findings reinforce the prior data combined from smaller series that CIC-rearranged sarcoma has distinct clinicopathologic features, which clearly warrant a stand-alone entity, separate from Ewing sarcoma. The challenging aspect for the future remains to design novel therapies, independent from Ewing sarcoma, possibly targeting the highly up-regulated downstream signature, such as the PEA3 family of transcription factors.
Supplementary Material
Acknowledgments
Supported in part by: P50 CA140146-01 (CRA, SS); P30-CA008748 (CRA, SS); Kristen Ann Carr Foundation (CRA); Cycle for Survival (CRA)
Footnotes
Conflicts of interest: none
References
- 1.Fletcher C, Bridge JA, Hogendoorn PC, et al. WHO Classification of Tumours of Soft Tissue and Bone. IARC; Lyon: 2013. [Google Scholar]
- 2.Delattre O, Zucman J, Plougastel B, et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature. 1992;359:162–165. doi: 10.1038/359162a0. [DOI] [PubMed] [Google Scholar]
- 3.Zucman J, Melot T, Desmaze C, et al. Combinatorial generation of variable fusion proteins in the Ewing family of tumours. Embo J. 1993;12:4481–4487. doi: 10.1002/j.1460-2075.1993.tb06137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sorensen PH, Lessnick SL, Lopez-Terrada D, et al. A second Ewing’s sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat Genet. 1994;6:146–151. doi: 10.1038/ng0294-146. [DOI] [PubMed] [Google Scholar]
- 5.Kawamura-Saito M, Yamazaki Y, Kaneko K, et al. Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Hum Mol Genet. 2006;15:2125–2137. doi: 10.1093/hmg/ddl136. [DOI] [PubMed] [Google Scholar]
- 6.Yoshimoto M, Graham C, Chilton-MacNeill S, et al. Detailed cytogenetic and array analysis of pediatric primitive sarcomas reveals a recurrent CIC-DUX4 fusion gene event. Cancer Genet Cytogenet. 2009;195:1–11. doi: 10.1016/j.cancergencyto.2009.06.015. [DOI] [PubMed] [Google Scholar]
- 7.Italiano A, Sung YS, Zhang L, et al. High prevalence of CIC fusion with double-homeobox (DUX4) transcription factors in EWSR1-negative undifferentiated small blue round cell sarcomas. Genes Chromosomes Cancer. 2012;51:207–218. doi: 10.1002/gcc.20945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Graham C, Chilton-MacNeill S, Zielenska M, et al. The CIC-DUX4 fusion transcript is present in a subgroup of pediatric primitive round cell sarcomas. Hum Pathol. 2012;43:180–189. doi: 10.1016/j.humpath.2011.04.023. [DOI] [PubMed] [Google Scholar]
- 9.Choi EY, Thomas DG, McHugh JB, et al. Undifferentiated small round cell sarcoma with t(4;19)(q35;q13.1) CIC-DUX4 fusion: a novel highly aggressive soft tissue tumor with distinctive histopathology. Am J Surg Pathol. 2013;37:1379–1386. doi: 10.1097/PAS.0b013e318297a57d. [DOI] [PubMed] [Google Scholar]
- 10.Sugita S, Arai Y, Tonooka A, et al. A novel CIC-FOXO4 gene fusion in undifferentiated small round cell sarcoma: a genetically distinct variant of Ewing-like sarcoma. Am J Surg Pathol. 2014;38:1571–1576. doi: 10.1097/PAS.0000000000000286. [DOI] [PubMed] [Google Scholar]
- 11.Solomon DA, Brohl AS, Khan J, et al. Clinicopathologic features of a second patient with Ewing-like sarcoma harboring CIC-FOXO4 gene fusion. Am J Surg Pathol. 2014;38:1724–1725. doi: 10.1097/PAS.0000000000000335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sankar S, Lessnick SL. Promiscuous partnerships in Ewing’s sarcoma. Cancer Genet. 2011;204:351–365. doi: 10.1016/j.cancergen.2011.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gambarotti M, Benini S, Gamberi G, et al. CIC-DUX4 fusion-positive round-cell sarcomas of soft tissue and bone: a single-institution morphological and molecular analysis of seven cases. Histopathology. 2016;69:624–634. doi: 10.1111/his.12985. [DOI] [PubMed] [Google Scholar]
- 14.Yoshida A, Goto K, Kodaira M, et al. CIC-rearranged Sarcomas: A Study of 20 Cases and Comparisons With Ewing Sarcomas. Am J Surg Pathol. 2016;40:313–323. doi: 10.1097/PAS.0000000000000570. [DOI] [PubMed] [Google Scholar]
- 15.Specht K, Sung YS, Zhang L, et al. Distinct transcriptional signature and immunoprofile of CIC-DUX4 fusion-positive round cell tumors compared to EWSR1-rearranged Ewing sarcomas: further evidence toward distinct pathologic entities. Genes Chromosomes Cancer. 2014;53:622–633. doi: 10.1002/gcc.22172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hung YP, Fletcher CD, Hornick JL. Evaluation of ETV4 and WT1 expression in CIC-rearranged sarcomas and histologic mimics. Mod Pathol. 2016;29:1324–1334. doi: 10.1038/modpathol.2016.140. [DOI] [PubMed] [Google Scholar]
- 17.Le Guellec S, Velasco V, Perot G, et al. ETV4 is a useful marker for the diagnosis of CIC-rearranged undifferentiated round-cell sarcomas: a study of 127 cases including mimicking lesions. Mod Pathol. 2016;29:1523–1531. doi: 10.1038/modpathol.2016.155. [DOI] [PubMed] [Google Scholar]
- 18.Jimenez G, Guichet A, Ephrussi A, et al. Relief of gene repression by torso RTK signaling: role of capicua in Drosophila terminal and dorsoventral patterning. Genes Dev. 2000;14:224–231. [PMC free article] [PubMed] [Google Scholar]
- 19.Bettegowda C, Agrawal N, Jiao Y, et al. Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science. 2011;333:1453–1455. doi: 10.1126/science.1210557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sahm F, Koelsche C, Meyer J, et al. CIC and FUBP1 mutations in oligodendrogliomas, oligoastrocytomas and astrocytomas. Acta Neuropathol. 2012;123:853–860. doi: 10.1007/s00401-012-0993-5. [DOI] [PubMed] [Google Scholar]
- 21.Dissanayake K, Toth R, Blakey J, et al. ERK/p90(RSK)/14-3-3 signalling has an impact on expression of PEA3 Ets transcription factors via the transcriptional repressor capicua. Biochem J. 2011;433:515–525. doi: 10.1042/BJ20101562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jimenez G, Shvartsman SY, Paroush Z. The Capicua repressor–a general sensor of RTK signaling in development and disease. J Cell Sci. 2012;125:1383–1391. doi: 10.1242/jcs.092965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang SC, Zhang L, Sung YS, et al. Recurrent CIC Gene Abnormalities in Angiosarcomas: A Molecular Study of 120 Cases With Concurrent Investigation of PLCG1, KDR, MYC, and FLT4 Gene Alterations. Am J Surg Pathol. 2016;40:645–655. doi: 10.1097/PAS.0000000000000582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sturm D, Orr BA, Toprak UH, et al. New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell. 2016;164:1060–1072. doi: 10.1016/j.cell.2016.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krom YD, Thijssen PE, Young JM, et al. Intrinsic epigenetic regulation of the D4Z4 macrosatellite repeat in a transgenic mouse model for FSHD. PLoS Genet. 2013;9:e1003415. doi: 10.1371/journal.pgen.1003415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van der Maarel SM, Tawil R, Tapscott SJ. Facioscapulohumeral muscular dystrophy and DUX4: breaking the silence. Trends Mol Med. 2011;17:252–258. doi: 10.1016/j.molmed.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Siegele B, Roberts J, Black JO, et al. DUX4 Immunohistochemistry is a Highly Sensitive and Specific Marker for CIC-DUX4 Fusion-positive Round Cell Tumor. Am J Surg Pathol. 2016 doi: 10.1097/PAS.0000000000000772. [In Press] [DOI] [PubMed] [Google Scholar]
- 28.van der Maarel SM, Frants RR. The D4Z4 repeat-mediated pathogenesis of facioscapulohumeral muscular dystrophy. Am J Hum Genet. 2005;76:375–386. doi: 10.1086/428361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang SC, Zhang L, Sung YS, et al. Recurrent CIC Gene Abnormalities in Angiosarcomas: A Molecular Study of 120 Cases With Concurrent Investigation of PLCG1, KDR, MYC, and FLT4 Gene Alterations. Am J Surg Pathol. 2016 doi: 10.1097/PAS.0000000000000582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holland PW, Booth HA, Bruford EA. Classification and nomenclature of all human homeobox genes. BMC Biol. 2007;5:47. doi: 10.1186/1741-7007-5-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.